Coordination of GPR40 and Ketogenesis Signaling by Medium Chain Fatty Acids Regulates Beta Cell Function


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell and Islet Culture
2.2. RNA Isolation and qPCR
2.3. Immunocytochemistry and Confocal Microscopy
2.4. IP1 Measurement Assay
2.5. Ketone Measurements
2.6. Palmitate β-Oxidation
2.7. Mitochondrial Membrane Potential
2.8. Insulin Secretion Assay
2.9. Animal Study
2.10. Statistics
3. Results
3.1. Medium Chain Fatty Acid Supplementation Improves β-Cell Function in Aged Rats
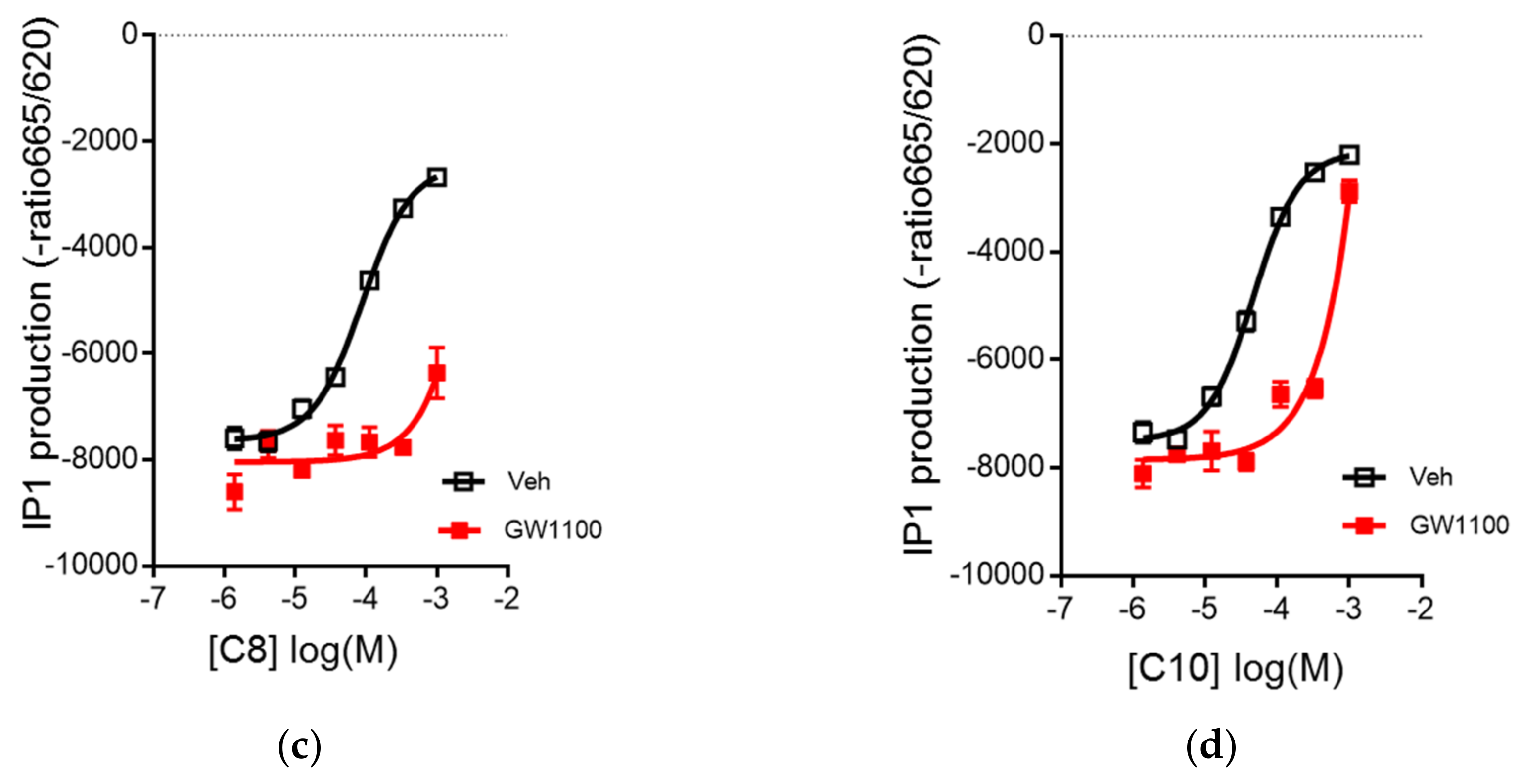
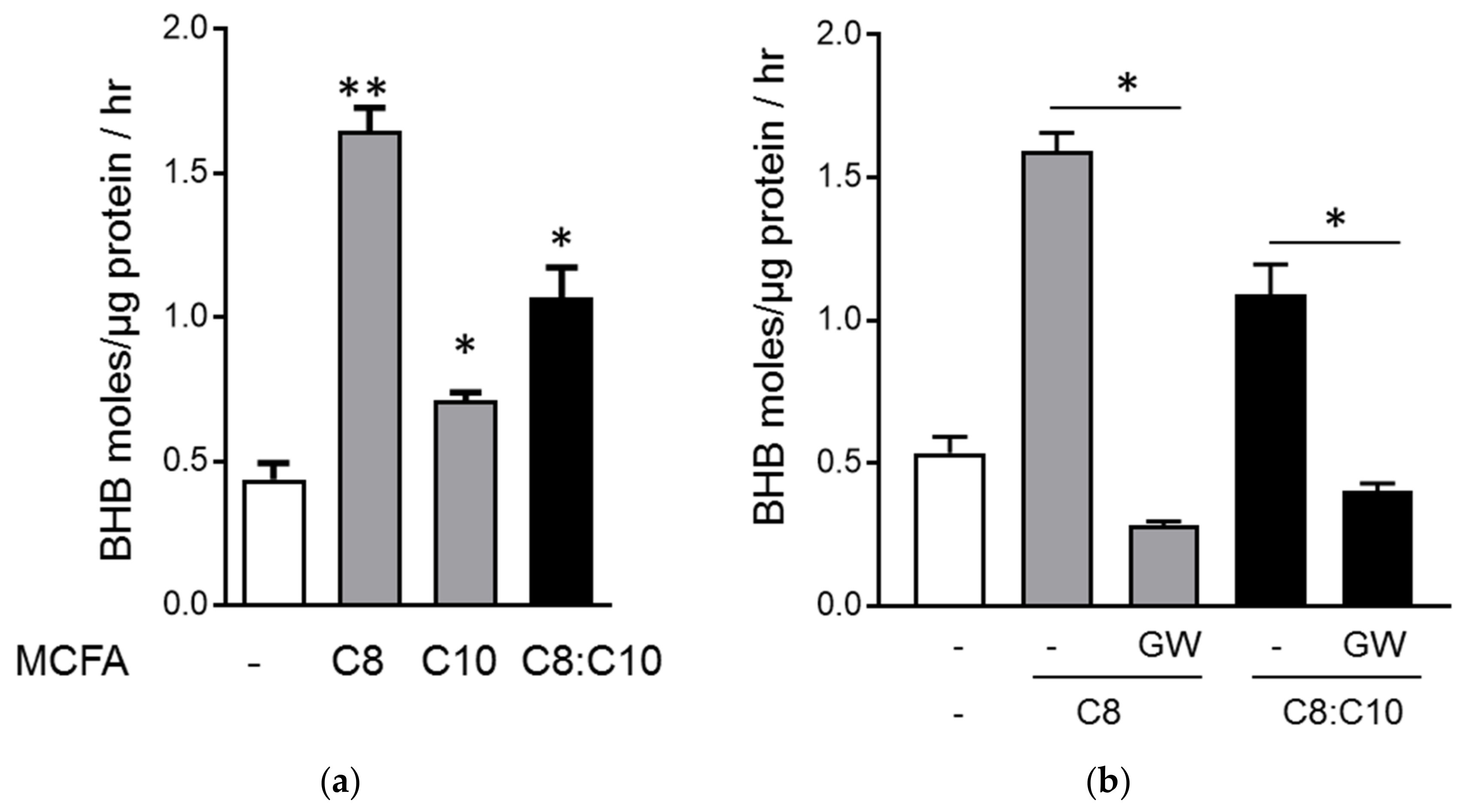
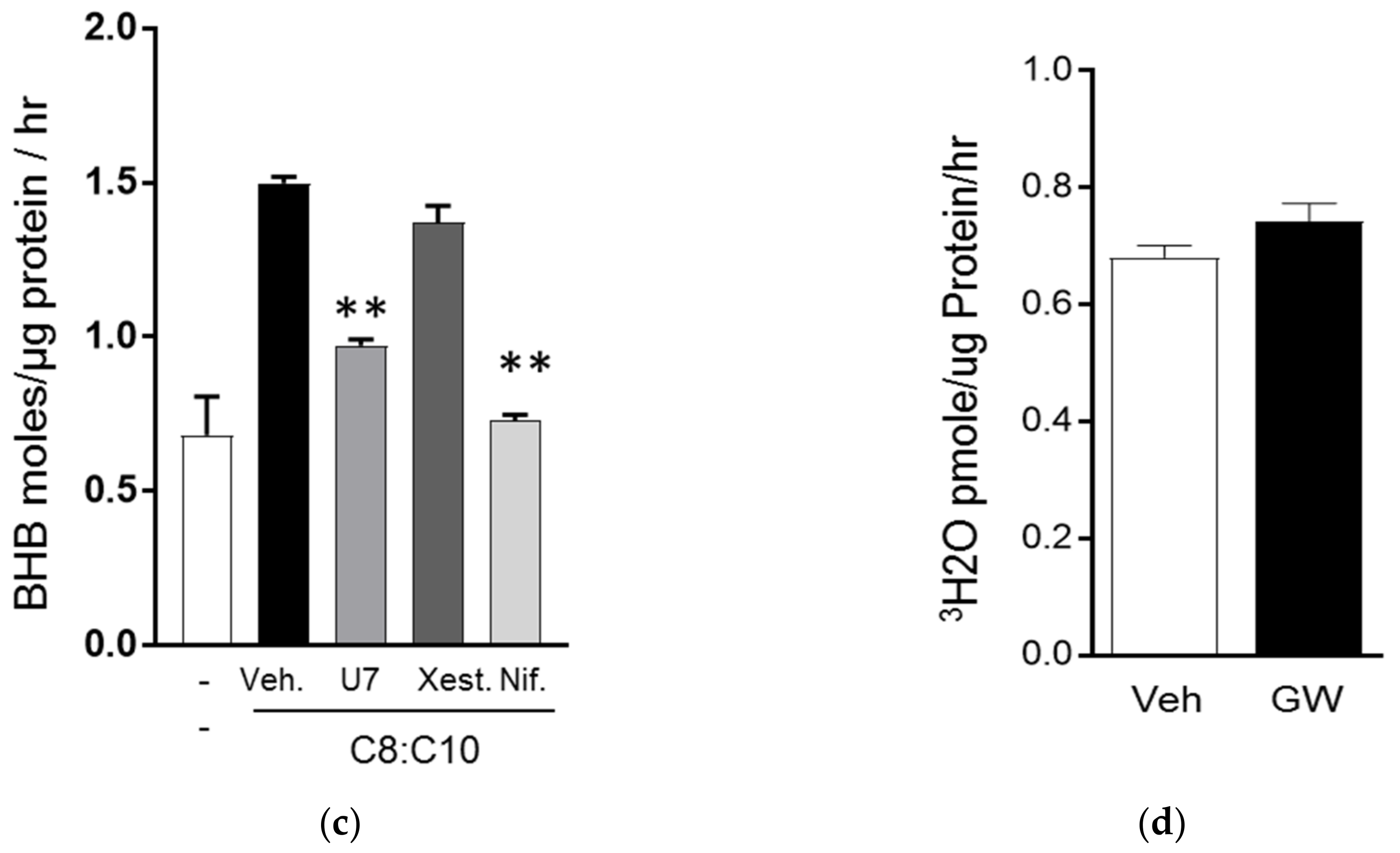
3.2. C8 and C10 Differentially Activate FFAR1/GPR40 and Ketogenesis in β Cells
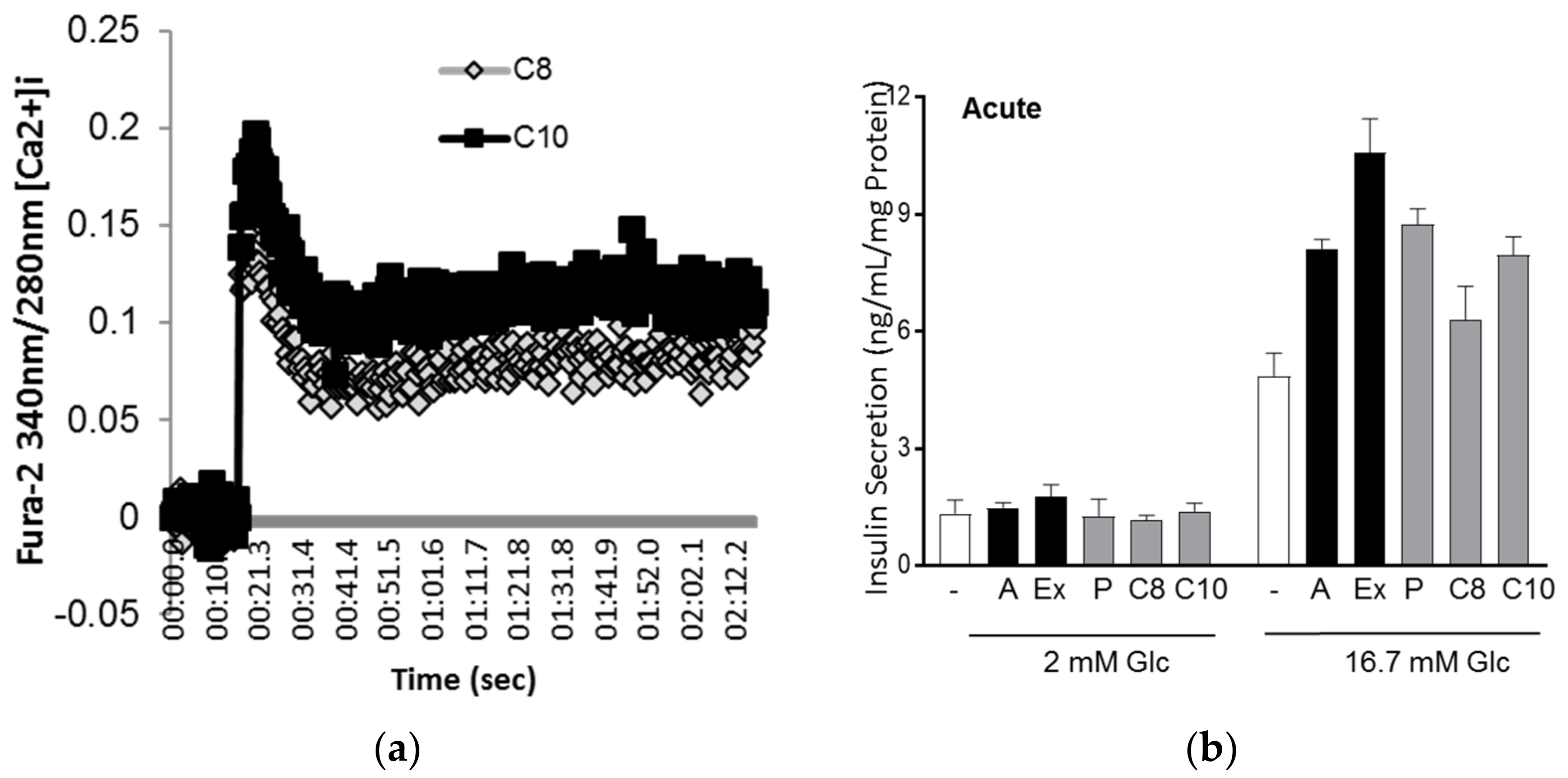
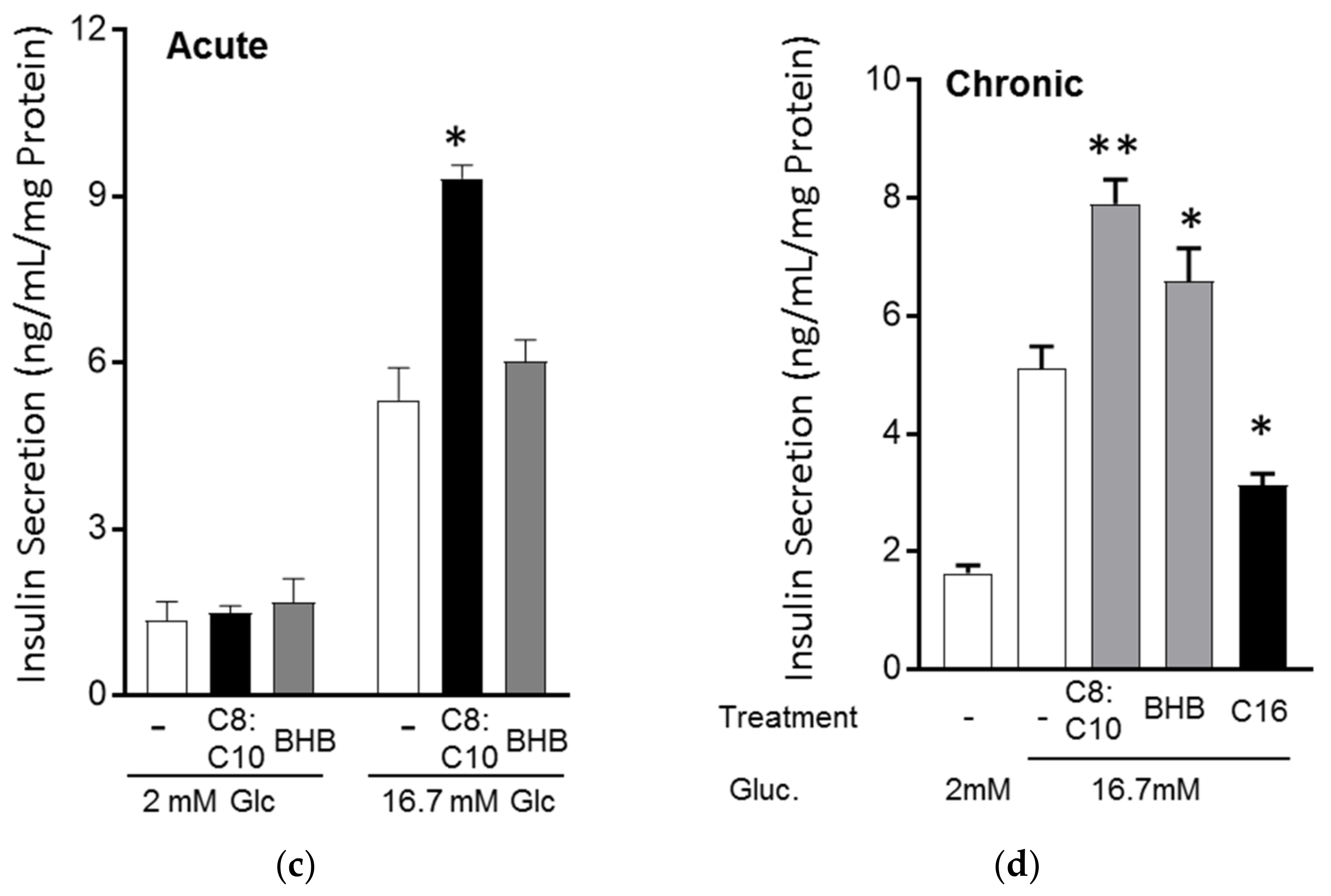
3.3. MCFA Acutely Increased Insulin Secretion with Beneficial Long-Term Effects
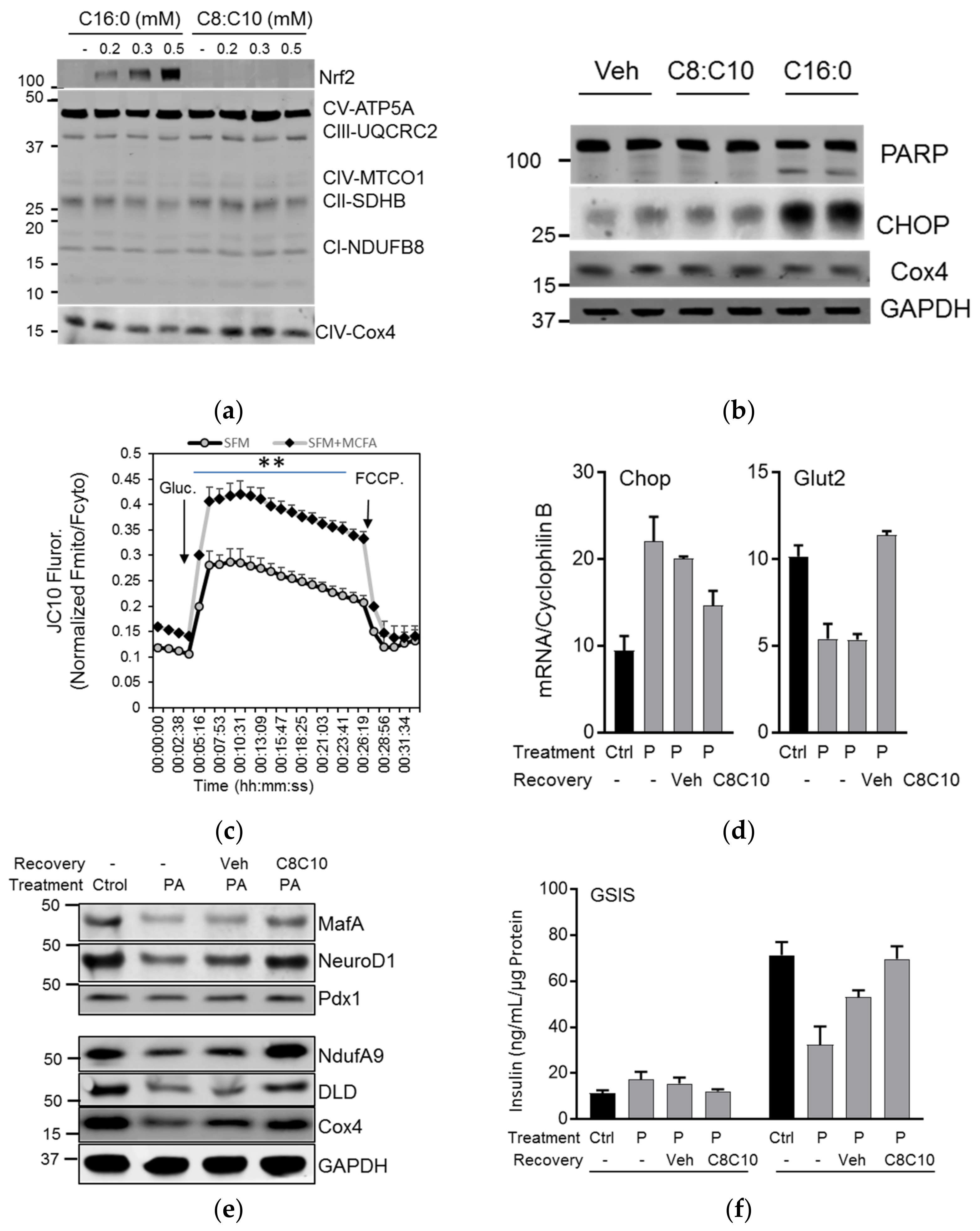
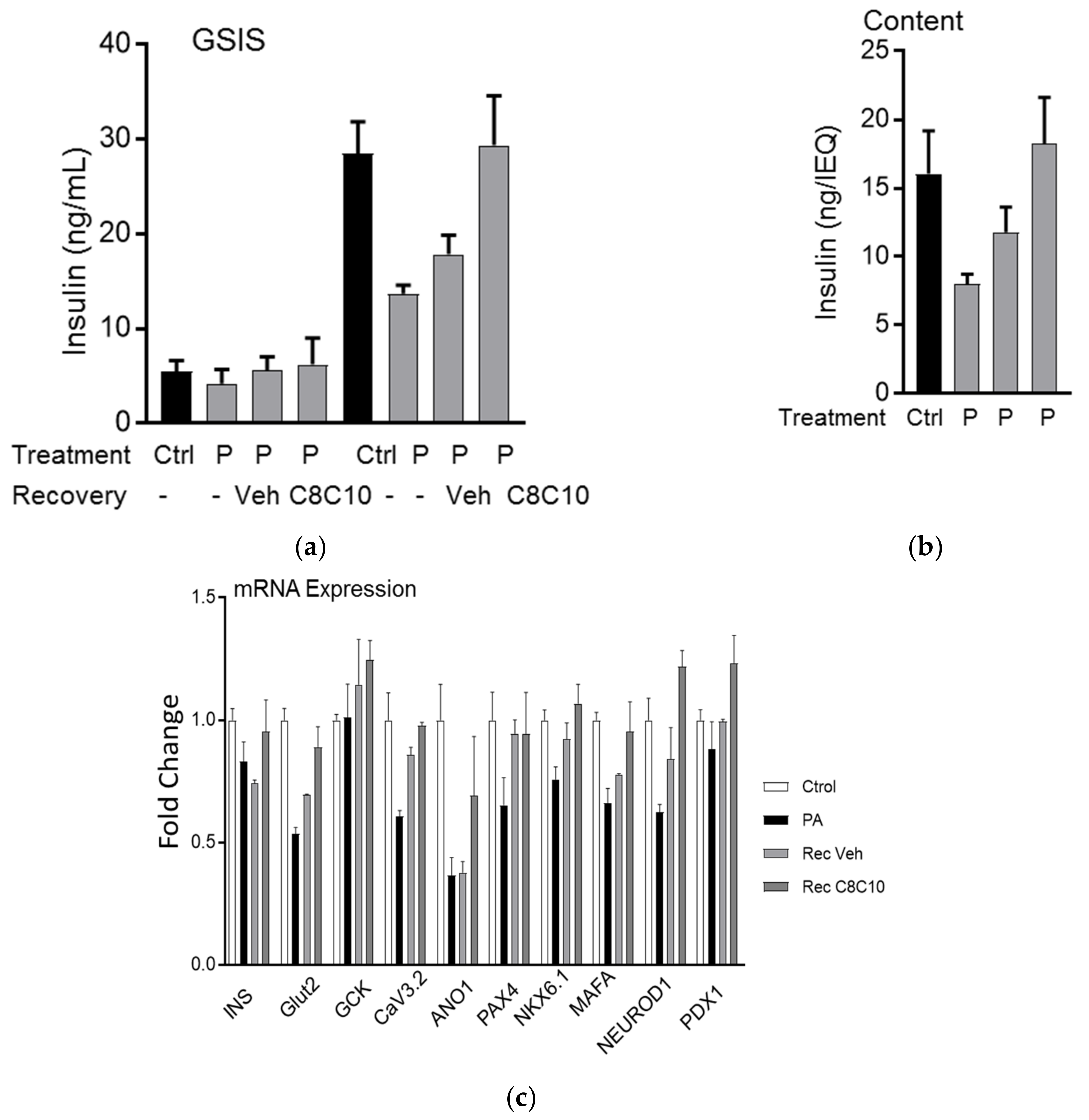
3.4. MCFA Help β-Cells Recover from Dysfunction
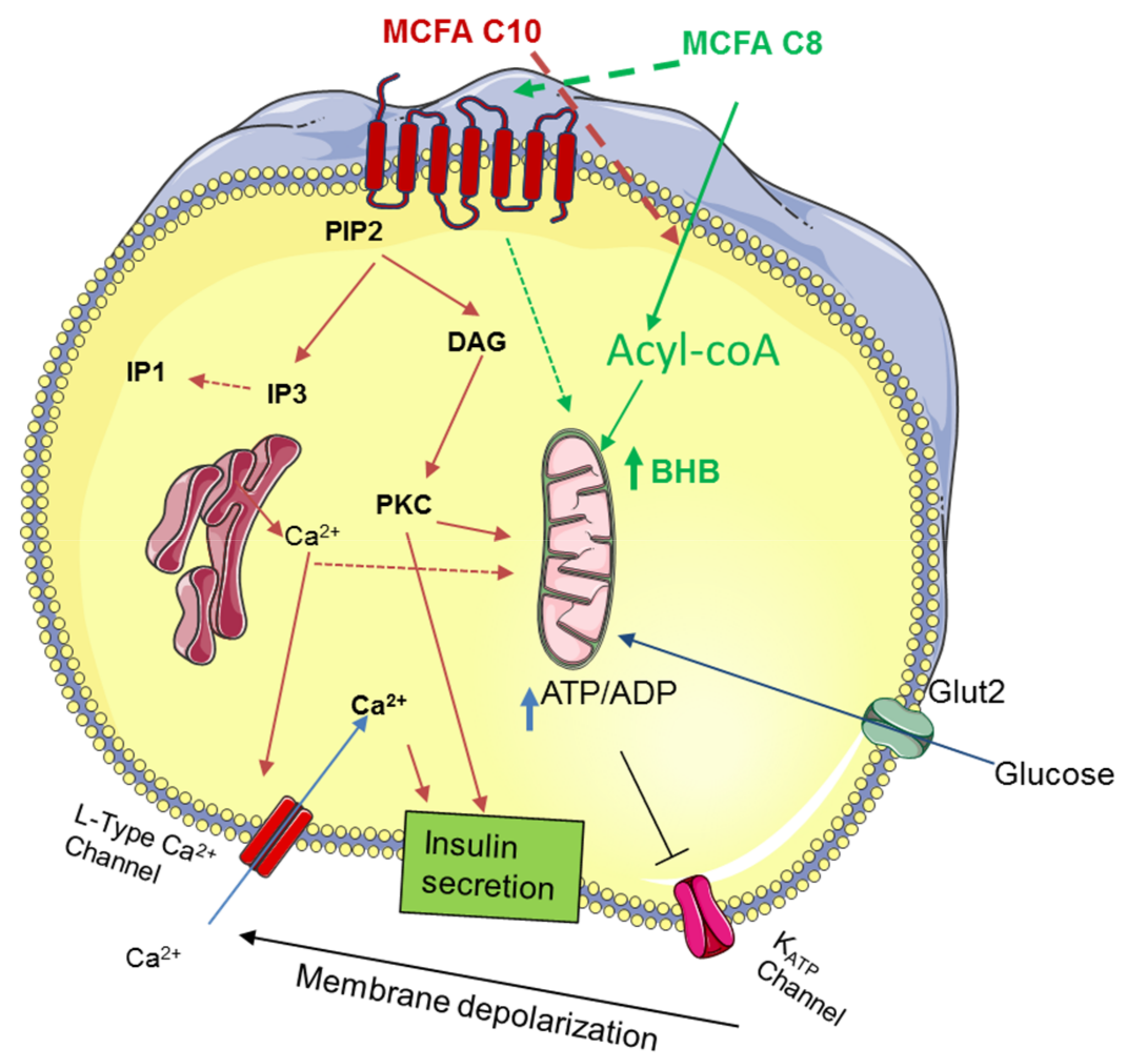
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cowie, C.C.; Rust, K.F.; Byrd-Holt, D.D.; Eberhardt, M.S.; Flegal, K.M.; Engelgau, M.M.; Saydah, S.H.; Williams, D.E.; Geiss, L.S.; Gregg, E.W. Prevalence of diabetes and impaired fasting glucose in adults in the U.S. population: National Health And Nutrition Examination Survey 1999–2002. Diabetes Care 2006, 29, 1263–1268. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.J.; Gregg, E.W.; Geiss, L.S.; Imperatore, G.; Williams, D.E.; Zhang, X.; Albright, A.L.; Cowie, C.C.; Klein, R.; Saaddine, J.B. Association of A1C and fasting plasma glucose levels with diabetic retinopathy prevalence in the U.S. population: Implications for diabetes diagnostic thresholds. Diabetes Care 2009, 32, 2027–2032. [Google Scholar] [CrossRef] [PubMed]
- Scheen, A.J. Diabetes mellitus in the elderly: Insulin resistance and/or impaired insulin secretion? Diabetes Metab. 2005, 31, 5S27–5S34. [Google Scholar] [CrossRef]
- Gunasekaran, U.; Gannon, M. Type 2 diabetes and the aging pancreatic beta cell. Aging (Albany NY) 2011, 3, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Weyer, C.; Bogardus, C.; Mott, D.M.; Pratley, R.E. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J. Clin. Investig. 1999, 104, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M.; Vidal, J.; Hull, R.L.; Utzschneider, K.M.; Carr, D.B.; Schraw, T.; Scherer, P.E.; Boyko, E.J.; Fujimoto, W.Y.; Kahn, S.E. Progressive loss of beta-cell function leads to worsening glucose tolerance in first-degree relatives of subjects with type 2 diabetes. Diabetes Care 2007, 30, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, F.M.; Rorsman, P. Diabetes mellitus and the beta cell: The last ten years. Cell 2012, 148, 1160–1171. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, P.; Beck-Nielsen, H.; Laakso, M.; Smith, U.; Yki-Jarvinen, H.; Ferrannini, E. Independent influence of age on basal insulin secretion in nondiabetic humans. European Group for the Study of Insulin Resistance. J. Clin. Endocrinol. Metab. 1999, 84, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Halban, P.A.; Polonsky, K.S.; Bowden, D.W.; Hawkins, M.A.; Ling, C.; Mather, K.J.; Powers, A.C.; Rhodes, C.J.; Sussel, L.; Weir, G.C. beta-cell failure in type 2 diabetes: Postulated mechanisms and prospects for prevention and treatment. Diabetes Care 2014, 37, 1751–1758. [Google Scholar] [CrossRef] [PubMed]
- Shanik, M.H.; Xu, Y.; Skrha, J.; Dankner, R.; Zick, Y.; Roth, J. Insulin resistance and hyperinsulinemia: Is hyperinsulinemia the cart or the horse? Diabetes Care 2008, 31, S262–S268. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Javaheri, A.; Godar, R.J.; Murphy, J.; Ma, X.; Rohatgi, N.; Mahadevan, J.; Hyrc, K.; Saftig, P.; Marshall, C.; et al. Intermittent fasting preserves beta-cell mass in obesity-induced diabetes via the autophagy-lysosome pathway. Autophagy 2017, 13, 1952–1968. [Google Scholar] [CrossRef] [PubMed]
- Ohneda, M.; Inman, L.R.; Unger, R.H. Caloric restriction in obese pre-diabetic rats prevents beta-cell depletion, loss of beta-cell GLUT 2 and glucose incompetence. Diabetologia 1995, 38, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Laffel, L. Ketone bodies: A review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab. Res. Rev. 1999, 15, 412–426. [Google Scholar] [CrossRef]
- Masuda, R.; Monahan, J.W.; Kashiwaya, Y. D-beta-hydroxybutyrate is neuroprotective against hypoxia in serum-free hippocampal primary cultures. J. Neurosci. Res. 2005, 80, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Thevenet, J.; De Marchi, U.; Domingo, J.S.; Christinat, N.; Bultot, L.; Lefebvre, G.; Sakamoto, K.; Descombes, P.; Masoodi, M.; Wiederkehr, A. Medium-chain fatty acids inhibit mitochondrial metabolism in astrocytes promoting astrocyte-neuron lactate and ketone body shuttle systems. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2016, 30, 1913–1926. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Mitchell, E.S. Cognition and Synaptic-Plasticity Related Changes in Aged Rats Supplemented with 8- and 10-Carbon Medium Chain Triglycerides. PLoS ONE 2016, 11, e0160159. [Google Scholar] [CrossRef] [PubMed]
- Neal, E.G.; Cross, J.H. Efficacy of dietary treatments for epilepsy. J. Hum. Nutr. Diet. 2010, 23, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Seaton, T.B.; Welle, S.L.; Warenko, M.K.; Campbell, R.G. Thermic effect of medium-chain and long-chain triglycerides in man. Am. J. Clin. Nutr. 1986, 44, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Bach, A.C.; Babayan, V.K. Medium-chain triglycerides: An update. Am. J. Clin. Nutr. 1982, 36, 950–962. [Google Scholar] [CrossRef] [PubMed]
- Marcovina, S.M.; Sirtori, C.; Peracino, A.; Gheorghiade, M.; Borum, P.; Remuzzi, G.; Ardehali, H. Translating the basic knowledge of mitochondrial functions to metabolic therapy: Role of l-carnitine. Transl. Res. 2013, 161, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Han, J.R.; Deng, B.; Sun, J.; Chen, C.G.; Corkey, B.E.; Kirkland, J.L.; Ma, J.; Guo, W. Effects of dietary medium-chain triglyceride on weight loss and insulin sensitivity in a group of moderately overweight free-living type 2 diabetic Chinese subjects. Metab. Clin. Exp. 2007, 56, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Gravena, C.; Mathias, P.C.; Ashcroft, S.J. Acute effects of fatty acids on insulin secretion from rat and human islets of Langerhans. J. Endocrinol. 2002, 173, 73–80. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Brown, A.J.; Goldsworthy, S.M.; Barnes, A.A.; Eilert, M.M.; Tcheang, L.; Daniels, D.; Muir, A.I.; Wigglesworth, M.J.; Kinghorn, I.; Fraser, N.J.; et al. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J. Biol. Chem. 2003, 278, 11312–11319. [Google Scholar] [CrossRef] [PubMed]
- Briscoe, C.P.; Tadayyon, M.; Andrews, J.L.; Benson, W.G.; Chambers, J.K.; Eilert, M.M.; Ellis, C.; Elshourbagy, N.A.; Goetz, A.S.; Minnick, D.T.; et al. The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J. Biol. Chem. 2003, 278, 11303–11311. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Ichimura, A.; Hirasawa, A. Therapeutic role and ligands of medium- to long-chain Fatty Acid receptors. Front. Endocrinol. 2014, 5, 83. [Google Scholar] [CrossRef] [PubMed]
- Hirasawa, A.; Tsumaya, K.; Awaji, T.; Katsuma, S.; Adachi, T.; Yamada, M.; Sugimoto, Y.; Miyazaki, S.; Tsujimoto, G. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat. Med. 2005, 11, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.P.; Feng, Y.; Zhou, Y.P.; Eiermann, G.J.; Petrov, A.; Zhou, C.; Lin, S.; Salituro, G.; Meinke, P.; Mosley, R.; et al. Selective small-molecule agonists of G protein-coupled receptor 40 promote glucose-dependent insulin secretion and reduce blood glucose in mice. Diabetes 2008, 57, 2211–2219. [Google Scholar] [CrossRef] [PubMed]
- Nagao, K.; Yanagita, T. Medium-chain fatty acids: Functional lipids for the prevention and treatment of the metabolic syndrome. Pharmacol. Res. 2010, 61, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, K.; Maekawa, F.; Yada, T. Oleic acid interacts with GPR40 to induce Ca2+ signaling in rat islet beta-cells: Mediation by PLC and l-type Ca2+ channel and link to insulin release. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E670–E677. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.D.; Kanabus, M.; Anderson, G.; Hargreaves, I.P.; Rutherford, T.; O’Donnell, M.; Cross, J.H.; Rahman, S.; Eaton, S.; Heales, S.J. The ketogenic diet component decanoic acid increases mitochondrial citrate synthase and complex I activity in neuronal cells. J. Neurochem. 2014, 129, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Arntfield, M.E.; van der Kooy, D. beta-Cell evolution: How the pancreas borrowed from the brain: The shared toolbox of genes expressed by neural and pancreatic endocrine cells may reflect their evolutionary relationship. Bioessays 2011, 33, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.C.; McGlynn, K.; Shao, C.; Duan, L.; Naziruddin, B.; Levy, M.F.; Cobb, M.H. Chromatin-bound mitogen-activated protein kinases transmit dynamic signals in transcription complexes in beta-cells. Proc. Natl. Acad. Sci. USA 2008, 105, 13315–13320. [Google Scholar] [CrossRef] [PubMed]
- Kalwat, M.A.; Wichaidit, C.; Nava Garcia, A.Y.; McCoy, M.K.; McGlynn, K.; Hwang, I.H.; MacMillan, J.B.; Posner, B.A.; Cobb, M.H. Insulin promoter-driven Gaussia luciferase-based insulin secretion biosensor assay for discovery of beta-cell glucose-sensing pathways. ACS Sens. 2016, 1, 1208–1212. [Google Scholar] [CrossRef] [PubMed]
- Hellemans, K.H.; Hannaert, J.C.; Denys, B.; Steffensen, K.R.; Raemdonck, C.; Martens, G.A.; Van Veldhoven, P.P.; Gustafsson, J.A.; Pipeleers, D. Susceptibility of pancreatic beta cells to fatty acids is regulated by LXR/PPARalpha-dependent stearoyl-coenzyme A desaturase. PLoS ONE 2009, 4, e7266. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.G.; Wang, H.D.; Qiao, L.; Yan, W.; Tan, Q.F.; Yin, H.X. The protective effect of the ketogenic diet on traumatic brain injury-induced cell death in juvenile rats. Brain Inj. 2009, 23, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Biden, T.J.; Taylor, K.W. Effects of ketone bodies on insulin release and islet-cell metabolism in the rat. Biochem. J. 1983, 212, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Busch, A.K.; Cordery, D.; Denyer, G.S.; Biden, T.J. Expression profiling of palmitate- and oleate-regulated genes provides novel insights into the effects of chronic lipid exposure on pancreatic beta-cell function. Diabetes 2002, 51, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Poitout, V.; Hagman, D.; Stein, R.; Artner, I.; Robertson, R.P.; Harmon, J.S. Regulation of the insulin gene by glucose and fatty acids. J. Nutr. 2006, 136, 873–876. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.L.; Hollingsworth, K.G.; Aribisala, B.S.; Chen, M.J.; Mathers, J.C.; Taylor, R. Reversal of type 2 diabetes: Normalisation of beta cell function in association with decreased pancreas and liver triacylglycerol. Diabetologia 2011, 54, 2506–2514. [Google Scholar] [CrossRef] [PubMed]
- Paoli, A.; Rubini, A.; Volek, J.S.; Grimaldi, K.A. Beyond weight loss: A review of the therapeutic uses of very-low-carbohydrate (ketogenic) diets. Eur. J. Clin. Nutr. 2013, 67, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Tetrick, M.A.; Greer, F.R.; Benevenga, N.J. Blood d-(-)-3-hydroxybutyrate concentrations after oral administration of trioctanoin, trinonanoin, or tridecanoin to newborn rhesus monkeys (Macaca mulatta). Comp. Med. 2010, 60, 486–490. [Google Scholar] [PubMed]
- Feinman, R.D.; Pogozelski, W.K.; Astrup, A.; Bernstein, R.K.; Fine, E.J.; Westman, E.C.; Accurso, A.; Frassetto, L.; Gower, B.A.; McFarlane, S.I.; et al. Dietary carbohydrate restriction as the first approach in diabetes management: Critical review and evidence base. Nutrition 2015, 31, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Scheuner, D.; Ron, D.; Pennathur, S.; Kaufman, R.J. Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J. Clin. Investig. 2008, 118, 3378–3389. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M.; Abdulkarim, B.; Bottu, G.; Cunha, D.A.; Igoillo-Esteve, M.; Masini, M.; Turatsinze, J.V.; Griebel, T.; Villate, O.; Santin, I.; et al. RNA sequencing identifies dysregulation of the human pancreatic islet transcriptome by the saturated fatty acid palmitate. Diabetes 2014, 63, 1978–1993. [Google Scholar] [CrossRef] [PubMed]
- De Tata, V. Age-related impairment of pancreatic Beta-cell function: Pathophysiological and cellular mechanisms. Front. Endocrinol. 2014, 5, 138. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.P.; Grill, V. Long term exposure to fatty acids and ketones inhibits B-cell functions in human pancreatic islets of Langerhans. J. Clin. Endocrinol. Metab. 1995, 80, 1584–1590. [Google Scholar] [PubMed]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [PubMed]










© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pujol, J.B.; Christinat, N.; Ratinaud, Y.; Savoia, C.; Mitchell, S.E.; Dioum, E.H.M. Coordination of GPR40 and Ketogenesis Signaling by Medium Chain Fatty Acids Regulates Beta Cell Function. Nutrients 2018, 10, 473. https://doi.org/10.3390/nu10040473
Pujol JB, Christinat N, Ratinaud Y, Savoia C, Mitchell SE, Dioum EHM. Coordination of GPR40 and Ketogenesis Signaling by Medium Chain Fatty Acids Regulates Beta Cell Function. Nutrients. 2018; 10(4):473. https://doi.org/10.3390/nu10040473
Chicago/Turabian StylePujol, Julien Benjamin, Nicolas Christinat, Yann Ratinaud, Claudia Savoia, Siobhan E. Mitchell, and El Hadji M Dioum. 2018. "Coordination of GPR40 and Ketogenesis Signaling by Medium Chain Fatty Acids Regulates Beta Cell Function" Nutrients 10, no. 4: 473. https://doi.org/10.3390/nu10040473
APA StylePujol, J. B., Christinat, N., Ratinaud, Y., Savoia, C., Mitchell, S. E., & Dioum, E. H. M. (2018). Coordination of GPR40 and Ketogenesis Signaling by Medium Chain Fatty Acids Regulates Beta Cell Function. Nutrients, 10(4), 473. https://doi.org/10.3390/nu10040473