Lysophosphatidic Acid Signaling in Obesity and Insulin Resistance



Abstract
1. Introduction
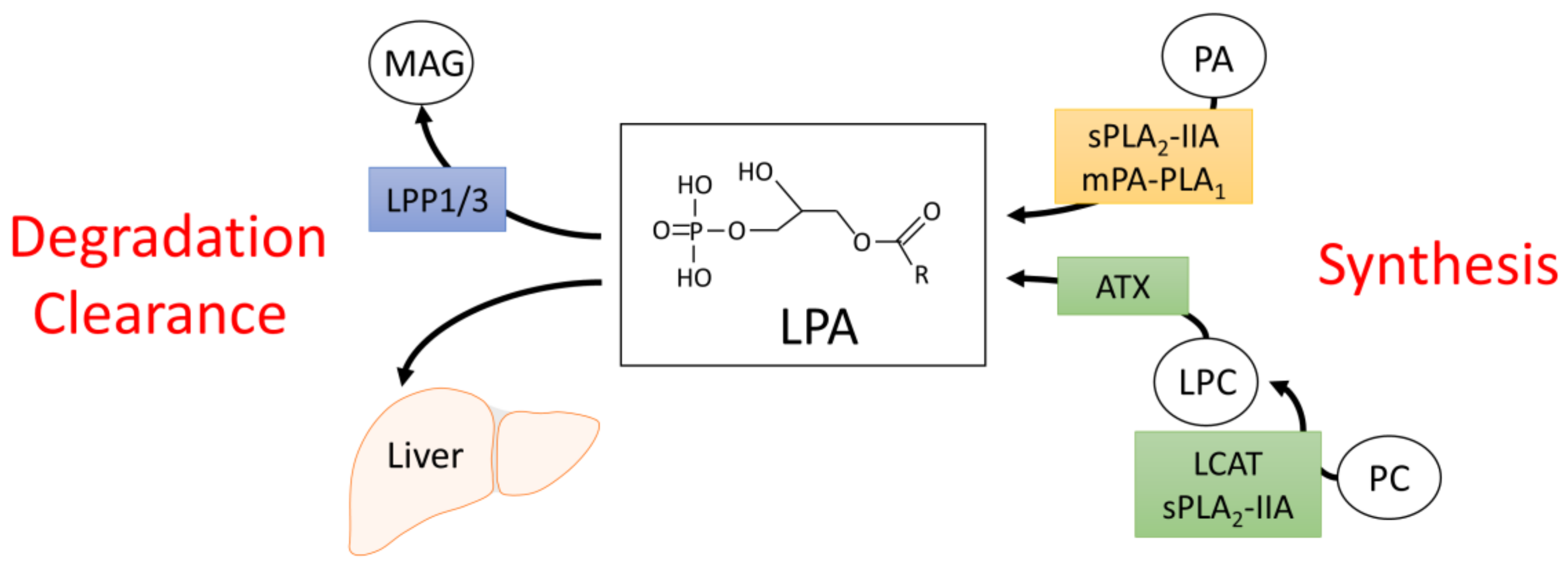
2. Synthesis and Degradation of LPA
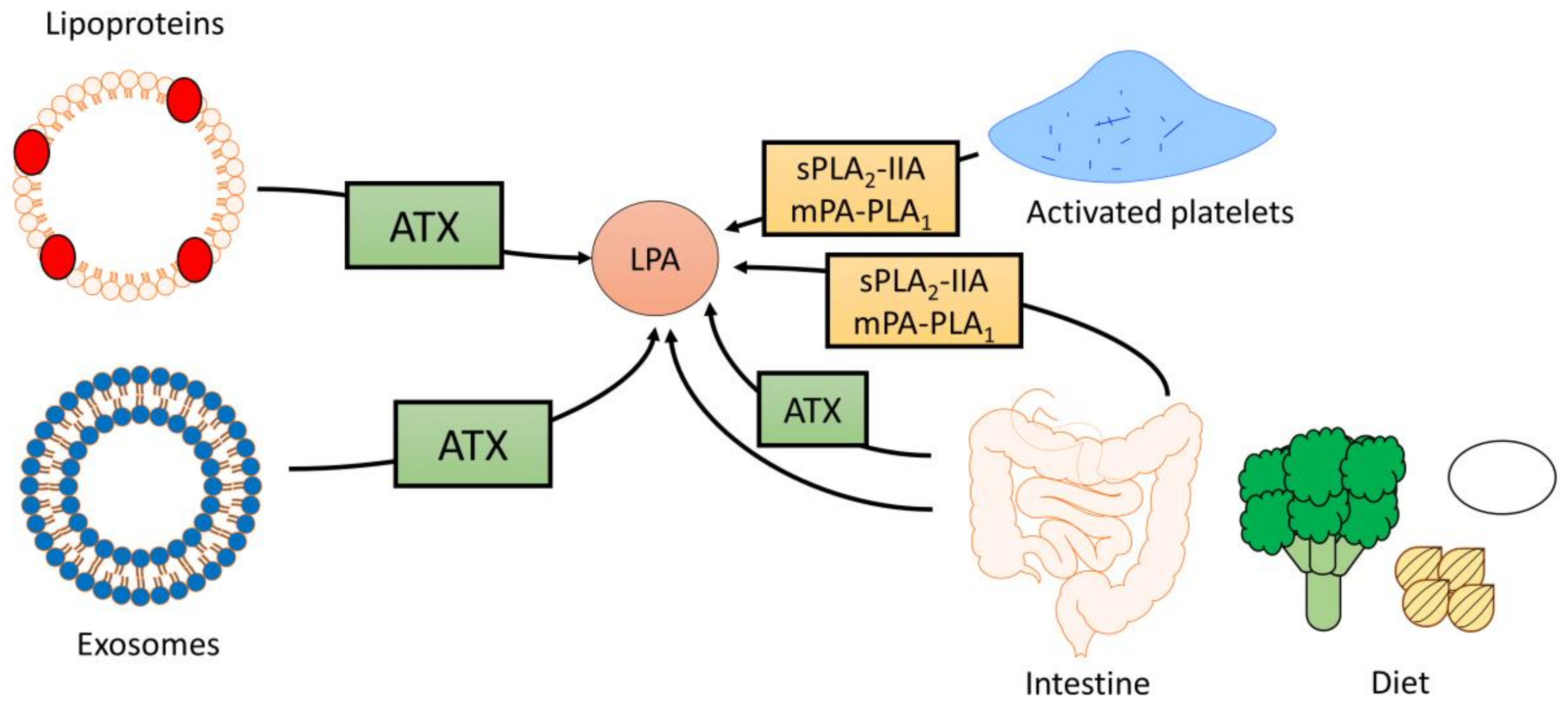
2.1. Sources of Circulating LPA
2.2. The Influence of Diet on LPA
3. ATX-LPA Signaling in Obesity
3.1. Role of the ATX–LPA Axis in Preadipocyte Proliferation and Differentiation
3.2. Role of ATX-LPA in Diet-Induced Obesity
4. Relationship between ATX-LPA and Insulin Signaling/Resistance
Potential Mechanisms by which the ATX–LPA Axis Influences Insulin Resistance
5. The ATX–LPA Axis—A Potential Link between Obesity/Insulin Resistance and Cardiovascular Disease
6. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Almena, M.; Merida, I. Shaping up the membrane: Diacylglycerol coordinates spatial orientation of signaling. Trends Biochem. Sci. 2011, 36, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Balla, T. Phosphoinositides: Tiny lipids with giant impact on cell regulation. Physiol. Rev. 2013, 93, 1019–1137. [Google Scholar] [CrossRef] [PubMed]
- Maceyka, M.; Harikumar, K.B.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012, 22, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Invest. 2006, 116, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Rutkowsky, J.M.; Snodgrass, R.G.; Ono-Moore, K.D.; Schneider, D.A.; Newman, J.W.; Adams, S.H.; Hwang, D.H. Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. J. Lipid Res. 2012, 53, 2002–2013. [Google Scholar] [CrossRef] [PubMed]
- Yore, M.M.; Syed, I.; Moraes-Vieira, P.M.; Zhang, T.; Herman, M.A.; Homan, E.A.; Patel, R.T.; Lee, J.; Chen, S.; Peroni, O.D.; et al. Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects. Cell 2014, 159, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Yung, Y.C.; Stoddard, N.C.; Chun, J. LPA receptor signaling: Pharmacology, physiology, and pathophysiology. J. Lipid Res. 2014, 55, 1192–1214. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Malchinkhuu, E.; Muraki, T.; Ishikawa, K.; Hayashi, K.; Tosaka, M.; Mochiduki, A.; Inoue, K.; Tomura, H.; Mogi, C.; et al. Identification of autotaxin as a neurite retraction-inducing factor of PC12 cells in cerebrospinal fluid and its possible sources. J. Neurochem. 2005, 92, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Houben, A.J.; van Wijk, X.M.; van Meeteren, L.A.; van Zeijl, L.; van de Westerlo, E.M.; Hausmann, J.; Fish, A.; Perrakis, A.; van Kuppevelt, T.H.; Moolenaar, W.H. The polybasic insertion in autotaxin alpha confers specific binding to heparin and cell surface heparan sulfate proteoglycans. J. Biol. Chem. 2013, 288, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Kaffe, E.; Katsifa, A.; Xylourgidis, N.; Ninou, I.; Zannikou, M.; Harokopos, V.; Foka, P.; Dimitriadis, A.; Evangelou, K.; Moulas, A.N.; et al. Hepatocyte autotaxin expression promotes liver fibrosis and cancer. Hepatology 2017, 65, 1369–1383. [Google Scholar] [CrossRef] [PubMed]
- Oikonomou, N.; Mouratis, M.A.; Tzouvelekis, A.; Kaffe, E.; Valavanis, C.; Vilaras, G.; Karameris, A.; Prestwich, G.D.; Bouros, D.; Aidinis, V. Pulmonary autotaxin expression contributes to the pathogenesis of pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Nikitopoulou, I.; Oikonomou, N.; Karouzakis, E.; Sevastou, I.; Nikolaidou-Katsaridou, N.; Zhao, Z.; Mersinias, V.; Armaka, M.; Xu, Y.; Masu, M.; et al. Autotaxin expression from synovial fibroblasts is essential for the pathogenesis of modeled arthritis. J. Exp. Med. 2012, 209, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Balood, M.; Zahednasab, H.; Siroos, B.; Mesbah-Namin, S.A.; Torbati, S.; Harirchian, M.H. Elevated serum levels of lysophosphatidic acid in patients with multiple sclerosis. Hum. Immunol. 2014, 75, 411–413. [Google Scholar] [CrossRef] [PubMed]
- Ray, U.; Roy Chowdhury, S.; Vasudevan, M.; Bankar, K.; Roychoudhury, S.; Roy, S.S. Gene regulatory networking reveals the molecular cue to lysophosphatidic acid-induced metabolic adaptations in ovarian cancer cells. Mol. Oncol. 2017, 11, 491–516. [Google Scholar] [CrossRef] [PubMed]
- Rancoule, C.; Attane, C.; Gres, S.; Fournel, A.; Dusaulcy, R.; Bertrand, C.; Vinel, C.; Treguer, K.; Prentki, M.; Valet, P.; et al. Lysophosphatidic acid impairs glucose homeostasis and inhibits insulin secretion in high-fat diet obese mice. Diabetologia 2013, 56, 1394–1402. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.; Hossain, I.; Perez, L.J.; Nzirorera, C.; Tozer, K.; D’Souza, K.; Trivedi, P.C.; Aguiar, C.; Yip, A.M.; Shea, J.; et al. Lysophosphatidic acid receptor mrna levels in heart and white adipose tissue are associated with obesity in mice and humans. PLoS ONE 2017, 12, e0189402. [Google Scholar] [CrossRef] [PubMed]
- Jean-Baptiste, G.; Yang, Z.; Khoury, C.; Greenwood, M.T. Lysophosphatidic acid mediates pleiotropic responses in skeletal muscle cells. Biochem. Biophys. Res. Commun. 2005, 335, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Bandoh, K.; Aoki, J.; Hosono, H.; Kobayashi, S.; Kobayashi, T.; Murakami-Murofushi, K.; Tsujimoto, M.; Arai, H.; Inoue, K. Molecular cloning and characterization of a novel human g-protein-coupled receptor, EDG7, for lysophosphatidic acid. J. Biol. Chem. 1999, 274, 27776–27785. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, K.; Ishii, S.; Shimizu, T. Identification of p2y9/GPR23 as a novel G protein-coupled receptor for lysophosphatidic acid, structurally distant from the Edg family. J. Biol. Chem. 2003, 278, 25600–25606. [Google Scholar] [CrossRef] [PubMed]
- Yanagida, K.; Masago, K.; Nakanishi, H.; Kihara, Y.; Hamano, F.; Tajima, Y.; Taguchi, R.; Shimizu, T.; Ishii, S. Identification and characterization of a novel lysophosphatidic acid receptor, p2y5/LPA6. J. Biol. Chem. 2009, 284, 17731–17741. [Google Scholar] [CrossRef] [PubMed]
- Bandoh, K.; Aoki, J.; Taira, A.; Tsujimoto, M.; Arai, H.; Inoue, K. Lysophosphatidic acid (LPA) receptors of the EDG family are differentially activated by LPA species. Structure-activity relationship of cloned LPA receptors. FEBS Lett. 2000, 478, 159–165. [Google Scholar] [CrossRef]
- Khandoga, A.L.; Fujiwara, Y.; Goyal, P.; Pandey, D.; Tsukahara, R.; Bolen, A.; Guo, H.; Wilke, N.; Liu, J.; Valentine, W.J.; et al. Lysophosphatidic acid-induced platelet shape change revealed through LPA(1–5) receptor-selective probes and albumin. Platelets 2008, 19, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.R.; Khandoga, A.L.; Goyal, P.; Fells, J.I.; Perygin, D.H.; Siess, W.; Parrill, A.L.; Tigyi, G.; Fujiwara, Y. Unique ligand selectivity of the GPR92/LPA5 lysophosphatidate receptor indicates role in human platelet activation. J. Biol. Chem. 2009, 284, 17304–17319. [Google Scholar] [CrossRef] [PubMed]
- Salous, A.K.; Panchatcharam, M.; Sunkara, M.; Mueller, P.; Dong, A.; Wang, Y.; Graf, G.A.; Smyth, S.S.; Morris, A.J. Mechanism of rapid elimination of lysophosphatidic acid and related lipids from the circulation of mice. J. Lipid Res. 2013, 54, 2775–2784. [Google Scholar] [CrossRef] [PubMed]
- Le Balle, F.; Simon, M.F.; Meijer, S.; Fourcade, O.; Chap, H. Membrane sidedness of biosynthetic pathways involved in the production of lysophosphatidic acid. Adv. Enzym. Regul. 1999, 39, 275–284. [Google Scholar] [CrossRef]
- Aoki, J.; Taira, A.; Takanezawa, Y.; Kishi, Y.; Hama, K.; Kishimoto, T.; Mizuno, K.; Saku, K.; Taguchi, R.; Arai, H. Serum lysophosphatidic acid is produced through diverse phospholipase pathways. J. Biol. Chem. 2002, 277, 48737–48744. [Google Scholar] [CrossRef] [PubMed]
- Van Meeteren, L.A.; Ruurs, P.; Stortelers, C.; Bouwman, P.; van Rooijen, M.A.; Pradere, J.P.; Pettit, T.R.; Wakelam, M.J.; Saulnier-Blache, J.S.; Mummery, C.L.; et al. Autotaxin, a secreted lysophospholipase D, is essential for blood vessel formation during development. Mol. Cell. Biol. 2006, 26, 5015–5022. [Google Scholar] [CrossRef] [PubMed]
- Ferry, G.; Tellier, E.; Try, A.; Gres, S.; Naime, I.; Simon, M.F.; Rodriguez, M.; Boucher, J.; Tack, I.; Gesta, S.; et al. Autotaxin is released from adipocytes, catalyzes lysophosphatidic acid synthesis, and activates preadipocyte proliferation. Up-regulated expression with adipocyte differentiation and obesity. J. Biol. Chem. 2003, 278, 18162–18169. [Google Scholar] [CrossRef] [PubMed]
- Pamuklar, Z.; Federico, L.; Liu, S.; Umezu-Goto, M.; Dong, A.; Panchatcharam, M.; Fulkerson, Z.; Berdyshev, E.; Natarajan, V.; Fang, X.; et al. Autotaxin/lysopholipase D and lysophosphatidic acid regulate murine hemostasis and thrombosis. J. Biol. Chem. 2009, 284, 7385–7394. [Google Scholar] [CrossRef] [PubMed]
- Dusaulcy, R.; Rancoule, C.; Gres, S.; Wanecq, E.; Colom, A.; Guigne, C.; van Meeteren, L.A.; Moolenaar, W.H.; Valet, P.; Saulnier-Blache, J.S. Adipose-specific disruption of autotaxin enhances nutritional fattening and reduces plasma lysophosphatidic acid. J. Lipid Res. 2011, 52, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, S.; Nagasaki, M.; Okudaira, S.; Aoki, J.; Ohmori, T.; Ohkawa, R.; Nakamura, K.; Igarashi, K.; Yamashita, H.; Eto, K.; et al. ENPP2 contributes to adipose tissue expansion in diet-induced obesity. Diabetes 2014, 63, 4154–4164. [Google Scholar] [CrossRef] [PubMed]
- Fotopoulou, S.; Oikonomou, N.; Grigorieva, E.; Nikitopoulou, I.; Paparountas, T.; Thanassopoulou, A.; Zhao, Z.; Xu, Y.; Kontoyiannis, D.L.; Remboutsika, E.; et al. Atx expression and lpa signalling are vital for the development of the nervous system. Dev. Biol. 2010, 339, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Katsifa, A.; Kaffe, E.; Nikolaidou-Katsaridou, N.; Economides, A.N.; Newbigging, S.; McKerlie, C.; Aidinis, V. The bulk of autotaxin activity is dispensable for adult mouse life. PLoS ONE 2015, 10, e0143083. [Google Scholar] [CrossRef] [PubMed]
- Gierse, J.; Thorarensen, A.; Beltey, K.; Bradshaw-Pierce, E.; Cortes-Burgos, L.; Hall, T.; Johnston, A.; Murphy, M.; Nemirovskiy, O.; Ogawa, S.; et al. A novel autotaxin inhibitor reduces lysophosphatidic acid levels in plasma and the site of inflammation. J. Pharmacol. Exp. Ther. 2010, 334, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Yukiura, H.; Kano, K.; Kise, R.; Inoue, A.; Aoki, J. Autotaxin overexpression causes embryonic lethality and vascular defects. PLoS ONE 2015, 10, e0126734. [Google Scholar] [CrossRef] [PubMed]
- Jasinska, R.; Zhang, Q.X.; Pilquil, C.; Singh, I.; Xu, J.; Dewald, J.; Dillon, D.A.; Berthiaume, L.G.; Carman, G.M.; Waggoner, D.W.; et al. Lipid phosphate phosphohydrolase-1 degrades exogenous glycerolipid and sphingolipid phosphate esters. Biochem. J. 1999, 340, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Benesch, M.G.; Dewald, J.; Zhao, Y.Y.; Patwardhan, N.; Santos, W.L.; Curtis, J.M.; McMullen, T.P.; Brindley, D.N. Lipid phosphate phosphatase-1 expression in cancer cells attenuates tumor growth and metastasis in mice. J. Lipid Res. 2014, 55, 2389–2400. [Google Scholar] [CrossRef] [PubMed]
- Sigal, Y.J.; McDermott, M.I.; Morris, A.J. Integral membrane lipid phosphatases/phosphotransferases: Common structure and diverse functions. Biochem. J. 2005, 387, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Tomsig, J.L.; Snyder, A.H.; Berdyshev, E.V.; Skobeleva, A.; Mataya, C.; Natarajan, V.; Brindley, D.N.; Lynch, K.R. Lipid phosphate phosphohydrolase type 1 (LPP1) degrades extracellular lysophosphatidic acid in vivo. Biochem. J. 2009, 419, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Escalante-Alcalde, D.; Hernandez, L.; Le Stunff, H.; Maeda, R.; Lee, H.S.; Gang, C., Jr.; Sciorra, V.A.; Daar, I.; Spiegel, S.; Morris, A.J.; et al. The lipid phosphatase LPP3 regulates extra-embryonic vasculogenesis and axis patterning. Development 2003, 130, 4623–4637. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Sundberg, J.P.; Gridley, T. Mice mutant for Ppap2c, a homolog of the germ cell migration regulator wunen, are viable and fertile. Genesis 2000, 27, 137–140. [Google Scholar] [CrossRef]
- Morris, K.E.; Schang, L.M.; Brindley, D.N. Lipid phosphate phosphatase-2 activity regulates s-phase entry of the cell cycle in Rat2 fibroblasts. J. Biol. Chem. 2006, 281, 9297–9306. [Google Scholar] [CrossRef] [PubMed]
- Brindley, D.N.; Pilquil, C. Lipid phosphate phosphatases and signaling. J. Lipid Res. 2009, 50, S225–S230. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.L.; Morrison, P.; Miller, B.; Riely, C.A.; Tolley, B.; Westermann, A.M.; Bonfrer, J.M.; Bais, E.; Moolenaar, W.H.; Tigyi, G. Plasma lysophosphatidic acid concentration and ovarian cancer. JAMA 2002, 287, 3081–3082. [Google Scholar] [CrossRef] [PubMed]
- Boucharaba, A.; Serre, C.M.; Gres, S.; Saulnier-Blache, J.S.; Bordet, J.C.; Guglielmi, J.; Clezardin, P.; Peyruchaud, O. Platelet-derived lysophosphatidic acid supports the progression of osteolytic bone metastases in breast cancer. J. Clin. Invest. 2004, 114, 1714–1725. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Subramanian, P.; Sevilmis, G.; Globke, B.; Soehnlein, O.; Karshovska, E.; Megens, R.; Heyll, K.; Chun, J.; Saulnier-Blache, J.S.; et al. Lipoprotein-derived lysophosphatidic acid promotes atherosclerosis by releasing CXCL1 from the endothelium. Cell Metab. 2011, 13, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Bouchareb, R.; Mahmut, A.; Nsaibia, M.J.; Boulanger, M.C.; Dahou, A.; Lepine, J.L.; Laflamme, M.H.; Hadji, F.; Couture, C.; Trahan, S.; et al. Autotaxin derived from lipoprotein(a) and valve interstitial cells promotes inflammation and mineralization of the aortic valve. Circulation 2015, 132, 677–690. [Google Scholar] [CrossRef] [PubMed]
- Mahmut, A.; Boulanger, M.C.; El Husseini, D.; Fournier, D.; Bouchareb, R.; Despres, J.P.; Pibarot, P.; Bosse, Y.; Mathieu, P. Elevated expression of lipoprotein-associated phospholipase A2 in calcific aortic valve disease: Implications for valve mineralization. J. Am. Coll. Cardiol. 2014, 63, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Jethwa, S.A.; Leah, E.J.; Zhang, Q.; Bright, N.A.; Oxley, D.; Bootman, M.D.; Rudge, S.A.; Wakelam, M.J. Exosomes bind to autotaxin and act as a physiological delivery mechanism to stimulate LPA receptor signalling in cells. J. Cell. Sci. 2016, 129, 3948–3957. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Kishimoto, T.; Ohkawa, R.; Okubo, S.; Tozuka, M.; Yokota, H.; Ikeda, H.; Ohshima, N.; Mizuno, K.; Yatomi, Y. Suppression of lysophosphatidic acid and lysophosphatidylcholine formation in the plasma in vitro: Proposal of a plasma sample preparation method for laboratory testing of these lipids. Anal. Biochem. 2007, 367, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Michalczyk, A.; Budkowska, M.; Dolegowska, B.; Chlubek, D.; Safranow, K. Lysophosphatidic acid plasma concentrations in healthy subjects: Circadian rhythm and associations with demographic, anthropometric and biochemical parameters. Lipids Health Dis. 2017, 16, 140. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Xu, Y. An extremely simple method for extraction of lysophospholipids and phospholipids from blood samples. J. Lipid Res. 2010, 51, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Navab, M.; Chattopadhyay, A.; Hough, G.; Meriwether, D.; Fogelman, S.I.; Wagner, A.C.; Grijalva, V.; Su, F.; Anantharamaiah, G.M.; Hwang, L.H.; et al. Source and role of intestinally derived lysophosphatidic acid in dyslipidemia and atherosclerosis. J. Lipid Res. 2015, 56, 871–887. [Google Scholar] [CrossRef] [PubMed]
- Smyth, S.S.; Mueller, P.; Yang, F.; Brandon, J.A.; Morris, A.J. Arguing the case for the autotaxin-lysophosphatidic acid-lipid phosphate phosphatase 3-signaling nexus in the development and complications of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 479–486. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, K.; Kane, D.A.; Touaibia, M.; Kershaw, E.E.; Pulinilkunnil, T.; Kienesberger, P.C. Autotaxin is regulated by glucose and insulin in adipocytes. Endocrinology 2017, 158, 791–803. [Google Scholar] [CrossRef] [PubMed]
- Ino, M.; Shimizu, Y.; Tanaka, T.; Tokumura, A. Alterations of plasma levels of lysophosphatidic acid in response to fasting of rats. Biol. Pharm. Bull. 2012, 35, 2059–2063. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rancoule, C.; Dusaulcy, R.; Treguer, K.; Gres, S.; Attane, C.; Saulnier-Blache, J.S. Involvement of autotaxin/lysophosphatidic acid signaling in obesity and impaired glucose homeostasis. Biochimie 2013, 96, 140–143. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Wang, R.; Song, J.; Guan, M.; Li, N.; Zhang, X.; Zhao, Z.; Zhang, J. Blocking gp130 signaling suppresses autotaxin expression in adipocytes and improves insulin sensitivity in diet-induced obesity. J. Lipid Res. 2017, 58, 2102–2113. [Google Scholar] [CrossRef] [PubMed]
- Navab, M.; Hough, G.; Buga, G.M.; Su, F.; Wagner, A.C.; Meriwether, D.; Chattopadhyay, A.; Gao, F.; Grijalva, V.; Danciger, J.S.; et al. Transgenic 6F tomatoes act on the small intestine to prevent systemic inflammation and dyslipidemia caused by western diet and intestinally derived lysophosphatidic acid. J. Lipid Res. 2013, 54, 3403–3418. [Google Scholar] [CrossRef] [PubMed]
- Nakane, S.; Tokumura, A.; Waku, K.; Sugiura, T. Hen egg yolk and white contain high amounts of lysophosphatidic acids, growth factor-like lipids: Distinct molecular species compositions. Lipids 2001, 36, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Horiuchi, G.; Matsuoka, M.; Hirano, K.; Tokumura, A.; Koike, T.; Satouchi, K. Formation of lysophosphatidic acid, a wound-healing lipid, during digestion of cabbage leaves. Biosci. Biotechnol. Biochem. 2009, 73, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.H.; Choi, S.H.; Kim, H.J.; Jung, S.W.; Kim, H.K.; Nah, S.Y. Plant lysophosphatidic acids: A rich source for bioactive lysophosphatidic acids and their pharmacological applications. Biol. Pharm. Bull. 2016, 39, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Adachi, M.; Shimizu, Y.; Tsutsumi, T.; Tokumura, A. Comparison of lysophospholipid levels in rat feces with those in a standard chow. J. Agric. Food Chem. 2011, 59, 7062–7067. [Google Scholar] [CrossRef] [PubMed]
- Eder, A.M.; Sasagawa, T.; Mao, M.; Aoki, J.; Mills, G.B. Constitutive and lysophosphatidic acid (LPA)-induced LPA production: Role of phospholipase D and phospholipase A2. Clin. Cancer Res. 2000, 6, 2482–2491. [Google Scholar] [PubMed]
- Chattopadhyay, A.; Navab, M.; Hough, G.; Grijalva, V.; Mukherjee, P.; Fogelman, H.R.; Hwang, L.H.; Faull, K.F.; Lusis, A.J.; Reddy, S.T.; et al. Tg6F ameliorates the increase in oxidized phospholipids in the jejunum of mice fed unsaturated LysoPC or WD. J. Lipid Res. 2016, 57, 832–847. [Google Scholar] [CrossRef] [PubMed]
- Rachakonda, V.P.; Reeves, V.L.; Aljammal, J.; Wills, R.C.; Trybula, J.S.; DeLany, J.P.; Kienesberger, P.C.; Kershaw, E.E. Serum autotaxin is independently associated with hepatic steatosis in women with severe obesity. Obesity (Silver Spring) 2015, 23, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Rancoule, C.; Dusaulcy, R.; Treguer, K.; Gres, S.; Guigne, C.; Quilliot, D.; Valet, P.; Saulnier-Blache, J.S. Depot-specific regulation of autotaxin with obesity in human adipose tissue. J. Physiol. Biochem. 2012, 68, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Reeves, V.L.; Trybula, J.S.; Wills, R.C.; Goodpaster, B.H.; Dubé, J.J.; Kienesberger, P.C.; Kershaw, E.E. Serum Autotaxin/ENPP2 correlates with insulin resistance in older humans with obesity. Obesity (Silver Spring) 2015, 23, 2371–2376. [Google Scholar] [CrossRef] [PubMed]
- Fayyaz, S.; Japtok, L.; Schumacher, F.; Wigger, D.; Schulz, T.J.; Haubold, K.; Gulbins, E.; Voller, H.; Kleuser, B. Lysophosphatidic acid inhibits insulin signaling in primary rat hepatocytes via the LPA3 receptor subtype and is increased in obesity. Cell. Physiol. Biochem. 2017, 43, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Jo, J.; Gavrilova, O.; Pack, S.; Jou, W.; Mullen, S.; Sumner, A.E.; Cushman, S.W.; Periwal, V. Hypertrophy and/or hyperplasia: Dynamics of adipose tissue growth. PLoS Comput. Biol. 2009, 5, e1000324. [Google Scholar] [CrossRef] [PubMed]
- Pages, C.; Daviaud, D.; An, S.; Krief, S.; Lafontan, M.; Valet, P.; Saulnier-Blache, J.S. Endothelial differentiation gene-2 receptor is involved in lysophosphatidic acid-dependent control of 3t3f442a preadipocyte proliferation and spreading. J. Biol. Chem. 2001, 276, 11599–11605. [Google Scholar] [CrossRef] [PubMed]
- Radhika, V.; Hee Ha, J.; Jayaraman, M.; Tsim, S.T.; Dhanasekaran, N. Mitogenic signaling by lysophosphatidic acid (LPA) involves galpha12. Oncogene 2005, 24, 4597–4603. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nobusue, H.; Kondo, D.; Yamamoto, M.; Kano, K. Effects of lysophosphatidic acid on the in vitro proliferation and differentiation of a novel porcine preadipocyte cell line. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2010, 157, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Holmstrom, T.E.; Mattsson, C.L.; Wang, Y.; Iakovleva, I.; Petrovic, N.; Nedergaard, J. Non-transactivational, dual pathways for LPA-induced Erk1/2 activation in primary cultures of brown pre-adipocytes. Exp. Cell Res. 2010, 316, 2664–2675. [Google Scholar] [CrossRef] [PubMed]
- Federico, L.; Ren, H.; Mueller, P.A.; Wu, T.; Liu, S.; Popovic, J.; Blalock, E.M.; Sunkara, M.; Ovaa, H.; Albers, H.M.; et al. Autotaxin and its product lysophosphatidic acid suppress brown adipose differentiation and promote diet-induced obesity in mice. Mol. Endocrinol. 2012, 26, 786–797. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.F.; Daviaud, D.; Pradere, J.P.; Gres, S.; Guigne, C.; Wabitsch, M.; Chun, J.; Valet, P.; Saulnier-Blache, J.S. Lysophosphatidic acid inhibits adipocyte differentiation via lysophosphatidic acid 1 receptor-dependent down-regulation of peroxisome proliferator-activated receptor gamma2. J. Biol. Chem. 2005, 280, 14656–14662. [Google Scholar] [CrossRef] [PubMed]
- Rancoule, C.; Viaud, M.; Gres, S.; Viguerie, N.; Decaunes, P.; Bouloumie, A.; Langin, D.; Bascands, J.L.; Valet, P.; Saulnier-Blache, J.S. Pro-fibrotic activity of lysophosphatidic acid in adipose tissue: In vivo and in vitro evidence. Biochim. Biophys. Acta 2014, 1841, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Yea, K.; Kim, J.; Lim, S.; Park, H.S.; Park, K.S.; Suh, P.G.; Ryu, S.H. Lysophosphatidic acid regulates blood glucose by stimulating myotube and adipocyte glucose uptake. J. Mol. Med. 2008, 86, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.; Quilliot, D.; Praderes, J.P.; Simon, M.F.; Gres, S.; Guigne, C.; Prevot, D.; Ferry, G.; Boutin, J.A.; Carpene, C.; et al. Potential involvement of adipocyte insulin resistance in obesity-associated up-regulation of adipocyte lysophospholipase D/autotaxin expression. Diabetologia 2005, 48, 569–577. [Google Scholar] [CrossRef] [PubMed]
- De Luca, C.; Olefsky, J.M. Inflammation and insulin resistance. FEBS Lett. 2008, 582, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Wieser, V.; Moschen, A.R.; Tilg, H. Inflammation, cytokines and insulin resistance: A clinical perspective. Arch. Immunol. Ther. Exp. 2013, 61, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Farquhar, M.J.; Humphreys, I.S.; Rudge, S.A.; Wilson, G.K.; Bhattacharya, B.; Ciaccia, M.; Hu, K.; Zhang, Q.; Mailly, L.; Reynolds, G.M.; et al. Autotaxin-lysophosphatidic acid receptor signalling regulates hepatitis C virus replication. J. Hepatol. 2017, 66, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Wick, G.; Grundtman, C.; Mayerl, C.; Wimpissinger, T.F.; Feichtinger, J.; Zelger, B.; Sgonc, R.; Wolfram, D. The immunology of fibrosis. Annu. Rev. Immunol. 2013, 31, 107–135. [Google Scholar] [CrossRef] [PubMed]
- Ueha, S.; Shand, F.H.; Matsushima, K. Cellular and molecular mechanisms of chronic inflammation-associated organ fibrosis. Front. Immunol. 2012, 3, 71. [Google Scholar] [CrossRef] [PubMed]
- Spencer, M.; Yao-Borengasser, A.; Unal, R.; Rasouli, N.; Gurley, C.M.; Zhu, B.; Peterson, C.A.; Kern, P.A. Adipose tissue macrophages in insulin-resistant subjects are associated with collagen VI and fibrosis and demonstrate alternative activation. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E1016–E1027. [Google Scholar] [CrossRef] [PubMed]
- Guglielmi, V.; Cardellini, M.; Cinti, F.; Corgosinho, F.; Cardolini, I.; D’Adamo, M.; Zingaretti, M.C.; Bellia, A.; Lauro, D.; Gentileschi, P.; et al. Omental adipose tissue fibrosis and insulin resistance in severe obesity. Nutr. Diabetes 2015, 5, e175. [Google Scholar] [CrossRef] [PubMed]
- Pradere, J.P.; Klein, J.; Gres, S.; Guigne, C.; Neau, E.; Valet, P.; Calise, D.; Chun, J.; Bascands, J.L.; Saulnier-Blache, J.S.; et al. LPA1 receptor activation promotes renal interstitial fibrosis. J. Am. Soc. Nephrol. 2007, 18, 3110–3118. [Google Scholar] [CrossRef] [PubMed]
- Castelino, F.V.; Seiders, J.; Bain, G.; Brooks, S.F.; King, C.D.; Swaney, J.S.; Lorrain, D.S.; Chun, J.; Luster, A.D.; Tager, A.M. Amelioration of dermal fibrosis by genetic deletion or pharmacologic antagonism of lysophosphatidic acid receptor 1 in a mouse model of scleroderma. Arthritis Rheum. 2011, 63, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
- Yoneshiro, T.; Aita, S.; Matsushita, M.; Okamatsu-Ogura, Y.; Kameya, T.; Kawai, Y.; Miyagawa, M.; Tsujisaki, M.; Saito, M. Age-related decrease in cold-activated brown adipose tissue and accumulation of body fat in healthy humans. Obesity (Silver Spring) 2011, 19, 1755–1760. [Google Scholar] [CrossRef] [PubMed]
- Yoneshiro, T.; Aita, S.; Matsushita, M.; Kameya, T.; Nakada, K.; Kawai, Y.; Saito, M. Brown adipose tissue, whole-body energy expenditure, and thermogenesis in healthy adult men. Obesity (Silver Spring) 2011, 19, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Orava, J.; Nuutila, P.; Lidell, M.E.; Oikonen, V.; Noponen, T.; Viljanen, T.; Scheinin, M.; Taittonen, M.; Niemi, T.; Enerback, S.; et al. Different metabolic responses of human brown adipose tissue to activation by cold and insulin. Cell Metab. 2011, 14, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Saito, M. Brown adipose tissue as a regulator of energy expenditure and body fat in humans. Diabetes Metab. J. 2013, 37, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Kontani, Y.; Wang, Y.; Kimura, K.; Inokuma, K.I.; Saito, M.; Suzuki-Miura, T.; Wang, Z.; Sato, Y.; Mori, N.; Yamashita, H. Ucp1 deficiency increases susceptibility to diet-induced obesity with age. Aging Cell 2005, 4, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Okamatsu-Ogura, Y.; Matsushita, M.; Watanabe, K.; Yoneshiro, T.; Nio-Kobayashi, J.; Iwanaga, T.; Miyagawa, M.; Kameya, T.; Nakada, K.; et al. High incidence of metabolically active brown adipose tissue in healthy adult humans: Effects of cold exposure and adiposity. Diabetes 2009, 58, 1526–1531. [Google Scholar] [CrossRef] [PubMed]
- Van Marken Lichtenbelt, W.D.; Vanhommerig, J.W.; Smulders, N.M.; Drossaerts, J.M.; Kemerink, G.J.; Bouvy, N.D.; Schrauwen, P.; Teule, G.J. Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med. 2009, 360, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Ouellet, V.; Routhier-Labadie, A.; Bellemare, W.; Lakhal-Chaieb, L.; Turcotte, E.; Carpentier, A.C.; Richard, D. Outdoor temperature, age, sex, body mass index, and diabetic status determine the prevalence, mass, and glucose-uptake activity of 18F-FDG-detected BAT in humans. J. Clin. Endocrinol. Metab. 2011, 96, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Smith, S.; Linderman, J.; Courville, A.B.; Brychta, R.J.; Dieckmann, W.; Werner, C.D.; Chen, K.Y.; Celi, F.S. Temperature-acclimated brown adipose tissue modulates insulin sensitivity in humans. Diabetes 2014, 63, 3686–3698. [Google Scholar] [CrossRef] [PubMed]
- Chondronikola, M.; Volpi, E.; Borsheim, E.; Porter, C.; Annamalai, P.; Enerback, S.; Lidell, M.E.; Saraf, M.K.; Labbe, S.M.; Hurren, N.M.; et al. Brown adipose tissue improves whole-body glucose homeostasis and insulin sensitivity in humans. Diabetes 2014, 63, 4089–4099. [Google Scholar] [CrossRef] [PubMed]
- Stanford, K.I.; Middelbeek, R.J.; Townsend, K.L.; An, D.; Nygaard, E.B.; Hitchcox, K.M.; Markan, K.R.; Nakano, K.; Hirshman, M.F.; Tseng, Y.H.; et al. Brown adipose tissue regulates glucose homeostasis and insulin sensitivity. J. Clin. Invest. 2013, 123, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Soccio, R.E.; Chen, E.R.; Lazar, M.A. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab. 2014, 20, 573–591. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; Spiegelman, B.M. Ppargamma: A nuclear regulator of metabolism, differentiation, and cell growth. J. Biol. Chem. 2001, 276, 37731–37734. [Google Scholar] [CrossRef] [PubMed]
- Barroso, I.; Gurnell, M.; Crowley, V.E.; Agostini, M.; Schwabe, J.W.; Soos, M.A.; Maslen, G.L.; Williams, T.D.; Lewis, H.; Schafer, A.J.; et al. Dominant negative mutations in human ppargamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature 1999, 402, 880–883. [Google Scholar] [CrossRef] [PubMed]
- Leonardini, A.; Laviola, L.; Perrini, S.; Natalicchio, A.; Giorgino, F. Cross-talk between ppargamma and insulin signaling and modulation of insulin sensitivity. PPAR Res. 2009, 2009, 818945. [Google Scholar] [CrossRef] [PubMed]
- Miles, P.D.; Romeo, O.M.; Higo, K.; Cohen, A.; Rafaat, K.; Olefsky, J.M. TNF-alpha-induced insulin resistance in vivo and its prevention by troglitazone. Diabetes 1997, 46, 1678–1683. [Google Scholar] [CrossRef] [PubMed]
- Okuno, A.; Tamemoto, H.; Tobe, K.; Ueki, K.; Mori, Y.; Iwamoto, K.; Umesono, K.; Akanuma, Y.; Fujiwara, T.; Horikoshi, H.; et al. Troglitazone increases the number of small adipocytes without the change of white adipose tissue mass in obese zucker rats. J. Clin. Invest. 1998, 101, 1354–1361. [Google Scholar] [CrossRef] [PubMed]
- Souza, S.C.; Yamamoto, M.T.; Franciosa, M.D.; Lien, P.; Greenberg, A.S. BRL 49653 blocks the lipolytic actions of tumor necrosis factor-alpha: A potential new insulin-sensitizing mechanism for thiazolidinediones. Diabetes 1998, 47, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.; Kataria, M.A.; Saini, V.; Yadav, A. Role of leptin and adiponectin in insulin resistance. Clin. Chim. Acta 2013, 417, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Hotta, K.; Funahashi, T.; Arita, Y.; Takahashi, M.; Matsuda, M.; Okamoto, Y.; Iwahashi, H.; Kuriyama, H.; Ouchi, N.; Maeda, K.; et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1595–1599. [Google Scholar] [CrossRef] [PubMed]
- Fruebis, J.; Tsao, T.S.; Javorschi, S.; Ebbets-Reed, D.; Erickson, M.R.; Yen, F.T.; Bihain, B.E.; Lodish, H.F. Proteolytic cleavage product of 30-kda adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc. Natl. Acad. Sci. USA 2001, 98, 2005–2010. [Google Scholar] [CrossRef] [PubMed]
- Berg, A.H.; Combs, T.P.; Du, X.; Brownlee, M.; Scherer, P.E. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat. Med. 2001, 7, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating amp-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Vella, C.A.; Burgos, X.; Ellis, C.J.; Zubia, R.Y.; Ontiveros, D.; Reyes, H.; Lozano, C. Associations of insulin resistance with cardiovascular risk factors and inflammatory cytokines in normal-weight hispanic women. Diabetes Care 2013, 36, 1377–1383. [Google Scholar] [CrossRef] [PubMed]
- Hanley, A.J.; Williams, K.; Stern, M.P.; Haffner, S.M. Homeostasis model assessment of insulin resistance in relation to the incidence of cardiovascular disease: The san antonio heart study. Diabetes Care 2002, 25, 1177–1184. [Google Scholar] [CrossRef] [PubMed]
- Steinberger, J.; Daniels, S.R.; American Heart Association Atherosclerosis, Hypertension and Obesity in the Young Committee; American Heart Association Diabetes Committee. Obesity, insulin resistance, diabetes, and cardiovascular risk in children: An american heart association scientific statement from the atherosclerosis, hypertension, and obesity in the young committee (council on cardiovascular disease in the young) and the diabetes committee (council on nutrition, physical activity, and metabolism). Circulation 2003, 107, 1448–1453. [Google Scholar] [PubMed]
- Abbasi, F.; Brown, B.W., Jr.; Lamendola, C.; McLaughlin, T.; Reaven, G.M. Relationship between obesity, insulin resistance, and coronary heart disease risk. J. Am. Coll. Cardiol. 2002, 40, 937–943. [Google Scholar] [CrossRef]
- Rother, E.; Brandl, R.; Baker, D.L.; Goyal, P.; Gebhard, H.; Tigyi, G.; Siess, W. Subtype-selective antagonists of lysophosphatidic acid receptors inhibit platelet activation triggered by the lipid core of atherosclerotic plaques. Circulation 2003, 108, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Dohi, T.; Miyauchi, K.; Ohkawa, R.; Nakamura, K.; Kurano, M.; Kishimoto, T.; Yanagisawa, N.; Ogita, M.; Miyazaki, T.; Nishino, A.; et al. Increased lysophosphatidic acid levels in culprit coronary arteries of patients with acute coronary syndrome. Atherosclerosis 2013, 229, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Siess, W.; Zangl, K.J.; Essler, M.; Bauer, M.; Brandl, R.; Corrinth, C.; Bittman, R.; Tigyi, G.; Aepfelbacher, M. Lysophosphatidic acid mediates the rapid activation of platelets and endothelial cells by mildly oxidized low density lipoprotein and accumulates in human atherosclerotic lesions. Proc. Natl. Acad. Sci. USA 1999, 96, 6931–6936. [Google Scholar] [CrossRef] [PubMed]
- Bot, M.; de Jager, S.C.; MacAleese, L.; Lagraauw, H.M.; van Berkel, T.J.; Quax, P.H.; Kuiper, J.; Heeren, R.M.; Biessen, E.A.; Bot, I. Lysophosphatidic acid triggers mast cell-driven atherosclerotic plaque destabilization by increasing vascular inflammation. J. Lipid Res. 2013, 54, 1265–1274. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, I.; Edsfeldt, A.; Ko, N.Y.; Grufman, H.; Berg, K.; Bjorkbacka, H.; Nitulescu, M.; Persson, A.; Nilsson, M.; Prehn, C.; et al. Evidence supporting a key role of Lp-PLA2-generated lysophosphatidylcholine in human atherosclerotic plaque inflammation. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.A.; Boyce, J.A. IL-4 regulates mek expression required for lysophosphatidic acid-mediated chemokine generation by human mast cells. J. Immunol. 2005, 175, 5430–5438. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ochoa, L.N.; Kagan, A.; Chai, H.; Liang, Z.; Lin, P.H.; Yao, Q. Lysophosphatidic acid causes endothelial dysfunction in porcine coronary arteries and human coronary artery endothelial cells. Atherosclerosis 2012, 222, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, Y.; Zhu, W.; Han, Y.; Han, B.; Xu, R.; Deng, L.; Cai, Y.; Cong, X.; Yang, Y.; et al. Specific lpa receptor subtype mediation of LPA-induced hypertrophy of cardiac myocytes and involvement of Akt and NFkappaB signal pathways. J. Cell Biochem. 2008, 103, 1718–1731. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Nie, Y.; Wang, F.; Hou, J.; Cong, X.; Hu, S.; Chen, X. Reciprocal regulation of miR-23a and lysophosphatidic acid receptor signaling in cardiomyocyte hypertrophy. Biochim. Biophys. Acta 2013, 1831, 1386–1394. [Google Scholar] [CrossRef] [PubMed]
- Chandra, M.; Escalante-Alcalde, D.; Bhuiyan, M.S.; Orr, A.W.; Kevil, C.; Morris, A.J.; Nam, H.; Dominic, P.; McCarthy, K.J.; Miriyala, S.; et al. Cardiac-specific inactivation of LPP3 in mice leads to myocardial dysfunction and heart failure. Redox Biol. 2017, 14, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yang, X.Y.; Wang, N.D.; Ding, C.; Yang, Y.J.; You, Z.J.; Su, Q.; Chen, J.H. Serum lysophosphatidic acid concentrations measured by dot immunogold filtration assay in patients with acute myocardial infarction. Scand. J. Clin. Lab. Invest. 2003, 63, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Liu, S.; Liu, X.; Yang, J.; Wang, F.; Cong, X.; Chen, X. Lysophosphatidic acid pretreatment attenuates myocardial ischemia/reperfusion injury in the immature hearts of rats. Front. Physiol. 2017, 8, 153. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kostic, I.; Fidalgo-Carvalho, I.; Aday, S.; Vazao, H.; Carvalheiro, T.; Graos, M.; Duarte, A.; Cardoso, C.; Goncalves, L.; Carvalho, L.; et al. Lysophosphatidic acid enhances survival of human CD34(+) cells in ischemic conditions. Sci. Rep. 2015, 5, 16406. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hou, J.; Shi, L.; Chen, J.; Sang, J.; Hu, S.; Cong, X.; Chen, X. Lysophosphatidic acid protects mesenchymal stem cells against ischemia-induced apoptosis in vivo. Stem Cells Dev. 2009, 18, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Stoddard, N.C.; Chun, J. Promising pharmacological directions in the world of lysophosphatidic acid signaling. Biomol. Ther. 2015, 23, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Llona-Minguez, S.; Ghassemian, A.; Helleday, T. Lysophosphatidic acid receptor (LPAR) modulators: The current pharmacological toolbox. Prog. Lipid Res. 2015, 58, 51–75. [Google Scholar] [CrossRef] [PubMed]
- Nikolaou, A.; Kokotou, M.G.; Limnios, D.; Psarra, A.; Kokotos, G. Autotaxin inhibitors: A patent review (2012–2016). Expert Opin. Ther. Pat. 2017, 27, 815–829. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Effect of ATX and/or LPA on: | Models | Ref. | |||
|---|---|---|---|---|---|
| Preadipocyte proliferation | Preadipocyte differentiation | Diet-induced adiposity | Diet-induced IR/GI | ||
| ↑ | ↓ | ↑ | ↑ | 3T3-L1 (pre)adipocytes, primary murine preadipocytes, ATX+/− mice, FATX−/− mice, fat-specific ATX-overexpressing mice | [31] |
| ↑ | n.d. | n.d. | n.d. | 3T3-F442A preadipocytes, NIH-3T3 fibroblasts | [71,72] |
| ↑ | ↓ | n.d. | n.d. | 3T3-L1 preadipocytes, DFAT-P preadipocytes | [73] |
| ↔ | ↓ | ↑ | ↔ | Primary murine brown preadipocytes, ATX-overexpressing mice | [75] |
| n.d. | ↓ | ↓ | n.d. | 3T3-F442A preadipocytes, SGBS preadipocytes, LPA1-KO mice, primary murine pre-adipocytes | [76] |
| n.d. | n.d. | ↓ | ↑ | FATX−/− mice | [30] |
| n.d. | n.d. | ↔ | ↑ | Chow-fed db/db mice treated with LPA1/3 antagonist (Ki16425) | [77] |
| n.d. | n.d. | n.d. | ↑ | Chow- and HFHS-fed WT mice treated with Ki16425 | [15] |
| n.d. | n.d. | ↔ | ↑ | 3T3-L1 adipocytes, chow- and high-fat diet-fed WT mice treated with Ki16425 | [58] |
| n.d. | n.d. | n.d. | ↔ | 3T3-L1 adipocytes treated with ATX inhibitor (PF-8380) | [55] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Souza, K.; Paramel, G.V.; Kienesberger, P.C. Lysophosphatidic Acid Signaling in Obesity and Insulin Resistance. Nutrients 2018, 10, 399. https://doi.org/10.3390/nu10040399
D’Souza K, Paramel GV, Kienesberger PC. Lysophosphatidic Acid Signaling in Obesity and Insulin Resistance. Nutrients. 2018; 10(4):399. https://doi.org/10.3390/nu10040399
Chicago/Turabian StyleD’Souza, Kenneth, Geena V. Paramel, and Petra C. Kienesberger. 2018. "Lysophosphatidic Acid Signaling in Obesity and Insulin Resistance" Nutrients 10, no. 4: 399. https://doi.org/10.3390/nu10040399
APA StyleD’Souza, K., Paramel, G. V., & Kienesberger, P. C. (2018). Lysophosphatidic Acid Signaling in Obesity and Insulin Resistance. Nutrients, 10(4), 399. https://doi.org/10.3390/nu10040399