Blood Ammonia as a Possible Etiological Agent for Alzheimer’s Disease


Abstract
:1. Introduction
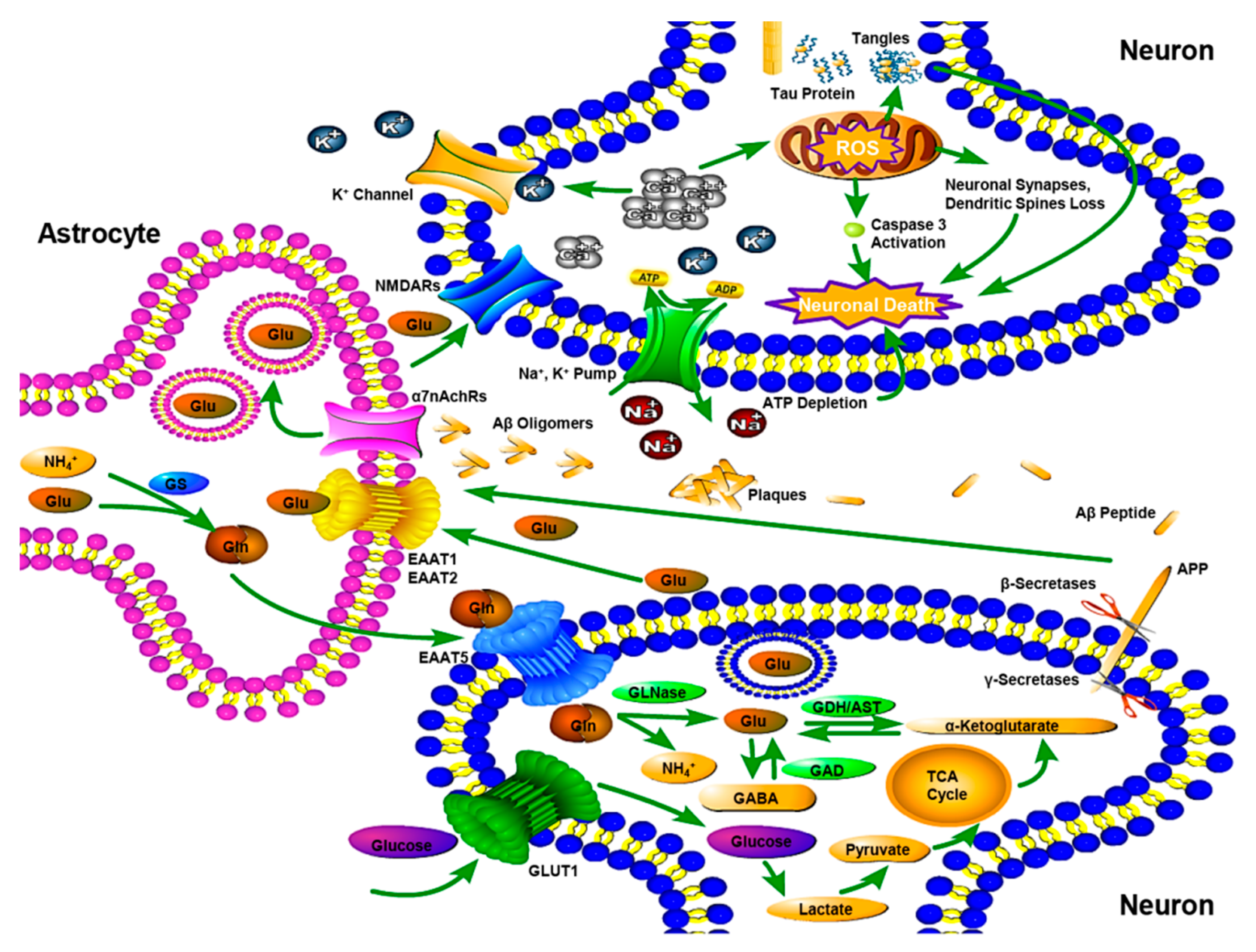
2. The Etiological Mechanism of AD
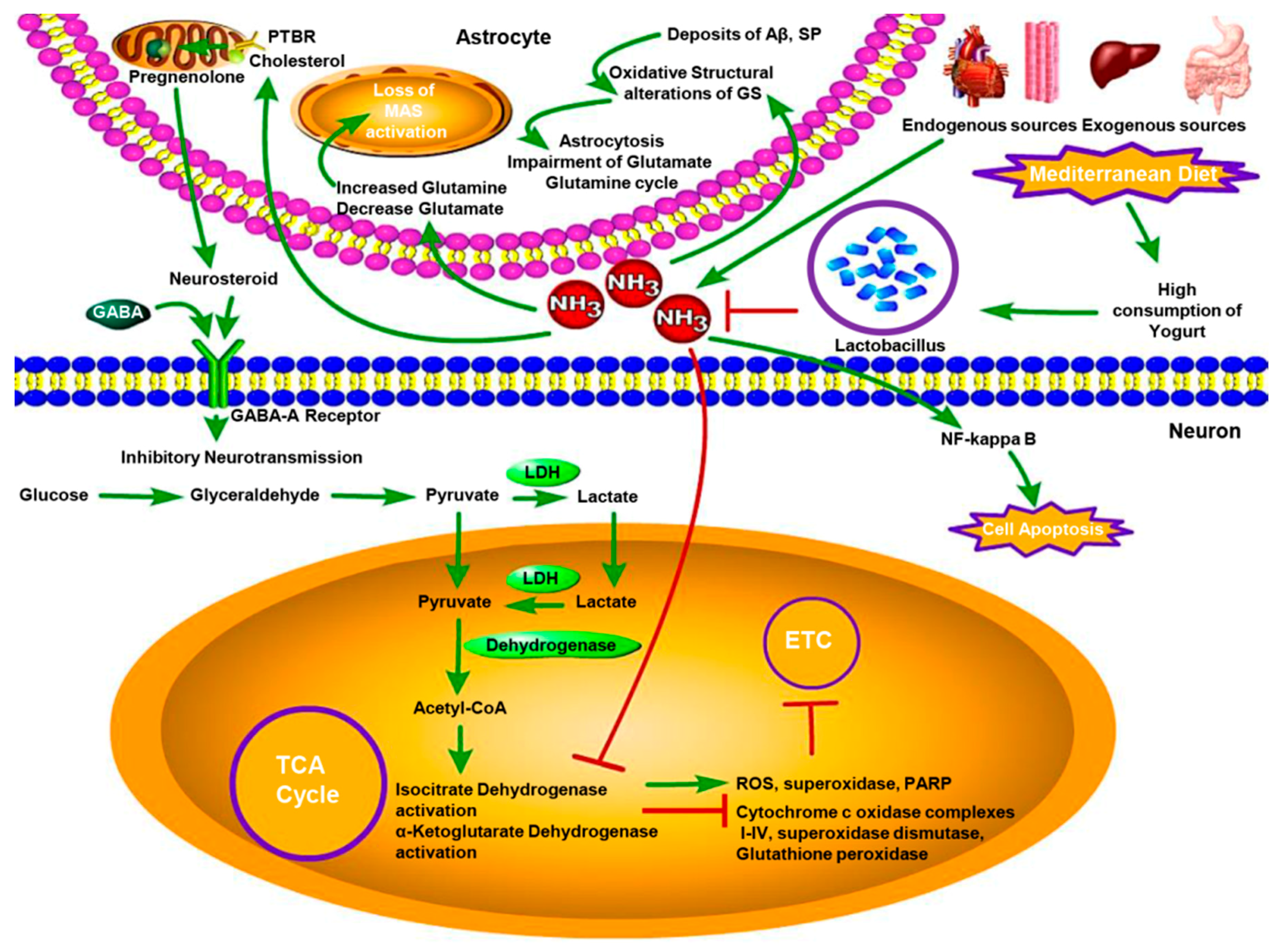
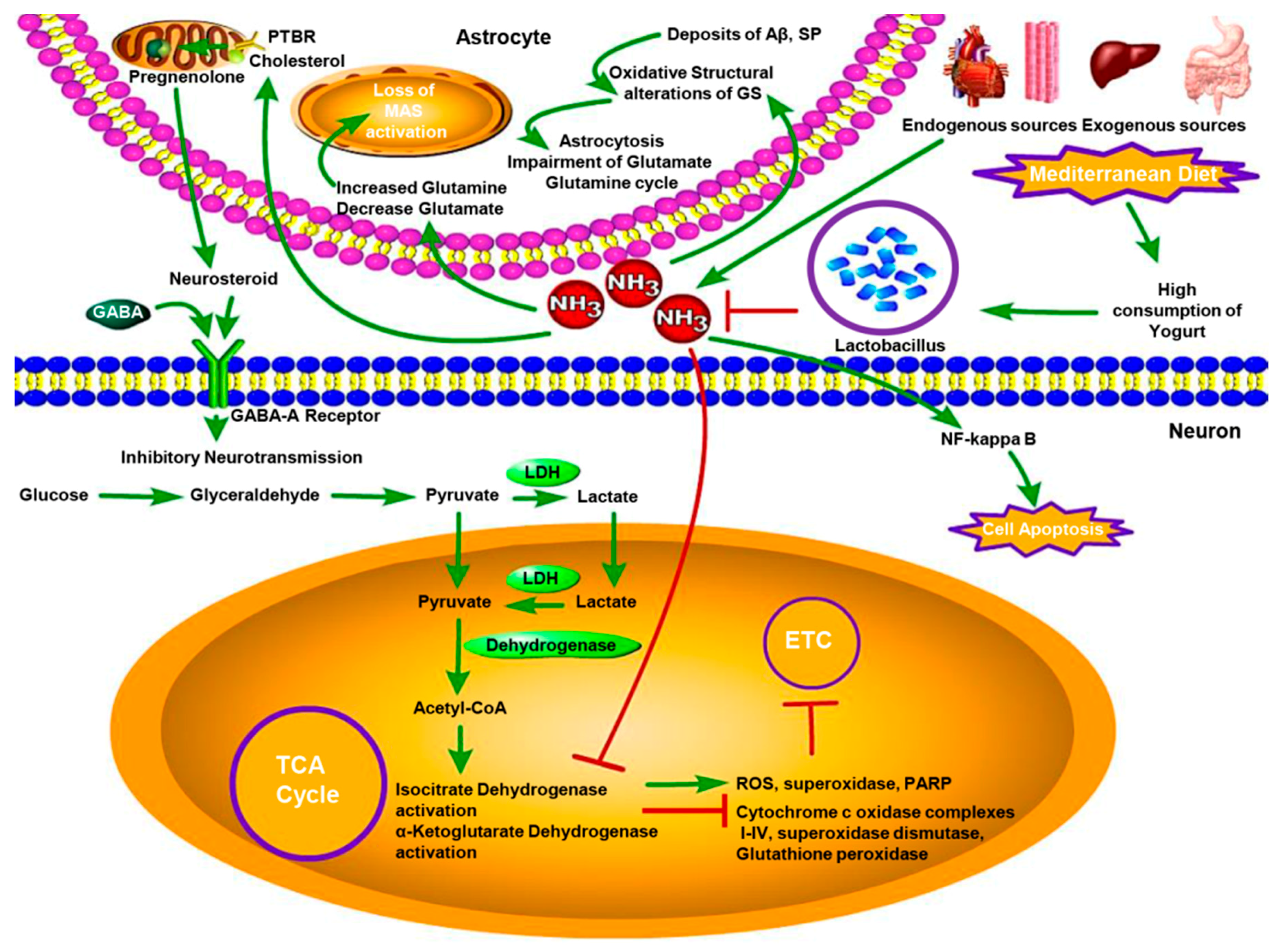
3. Association of AD with Low Degree of Chronic Hyperammonemia
4. Effect of Lactobacilli on Lowering Blood Ammonia Levels
5. Consumption of a Large Quantity of Lactobacilli in the Medi as a Reason for a Lowered Incidence Rate of AD in Mediterranean
6. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Islam, M.A.; Khandker, S.S.; Alam, F.; Khalil, M.I.; Kamal, M.A.; Gan, S.H. Alzheimer’s disease and natural products: Future regimens emerging from nature. Curr. Top. Med. Chem. 2017, 17, 1408–1428. [Google Scholar] [CrossRef] [PubMed]
- Burns, A.; Iliffe, S. Alzheimer’s disease. BMJ 2009, 338, b158. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Agostinho, P.; Cunha, R.A.; Oliveira, C. Neuroinflammation, oxidative stress and the pathogenesis of Alzheimer’s disease. Curr. Pharm. Des. 2010, 16, 2766–2778. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.S.; Anderson, G.M.; Fischer, H.D.; Bell, C.M.; Li, P.; Normand, S.L.; Rochon, P.A. Syncope and its consequences in patients with dementia receiving cholinesterase inhibitors: A population-based cohort study. Arch. Intern. Med. 2009, 169, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Fisman, M.; Ball, M.; Blume, W. Hyperammonemia and Alzheimer’s disease. J. Am. Geriatr. Soc. 1989, 37, 1102. [Google Scholar] [CrossRef] [PubMed]
- Fisman, M.; Gordon, B.; Feleki, V.; Helmes, E.; Appell, J.; Rabheru, K. Hyperammonemia in Alzheimer’s disease. Am. J. Psychiatry 1985, 142, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Alhasson, F.; Das, S.; Seth, R.; Dattaroy, D.; Chandrashekaran, V.; Ryan, C.N.; Chan, L.S.; Testerman, T.; Burch, J.; Hofseth, L.J.; et al. Altered gut microbiome in a mouse model of gulf war illness causes neuroinflammation and intestinal injury via leaky gut and tlr4 activation. PLoS ONE 2017, 12, e0172914. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; D’Souza, R.; Hong, S.T. The role of gut microbiota in the gut-brain axis: Current challenges and perspectives. Protein Cell 2013, 4, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Diamanti, A.P.; Manuela Rosado, M.; Lagana, B.; D’Amelio, R. Microbiota and chronic inflammatory arthritis: An interwoven link. J. Transl. Med. 2016, 14, 233. [Google Scholar] [CrossRef] [PubMed]
- Ghaisas, S.; Maher, J.; Kanthasamy, A. Gut microbiome in health and disease: Linking the microbiome-gut-brain axis and environmental factors in the pathogenesis of systemic and neurodegenerative diseases. Pharmacol. Ther. 2016, 158, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Tremlett, H.; Bauer, K.C.; Appel-Cresswell, S.; Finlay, B.B.; Waubant, E. The gut microbiome in human neurological disease: A review. Ann. Neurol. 2017, 81, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Umbrello, G.; Esposito, S. Microbiota and neurologic diseases: Potential effects of probiotics. J. Transl. Med. 2016, 14, 298. [Google Scholar] [CrossRef] [PubMed]
- Yarandi, S.S.; Peterson, D.A.; Treisman, G.J.; Moran, T.H.; Pasricha, P.J. Modulatory effects of gut microbiota on the central nervous system: How gut could play a role in neuropsychiatric health and diseases. J. Neurogastroenterol. Motil. 2016, 22, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Cruchaga, C.; Haller, G.; Chakraverty, S.; Mayo, K.; Vallania, F.L.; Mitra, R.D.; Faber, K.; Williamson, J.; Bird, T.; Diaz-Arrastia, R.; et al. Rare variants in app, psen1 and psen2 increase risk for ad in late-onset Alzheimer’s disease families. PLoS ONE 2012, 7, e31039. [Google Scholar] [CrossRef]
- Scahill, R.I.; Ridgway, G.R.; Bartlett, J.W.; Barnes, J.; Ryan, N.S.; Mead, S.; Beck, J.; Clarkson, M.J.; Crutch, S.J.; Schott, J.M.; et al. Genetic influences on atrophy patterns in familial Alzheimer’s disease: A comparison of app and psen1 mutations. J. Alzheimers Dis. 2013, 35, 199–212. [Google Scholar] [PubMed]
- Anand, R.; Gill, K.D.; Mahdi, A.A. Therapeutics of Alzheimer’s disease: Past, present and future. Neuropharmacology 2014, 76 Pt A, 27–50. [Google Scholar] [CrossRef] [PubMed]
- Dal Pra, I.; Chiarini, A.; Gui, L.; Chakravarthy, B.; Pacchiana, R.; Gardenal, E.; Whitfield, J.F.; Armato, U. Do astrocytes collaborate with neurons in spreading the “infectious” abeta and tau drivers of Alzheimer’s disease? Neuroscientist 2015, 21, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.H.; Nagao, T.; Gouras, G.K. Plaque formation and the intraneuronal accumulation of beta-amyloid in Alzheimer’s disease. Pathol. Int. 2017, 67, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J. The amyloid hypothesis for Alzheimer’s disease: A critical reappraisal. J. Neurochem. 2009, 110, 1129–1134. [Google Scholar] [CrossRef] [PubMed]
- Priller, C.; Bauer, T.; Mitteregger, G.; Krebs, B.; Kretzschmar, H.A.; Herms, J. Synapse formation and function is modulated by the amyloid precursor protein. J. Neurosci. 2006, 26, 7212–7221. [Google Scholar] [CrossRef] [PubMed]
- Salomone, S.; Caraci, F.; Leggio, G.M.; Fedotova, J.; Drago, F. New pharmacological strategies for treatment of Alzheimer’s disease: Focus on disease modifying drugs. Br. J. Clin. Pharmacol. 2012, 73, 504–517. [Google Scholar] [CrossRef] [PubMed]
- Talantova, M.; Sanz-Blasco, S.; Zhang, X.; Xia, P.; Akhtar, M.W.; Okamoto, S.; Dziewczapolski, G.; Nakamura, T.; Cao, G.; Pratt, A.E.; et al. Abeta induces astrocytic glutamate release, extrasynaptic nmda receptor activation, and synaptic loss. Proc. Natl. Acad. Sci. USA 2013, 110, E2518–E2527. [Google Scholar] [CrossRef] [PubMed]
- Tuppo, E.E.; Arias, H.R. The role of inflammation in Alzheimer’s disease. Int. J. Biochem. Cell Biol. 2005, 37, 289–305. [Google Scholar] [CrossRef] [PubMed]
- Kurz, A.; Perneczky, R. Novel insights for the treatment of Alzheimer’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Marcaida, G.; Felipo, V.; Hermenegildo, C.; Minana, M.D.; Grisolia, S. Acute ammonia toxicity is mediated by the nmda type of glutamate receptors. FEBS Lett. 1992, 296, 67–68. [Google Scholar] [CrossRef]
- Zempel, H.; Mandelkow, E. Lost after translation: Missorting of tau protein and consequences for Alzheimer disease. Trends Neurosci. 2014, 37, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Mudher, A.; Lovestone, S. Alzheimer’s disease-do tauists and baptists finally shake hands? Trends Neurosci. 2002, 25, 22–26. [Google Scholar] [CrossRef]
- Wang, J.Z.; Xia, Y.Y.; Grundke-Iqbal, I.; Iqbal, K. Abnormal hyperphosphorylation of tau: Sites, regulation, and molecular mechanism of neurofibrillary degeneration. J. Alzheimers Dis. 2013, 33 (Suppl. 1), S123–S139. [Google Scholar] [PubMed]
- Iqbal, K.; Alonso Adel, C.; Chen, S.; Chohan, M.O.; El-Akkad, E.; Gong, C.X.; Khatoon, S.; Li, B.; Liu, F.; Rahman, A.; et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta 2005, 1739, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Roquilly, A.; Perbet, S.; Simonneau, F.; Cinotti, R.; Sebille, V.; Volteau, C.; Gratas, C.; Minet-Quinard, R.; Loutrel, O.; Rozec, B.; et al. Ammonia plasma concentration and prolonged infusion of remifentanil in patients with acute kidney injury. Miner. Anestesiol. 2013, 79, 884–890. [Google Scholar]
- Cooper, A.J.; Plum, F. Biochemistry and physiology of brain ammonia. Physiol. Rev. 1987, 67, 440–519. [Google Scholar] [CrossRef] [PubMed]
- Norenberg, M.D.; Martinez-Hernandez, A. Fine structural localization of glutamine synthetase in astrocytes of rat brain. Brain Res. 1979, 161, 303–310. [Google Scholar] [CrossRef]
- Robinson, S.R. Neuronal expression of glutamine synthetase in Alzheimer’s disease indicates a profound impairment of metabolic interactions with astrocytes. Neurochem. Int. 2000, 36, 471–482. [Google Scholar] [CrossRef]
- Suarez, I.; Bodega, G.; Fernandez, B. Glutamine synthetase in brain: Effect of ammonia. Neurochem. Int. 2002, 41, 123–142. [Google Scholar] [CrossRef]
- Branconnier, R.J.; Dessain, E.C.; McNiff, M.E.; Cole, J.O. Blood ammonia and Alzheimer’s disease. Am. J. Psychiatry 1986, 143, 1313–1314. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.I.; Nunomura, A.; Nakamura, M.; Takeda, A.; Shenk, J.C.; Aliev, G.; Smith, M.A.; Perry, G. Nucleic acid oxidation in Alzheimer disease. Free Radic. Biol. Med. 2008, 44, 1493–1505. [Google Scholar] [CrossRef] [PubMed]
- Esteves, A.R.; Arduino, D.M.; Swerdlow, R.H.; Oliveira, C.R.; Cardoso, S.M. Oxidative stress involvement in alpha-synuclein oligomerization in parkinson’s disease cybrids. Antioxid. Redox Signal. 2009, 11, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Kosenko, E.A.; Solomadin, I.N.; Tikhonova, L.A.; Reddy, V.P.; Aliev, G.; Kaminsky, Y.G. Pathogenesis of Alzheimer disease: Role of oxidative stress, amyloid-beta peptides, systemic ammonia and erythrocyte energy metabolism. CNS Neurol. Disord. Drug Targets 2014, 13, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Cadonic, C.; Sabbir, M.G.; Albensi, B.C. Mechanisms of mitochondrial dysfunction in Alzheimer’s disease. Mol. Neurobiol. 2016, 53, 6078–6090. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Perry, G.; Smith, M.A.; Wang, X. Abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J. Alzheimers Dis. 2013, 33 (Suppl. 1), S253–S262. [Google Scholar] [PubMed]
- Bubber, P.; Haroutunian, V.; Fisch, G.; Blass, J.P.; Gibson, G.E. Mitochondrial abnormalities in Alzheimer brain: Mechanistic implications. Ann. Neurol. 2005, 57, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Tikhonova, L.A.; Kaminsky, Y.G.; Reddy, V.P.; Li, Y.; Solomadin, I.N.; Kosenko, E.A.; Aliev, G. Impact of amyloid beta25-35 on membrane stability, energy metabolism, and antioxidant enzymes in erythrocytes. Am. J. Alzheimers Dis. Other Demen. 2014, 29, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L.; Kala, G. Energy metabolism in brain cells: Effects of elevated ammonia concentrations. Metab. Brain Dis. 2007, 22, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Rama Rao, K.V.; Jayakumar, A.R.; Tong, X.; Alvarez, V.M.; Norenberg, M.D. Marked potentiation of cell swelling by cytokines in ammonia-sensitized cultured astrocytes. J. Neuroinflamm. 2010, 7, 66. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, R.F. The astrocytic (“peripheral-type”) benzodiazepine receptor: Role in the pathogenesis of portal-systemic encephalopathy. Neurochem. Int. 2000, 36, 411–416. [Google Scholar] [CrossRef]
- Norenberg, M.D. Astroglial dysfunction in hepatic encephalopathy. Metab. Brain Dis. 1998, 13, 319–335. [Google Scholar] [CrossRef] [PubMed]
- Felipo, V.; Butterworth, R.F. Neurobiology of ammonia. Prog. Neurobiol. 2002, 67, 259–279. [Google Scholar] [CrossRef]
- Buzanska, L.; Zablocka, B.; Dybel, A.; Domanska-Janik, K.; Albrecht, J. Delayed induction of apoptosis by ammonia in c6 glioma cells. Neurochem. Int. 2000, 37, 287–297. [Google Scholar] [CrossRef]
- Shi, C.; Zhu, X.; Wang, J.; Long, D. Intromitochondrial ikappab/nf-kappab signaling pathway is involved in amyloid beta peptide-induced mitochondrial dysfunction. J. Bioenerg. Biomembr. 2014, 46, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Sinke, A.P.; Jayakumar, A.R.; Panickar, K.S.; Moriyama, M.; Reddy, P.V.; Norenberg, M.D. Nfkappab in the mechanism of ammonia-induced astrocyte swelling in culture. J. Neurochem. 2008, 106, 2302–2311. [Google Scholar] [PubMed]
- O’Donnell, M.J. Mechanisms of excretion and ion transport in invertebrates. In Comprehensive Physiology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010. [Google Scholar]
- Marcaggi, P.; Coles, J.A. Ammonium in nervous tissue: Transport across cell membranes, fluxes from neurons to glial cells, and role in signalling. Prog. Neurobiol. 2001, 64, 157–183. [Google Scholar] [CrossRef]
- Back, A.; Tupper, K.Y.; Bai, T.; Chiranand, P.; Goldenberg, F.D.; Frank, J.I.; Brorson, J.R. Ammonia-induced brain swelling and neurotoxicity in an organotypic slice model. Neurol. Res. 2011, 33, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Marteau, P. Butyrate-producing bacteria as pharmabiotics for inflammatory bowel disease. Gut 2013, 62, 1673. [Google Scholar] [CrossRef] [PubMed]
- Agostini, L.; Down, P.F.; Murison, J.; Wrong, O.M. Faecal ammonia and ph during lactulose administration in man: Comparison with other cathartics. Gut 1972, 13, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Itzhak, Y.; Roig-Cantisano, A.; Dombro, R.S.; Norenberg, M.D. Acute liver failure and hyperammonemia increase peripheral-type benzodiazepine receptor binding and pregnenolone synthesis in mouse brain. Brain Res. 1995, 705, 345–348. [Google Scholar] [CrossRef]
- Jia, L.; Zhang, M.-H. Comparison of probiotics and lactulose in the treatment of minimal hepatic encephalopathy in rats. WJG 2005, 11, 908–911. [Google Scholar] [CrossRef] [PubMed]
- Nicaise, C.; Prozzi, D.; Viaene, E.; Moreno, C.; Gustot, T.; Quertinmont, E.; Demetter, P.; Suain, V.; Goffin, P.; Deviere, J.; et al. Control of acute, chronic, and constitutive hyperammonemia by wild-type and genetically engineered lactobacillus plantarum in rodents. Hepatology 2008, 48, 1184–1192. [Google Scholar] [CrossRef] [PubMed]
- Solga, S.F. Probiotics can treat hepatic encephalopathy. Med. Hypotheses 2003, 61, 307–313. [Google Scholar] [CrossRef]
- Luo, J.; Wang, T.; Liang, S.; Hu, X.; Li, W.; Jin, F. Ingestion of lactobacillus strain reduces anxiety and improves cognitive function in the hyperammonemia rat. Sci. China Life Sci. 2014, 57, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Akbari, E.; Asemi, Z.; Daneshvar Kakhaki, R.; Bahmani, F.; Kouchaki, E.; Tamtaji, O.R.; Hamidi, G.A.; Salami, M. Effect of probiotic supplementation on cognitive function and metabolic status in Alzheimer’s disease: A. randomized, double-blind and controlled trial. Front Aging Neurosci. 2016, 8, 256. [Google Scholar] [CrossRef] [PubMed]
- Loguercio, C.; Abbiati, R.; Rinaldi, M.; Romano, A.; Del Vecchio Blanco, C.; Coltorti, M. Long-term effects of enterococcus faecium sf68 versus lactulose in the treatment of patients with cirrhosis and grade 1-2 hepatic encephalopathy. J. Hepatol. 1995, 23, 39–46. [Google Scholar] [CrossRef]
- Liu, Q.; Duan, Z.P.; Ha, D.K.; Bengmark, S.; Kurtovic, J.; Riordan, S.M. Synbiotic modulation of gut flora: Effect on minimal hepatic encephalopathy in patients with cirrhosis. Hepatology 2004, 39, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Mittal, V.V.; Sharma, B.C.; Sharma, P.; Sarin, S.K. A randomized controlled trial comparing lactulose, probiotics, and l-ornithine l-aspartate in treatment of minimal hepatic encephalopathy. Eur. J. Gastroenterol. Hepatol. 2011, 23, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.S.; Saeian, K.; Christensen, K.M.; Hafeezullah, M.; Varma, R.R.; Franco, J.; Pleuss, J.A.; Krakower, G.; Hoffmann, R.G.; Binion, D.G. Probiotic yogurt for the treatment of minimal hepatic encephalopathy. Am. J. Gastroenterol. 2008, 103, 1707–1715. [Google Scholar] [CrossRef] [PubMed]
- Willett, W.C.; Sacks, F.; Trichopoulou, A.; Drescher, G.; Ferro-Luzzi, A.; Helsing, E.; Trichopoulos, D. Mediterranean diet pyramid: A cultural model for healthy eating. Am. J. Clin. Nutr. 1995, 61, 1402s–1406s. [Google Scholar] [CrossRef] [PubMed]
- Trichopoulou, A.; Costacou, T.; Bamia, C.; Trichopoulos, D. Adherence to a mediterranean diet and survival in a greek population. N. Engl. J. Med. 2003, 348, 2599–2608. [Google Scholar] [CrossRef] [PubMed]
- Thaipisuttikul, P.; Galvin, J.E. Use of medical foods and nutritional approaches in the treatment of Alzheimer’s disease. Clin. Pract. 2012, 9, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Vascular factors and risk of dementia: Design of the three-city study and baseline characteristics of the study population. Neuroepidemiology 2003, 22, 316–325.
- Feart, C.; Samieri, C.; Rondeau, V.; Amieva, H.; Portet, F.; Dartigues, J.F.; Scarmeas, N.; Barberger-Gateau, P. Adherence to a mediterranean diet, cognitive decline, and risk of dementia. JAMA 2009, 302, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Dontas, A.S.; Zerefos, N.S.; Panagiotakos, D.B.; Valis, D.A. Mediterranean diet and prevention of coronary heart disease in the elderly. Clin. Intervent. Aging 2007, 2, 109–115. [Google Scholar] [CrossRef]
- Scarmeas, N.; Stern, Y.; Mayeux, R.; Manly, J.; Schupf, N.; Luchsinger, J.A. Mediterranean diet and mild cognitive impairment. Arch. Neurol. 2009, 66, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, J.A.; Mayeux, R. Dietary factors and Alzheimer’s disease. Lancet Neurol. 2004, 3, 579–587. [Google Scholar] [CrossRef]
- Mattson, M.P. Emerging neuroprotective strategies for Alzheimer’s disease: Dietary restriction, telomerase activation, and stem cell therapy. Exp. Gerontol. 2000, 35, 489–502. [Google Scholar] [CrossRef]
- Morris, M.C.; Tangney, C.C.; Wang, Y.; Sacks, F.M.; Bennett, D.A.; Aggarwal, N.T. Mind diet associated with reduced incidence of Alzheimer’s disease. Alzheimers Dement. 2015, 11, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Scarmeas, N.; Stern, Y.; Tang, M.-X.; Mayeux, R.; Luchsinger, J.A. Mediterranean diet and risk for Alzheimer’s disease. Ann. Neurol. 2006, 59, 912–921. [Google Scholar] [CrossRef] [PubMed]
- Tsivgoulis, G.; Judd, S.; Letter, A.J.; Alexandrov, A.V.; Howard, G.; Nahab, F.; Unverzagt, F.W.; Moy, C.; Howard, V.J.; Kissela, B.; et al. Adherence to a mediterranean diet and risk of incident cognitive impairment. Neurology 2013, 80, 1684–1692. [Google Scholar] [CrossRef] [PubMed]
- Tangney, C.C.; Kwasny, M.J.; Li, H.; Wilson, R.S.; Evans, D.A.; Morris, M.C. Adherence to a mediterranean-type dietary pattern and cognitive decline in a community population. Am. J. Clin. Nutr. 2011, 93, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Perez, J.M.; Morillas-Ruiz, J.M. A review: Inflammatory process in Alzheimer’s disease, role of cytokines. Sci. World J. 2012, 2012, 756357. [Google Scholar] [CrossRef] [PubMed]
- Gardener, S.; Gu, Y.; Rainey-Smith, S.R.; Keogh, J.B.; Clifton, P.M.; Mathieson, S.L.; Taddei, K.; Mondal, A.; Ward, V.K.; Scarmeas, N.; et al. Adherence to a mediterranean diet and Alzheimer’s disease risk in an australian population. Transl. Psychiatry 2012, 2, e164. [Google Scholar] [CrossRef] [PubMed]
- Chrysohoou, C.; Panagiotakos, D.B.; Pitsavos, C.; Das, U.N.; Stefanadis, C. Adherence to the mediterranean diet attenuates inflammation and coagulation process in healthy adults: The attica study. J. Am. Coll. Cardiol. 2004, 44, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Ischiropoulos, H.; Lee, V.M.; Trojanowski, J.Q. The relationship between oxidative/nitrative stress and pathological inclusions in Alzheimer’s and parkinson’s diseases. Free Radic. Biol. Med. 2002, 32, 1264–1275. [Google Scholar] [CrossRef]
- Adlimoghaddam, A.; Sabbir, M.G.; Albensi, B.C. Ammonia as a potential neurotoxic factor in Alzheimer’s disease. Front. Mol. Neurosci. 2016, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Kosenko, E.A.; Venediktova, N.I.; Kaminsky, Y.G. Calcium and ammonia stimulate monoamine oxidase a activity in brain mitochondria. Biol. Bull. Russ. Acad. Sci. 2003, 30, 449–452. [Google Scholar] [CrossRef]
- Tabernero, M.; Venema, K.; Maathuis, A.J.; Saura-Calixto, F.D. Metabolite production during in vitro colonic fermentation of dietary fiber: Analysis and comparison of two european diets. J. Agric. Food Chem. 2011, 59, 8968–8975. [Google Scholar] [CrossRef] [PubMed]
- Trichopoulou, A.; Lagiou, P. Healthy traditional mediterranean diet: An expression of culture, history, and lifestyle. Nutr. Rev. 1997, 55, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Walker, W.A. Role of nutrients and bacterial colonization in the development of intestinal host defense. J. Pediatr. Gastroenterol. Nutr. 2000, 30 (Suppl. 2), S2–S7. [Google Scholar] [CrossRef] [PubMed]
- Imani Fooladi, A.A.; Mahmoodzadeh Hosseini, H.; Nourani, M.R.; Khani, S.; Alavian, S.M. Probiotic as a novel treatment strategy against liver disease. Hepatitis Mon. 2013, 13, e7521. [Google Scholar] [CrossRef] [PubMed]
- Alfawaz, H.A.; Aljumah, A.A. What improves minimal hepatic encephalopathy: Probiotic yogurt, protein restriction or nonabsorbable disaccharides? Saudi J. Gastroenterol. 2012, 18, 153–154. [Google Scholar] [PubMed]

{kind=link}
{kind=link}
| Method | Subjects | Treatment | Duration of Treatment | Result | References |
|---|---|---|---|---|---|
| RCT | 40 | Enterococcus faecium SF68 or lactulose | Three periods of 4 weeks with 2 weeks of drug-free intervals | Reduction in blood ammonia levels, improved neurocoginitive (Reitan’s) test | [63] |
| RCT | 55 | synbiotic preparation (n = 20), fermentable fiber alone (n = 20), or placebo (n = 15) | 30 days | Increment in the fecal content of non-urease-producing Lactobacillus species, significant reduction in blood ammonia levels and reversal of minimal hepatic encephalopathy (MHE) in 50% of patients | [64] |
| RCT | 160 | lactulose, probiotics and L-ornithine L-aspartate (LOLA) | 3 months | Reduction in blood ammonia levels, significantly improved MHE | [65] |
| RCT | 25 | probiotic yogurt | 2 months | significant rate of MHE reversal | [66] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, Y.Y.; Singh, P.; Chung, H.-J.; Hong, S.-T. Blood Ammonia as a Possible Etiological Agent for Alzheimer’s Disease. Nutrients 2018, 10, 564. https://doi.org/10.3390/nu10050564
Jin YY, Singh P, Chung H-J, Hong S-T. Blood Ammonia as a Possible Etiological Agent for Alzheimer’s Disease. Nutrients. 2018; 10(5):564. https://doi.org/10.3390/nu10050564
Chicago/Turabian StyleJin, Yan Yan, Parul Singh, Hea-Jong Chung, and Seong-Tschool Hong. 2018. "Blood Ammonia as a Possible Etiological Agent for Alzheimer’s Disease" Nutrients 10, no. 5: 564. https://doi.org/10.3390/nu10050564
APA StyleJin, Y. Y., Singh, P., Chung, H.-J., & Hong, S.-T. (2018). Blood Ammonia as a Possible Etiological Agent for Alzheimer’s Disease. Nutrients, 10(5), 564. https://doi.org/10.3390/nu10050564