l-Glutamine Attenuates DSS-Induced Colitis via Induction of MAPK Phosphatase-1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Reagents
2.3. Colitis Mouse Model
2.4. Histological Examination
2.5. Measurement of TNF-α, Leukotriene B4
2.6. Western Blotting
2.7. Small Interfering RNA (siRNA) Interference
2.8. Statistical Analysis
3. Results
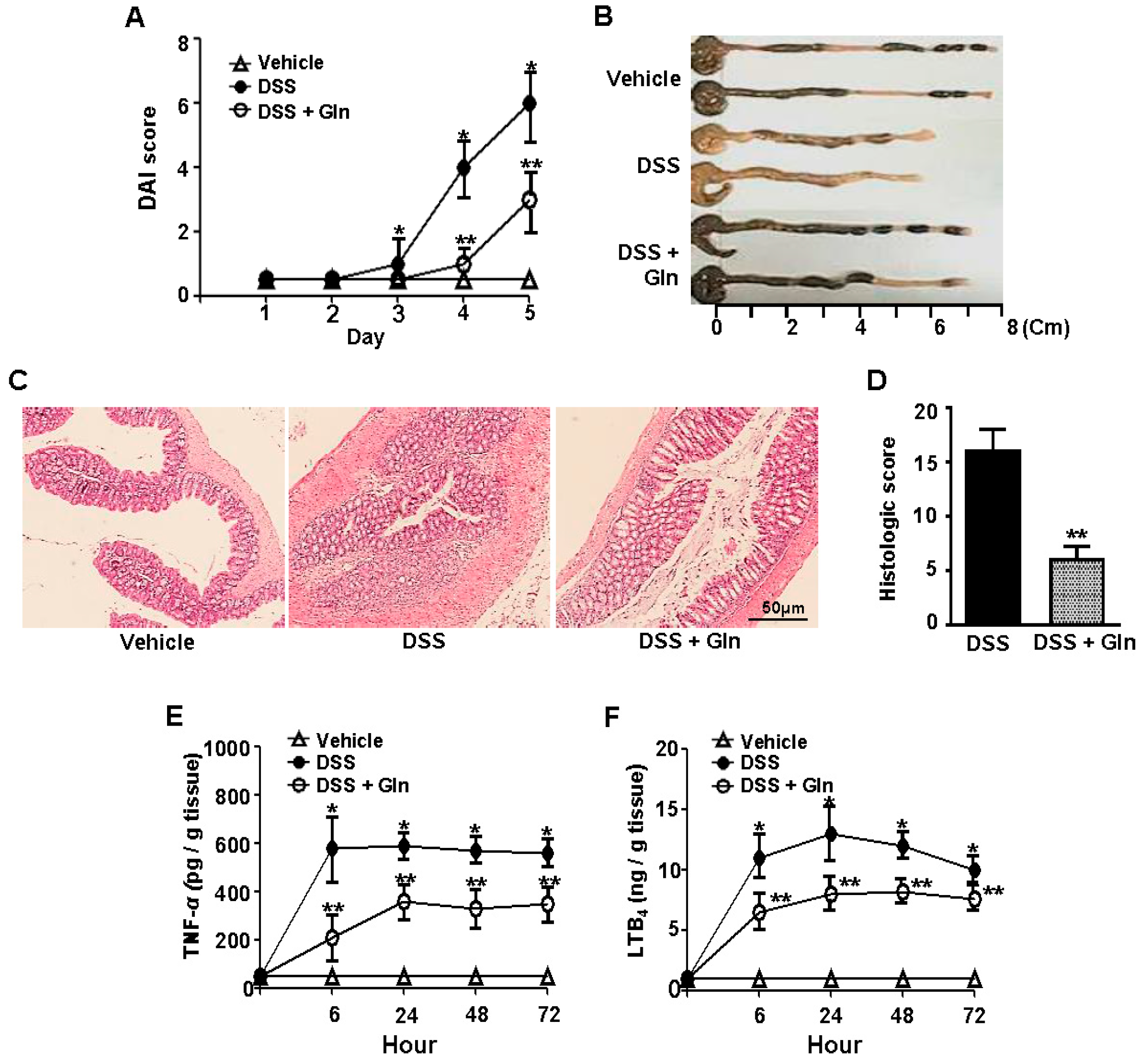
3.1. Oral Gln Intake Improves DSS-Induced Acute Colitis
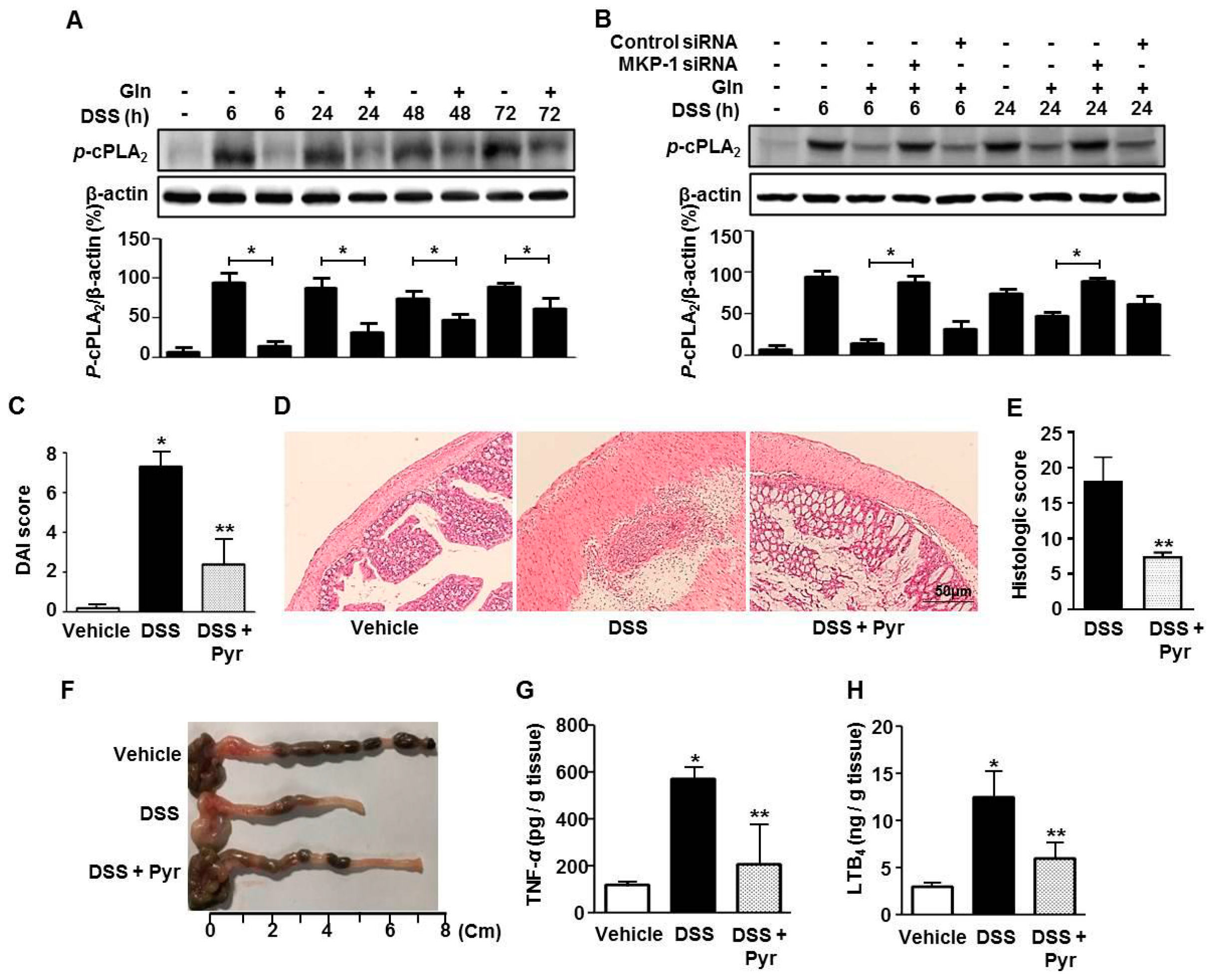
3.2. MKP-1 Induction is Crucial for the Gln’s Beneficial Effect
3.3. MKP-1 Inhibition of cPLA2 Is Associated with the Beneficial Effect of Gln
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Buhner, S.; Buning, C.; Genschel, J.; Kling, K.; Herrmann, D.; Dignass, A.; Kuechler, I.; Krueger, S.; Schmidt, H.H.; Lochs, H. Genetic basis for increased intestinal permeability in families with Crohn’s disease: Role of CARD15 3020insC mutation? Gut 2006, 55, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Wehkamp, J.; Salzman, N.H.; Porter, E.; Nuding, S.; Weichenthal, M.; Petras, R.E.; Shen, B.; Schaeffeler, E.; Schwab, M.; Linzmeier, R.; et al. Reduced Paneth cell α-defensins in ileal Crohn’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 18129–18134. [Google Scholar] [CrossRef] [PubMed]
- Cobrin, G.M.; Abreu, M.T. Defects in mucosal immunity leading to Crohn’s disease. Immunol. Rev. 2005, 206, 277–295. [Google Scholar] [CrossRef] [PubMed]
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Darfeuille-Michaud, A.; Boudeau, J.; Bulois, P.; Neut, C.; Glasser, A.L.; Barnich, N.; Bringer, M.A.; Swidsinski, A.; Beaugerie, L.; Colombel, J.F. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology 2004, 127, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Hoivik, M.L.; Moum, B.; Solberg, I.C.; Cvancarova, M.; Hoie, O.; Vatn, M.H.; Bernklev, T. IBSEN Study Group, Health-related quality of life in patients with ulcerative colitis after a 10-year disease course: Results from the IBSEN study. Inflamm. Bowel Dis. 2012, 18, 1540–1549. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Mitchell, R. Impact of inflammatory bowel disease on quality of life: Results of the European Federation of Crohn’s and Ulcerative Colitis Associations (EFCCA) patient survey. J. Crohns Colitis 2007, 1, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.W.; Qiao, S.Y.; Li, D.F. Amino acids and gut function. Amino Acids 2008, 37, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Windmueller, H.G. Glutamine utilization by the small intestine. Adv. Enzymol. 1982, 53, 202–237. [Google Scholar]
- Windmueller, H.G.; Spaeth, A.E. Uptake and metabolism of plasma glutamine by the small intestine. J. Biol. Chem. 1980, 249, 5070–5079. [Google Scholar] [CrossRef]
- Souba, W.W.; Klimberg, V.S.; Plumley, D.A.; Salloum, R.M.; Flynn, T.C.; Bland, K.I.; Copeland, E.M., II. The role of glutamine in maintaining the gut and supporting the metabolic response to injury and infection. J. Surg. Res. 1990, 48, 383–391. [Google Scholar] [CrossRef]
- Roediger, W.E. Utilization of nutrients by isolated epithelial cells of the rat colon. Gastroenterology 1982, 83, 424–429. [Google Scholar] [PubMed]
- Jacobs, D.O.; Evans, D.A.; O’Dwyer, S.T.; Smith, R.J.; Wilmore, D.W. Disparate effect of fluorouracil on the ileum and colon of enterally fed rats with protection by dietary glutamine. Surg. Forum 1987, 38, 45–49. [Google Scholar]
- Lai, Y.N.; Yeh, S.L.; Lin, M.T.; Shang, H.F.; Yeh, C.L.; Chen, W.J. Glutamine supplementation enhances mucosal immunity in rats with Gut-Derived sepsis. Nutrition 2004, 20, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Vicario, M.; Amat, C.; Rivero, M.; Moretó, M.; Pelegrí, C. Dietary glutamine affects mucosal functions in rats with mild DSS-induced colitis. J. Nutr. 2007, 137, 1931–1937. [Google Scholar] [CrossRef] [PubMed]
- Kaya, E.; Gür, E.S.; Ozgüç, H.; Bayer, A.; Tokyay, R. l-glutamine enemas attenuate mucosal injury in experimental colitis. Dis. Colon Rectum 1999, 42, 1209–1215. [Google Scholar] [CrossRef] [PubMed]
- Crespo, I.; San-Miguel, B.; Prause, C.; Marroni, N.; Cuevas, M.J.; González-Gallego, J.; Tuñón, M.J. Glutamine treatment attenuates endoplasmic reticulum stress and apoptosis in TNBS-induced colitis. PLoS ONE 2012, 7, e50407. [Google Scholar] [CrossRef] [PubMed]
- Fillmann, H.; Kretzmann, N.A.; San-Miguel, B.; Llesuy, S.; Marroni, N.; González-Gallego, J.; Tuñón, M.J. Glutamine inhibits over-expression of pro-inflammatory genes and down-regulates the nuclear factor κB pathway in an experimental model of colitis in the rat. Toxicology 2007, 236, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Funakoshi, T.; Yamashita, K.; Ichikawa, N.; Fukai, M.; Suzuki, T.; Goto, R.; Oura, T.; Kobayashi, N.; Katsurada, T.; Ichihara, S.; et al. A novel NF-κB inhibitor, dehydroxymethylepoxyquinomicin, ameliorates inflammatory colonic injury in mice. J. Crohns Colitis 2012, 6, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.M.; Oh, S.H.; Bang, H.S.; Kang, N.I.; Cho, B.H.; Im, S.Y.; Lee, H.K. Glutamine protects mice from lethal endotoxic shock via a rapid induction of MAPK phosphatase-1. J. Immunol. 2009, 182, 7957–7962. [Google Scholar] [CrossRef] [PubMed]
- Franklin, C.C.; Kraft, A.S. Conditional expression of the mitogen-activated protein kinase (MAPK) phosphatase MKP-1 preferentially inhibits p38 MAPK and stress-activated protein kinase in U937 cells. J. Biol. Chem. 1997, 272, 16917–16923. [Google Scholar] [CrossRef] [PubMed]
- Hammer, M.; Mages, J.; Dietrich, H.; Servatius, A.; Howells, N.; Cato, A.C.; Lang, R. Dual specificity phosphatase 1 (DUSP1) regulates a subset of LPS-induced genes and protects mice from lethal endotoxin shock. J. Exp. Med. 2006, 203, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Kim, H.K.; Kim, J.M.; Ayush, O.; Im, S.Y.; Oh, D.K.; Lee, H.K. Gutamine suppresses airway neutrophilia by blocking cytosolic phospholipase A(2) via an induction of MAPK phosphatase-1. J. Immunol. 2012, 189, 5139–5146. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Kim, H.K.; Jeong, J.S.; Lee, Y.D.; Jin, Z.W.; Im, S.Y.; Lee, H.K. Mechanism of glutamine inhibition of cytosolic phospholipase A2 (cPLA2): Evidence of physical interaction between glutamine-Induced mitogen-activated protein kinase phosphatase-1 and cPLA2. Clin. Exp. Immunol. 2015, 180, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Leslie, C.C. Regulation of arachidonic acid availability for eicosanoid production. Biochem. Cell Biol. 2004, 82, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, B.; Dhalen, S.-E.; Lindgren, J.A.; Rouzer, C.A.; Serhan, C.N. Leukotrienes and lipoxins: Structures, biosynthesis, and biological effects. Science 1987, 237, 1171–1176. [Google Scholar] [CrossRef] [PubMed]
- Niknami, M.; Patel, M.; Witting, P.K.; Dong, Q. Molecules in focus: Cytosolic phospholipase A2-α. Int. J. Biochem. Cell Boil. 2009, 41, 994–997. [Google Scholar] [CrossRef] [PubMed]
- Rosengarten, M.; Hadad, N.; Solomonov, Y.; Lamprecht, S.; Levy, R. Cytosolic phospholipase A2 α has a crucial role in the pathogenesis of DSS-induced colitis in mice. Eur. J. Immunol. 2016, 46, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Cooper, H.S.; Murthy, S.N.; Shah, R.S.; Sedergran, D.J. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab. Investig. 1993, 69, 238–249. [Google Scholar] [PubMed]
- Dieleman, L.A.; Palmen, M.J.; Akol, H.; Bloemena, E.; Peña, A.S.; Meuwissen, S.G.; Van Rees, E.P. Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clin. Exp. Immunol. 1998, 114, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Bjork, J.; Hedqvist, P.; Arfors, K.E. Increase in vascular permeability induced by leukotriene B4 and the role of polymorphonuclear leukocytes. Inflammation 1982, 6, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Di Gennaro, A.; Kenne, E.; Wan, M.; Soehnlein, O.; Lindbom, L.; Haeggström, J.Z. Leukotriene B4-induced changes in vascular permeability are mediated by neutrophil release of heparin-binding protein (HBP/CAP37/azurocidin). FASEB J. 2009, 23, 1750–1757. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.; Barry, S.P.; Roth, R.J.; Wu, J.J.; Jones, E.A.; Bennett, A.M.; Flavell, R.A. Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 2274–2279. [Google Scholar] [CrossRef] [PubMed]
- Kyriakis, J.M.; Avruch, J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 2001, 81, 807–869. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, R.Z.; Small, G.W.; Shi, Y.Y. Evidence that inhibition of p44/42 mitogen-activated protein kinase signaling is a factor in proteasome inhibitor-mediated apoptosis. J. Biol. Chem. 2002, 277, 27864–27871. [Google Scholar] [CrossRef] [PubMed]
- Keyse, S.M.; Emslie, E.A. Oxidative stress and heat shock induce a human gene encoding a protein-tyrosine phosphatase. Nature 1992, 359, 644–647. [Google Scholar] [CrossRef] [PubMed]
- Franklin, C.C.; Srikanth, S.; Kraft, A.S. Conditional expression of mitogen-activated protein kinase phosphatase-1, MKP-1, is cytoprotective against UV-induced apoptosis. Proc. Natl. Acad. Sci. USA 1998, 95, 3014–3019. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xu, J.; Zhou, J.Y.; Liu, Y.; Wu, G.S. Mitogen-activated protein kinase phosphatase-1 is required for cisplatin resistance. Cancer Res. 2006, 66, 8870–8877. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, S.; Machado-Pinilla, R.; Manguan-Garcia, C.; Belda-Iniesta, C.; Moratilla, C.; Cejas, P.; Fresno-Vara, J.A.; de Castro-Carpeno, J.; Casado, E.; Nistal, M.; et al. MKP1/CL100 controls tumor growth and sensitivity to cisplatin in non-small-cell lung cancer. Oncogene 2006, 25, 3335–3345. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yin, D.P.; Liu, Y.X.; Baer, R.; Yin, Y. Dual specificity phosphatase 1/CL100 is a direct transcriptional target of E2F-1 in the apoptotic response to oxidative stress. Cancer Res. 2007, 67, 6737–6744. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gorospe, M.; Hutter, D.; Barnes, J.; Keyse, S.M.; Liu, Y. Transcriptional induction of MKP-1 in response to stress is associated with histone H3 phosphorylation-acetylation. Mol. Cell Biol. 2001, 21, 8213–8224. [Google Scholar] [CrossRef] [PubMed]
- Brondello, J.M.; Pouyssegur, J.; McKenzie, F.R. Reduced MAP kinase phosphatase-1 degradation after p42/p44MAPK-dependent phosphorylation. Science 1999, 286, 2514–2517. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.W.; Yang, J.L. Cooperation of ERK and SCFSkp2 for MKP-1 destruction provides a positive feedback regulation of proliferating signaling. J. Biol. Chem. 2006, 281, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Ayush, O.; Jin, Z.W.; Kim, H.K.; Shin, Y.; Im, S.Y.; Lee, H.K. Glutamine up-regulates MAPK phosphatase-1 induction via activation of Ca2+→ERK cascade pathway. Biochem. Biophys. Rep. 2016, 7, 10–19. [Google Scholar] [PubMed]
- Sklyarov, A.Y.; Panasyuk, N.B.; Fomenko, I.S. Role of nitric oxide-synthase and cyclooxygenase/lipooxygenase systems in development of experimental ulcerative colitis. J. Physiol. Pharmacol. 2011, 62, 65–73. [Google Scholar] [PubMed]
- Wang, D.; Dubois, R.N. The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene 2010, 29, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Karin, M. Mitogen-activated protein kinase cascades and regulation of gene expression. Curr. Opin. Immunol. 1996, 8, 402–411. [Google Scholar] [CrossRef]
- Shmelzer, Z.; Karter, M.; Eisenstein, M.; Leto, T.L.; Hadad, N.; Ben-Menahem, D.; Gitler, D.; Banani, S.; Wolach, B.; Rotem, M.; et al. Cytosolic phospholipase A2α is targeted to the p47phox-PX domain of the assembled NADPH oxidase via a novel binding site in its C2 domain. J. Biol. Chem. 2008, 283, 31898–31908. [Google Scholar] [CrossRef] [PubMed]
- Kukreja, R.C.; Kontos, H.A.; Hess, M.L.; Ellis, E.F. PGH synthase and lipoxygenase generate superoxide in the presence of NADH or NADPH. Circ. Res. 1986, 59, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Laurent, B.; Raymond, A. Reactive oxygen species: Production and role in the kidney. Am. J. Physiol. 1986, 251, F765–F776. [Google Scholar]
- Okabe, E.; Kato, Y.; Kohno, H.; Hess, M.L.; Ito, H. Inhibition by free radical scavengers and by cyclooxygenase inhibitors of the effect of acidosis on calcium transport by masseter muscle sarcoplasmic reticulum. Biochem. Pharmacol. 1985, 34, 961–968. [Google Scholar] [CrossRef]
- Wei, E.P.; Kontos, H.A.; Dietrich, W.D.; Povlishock, J.T.; Ellis, E.F. Inhibition by free radical scavengers and by cyclooxygenase inhibitors of pial arteriolar abnormalities from concussive brain injury in cats. Circ. Res. 1981, 48, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, S.-Y.; Im, Y.N.; Youm, J.Y.; Lee, H.-K.; Im, S.-Y. l-Glutamine Attenuates DSS-Induced Colitis via Induction of MAPK Phosphatase-1. Nutrients 2018, 10, 288. https://doi.org/10.3390/nu10030288
Jeong S-Y, Im YN, Youm JY, Lee H-K, Im S-Y. l-Glutamine Attenuates DSS-Induced Colitis via Induction of MAPK Phosphatase-1. Nutrients. 2018; 10(3):288. https://doi.org/10.3390/nu10030288
Chicago/Turabian StyleJeong, Soo-Yeon, Yoo Na Im, Ji Young Youm, Hern-Ku Lee, and Suhn-Young Im. 2018. "l-Glutamine Attenuates DSS-Induced Colitis via Induction of MAPK Phosphatase-1" Nutrients 10, no. 3: 288. https://doi.org/10.3390/nu10030288
APA StyleJeong, S.-Y., Im, Y. N., Youm, J. Y., Lee, H.-K., & Im, S.-Y. (2018). l-Glutamine Attenuates DSS-Induced Colitis via Induction of MAPK Phosphatase-1. Nutrients, 10(3), 288. https://doi.org/10.3390/nu10030288