Thromboxane-Dependent Platelet Activation in Obese Subjects with Prediabetes or Early Type 2 Diabetes: Effects of Liraglutide- or Lifestyle Changes-Induced Weight Loss †




Abstract
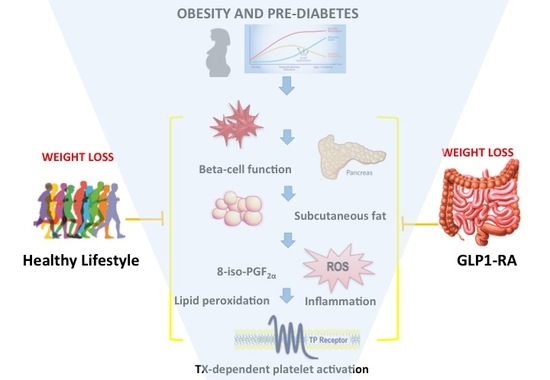
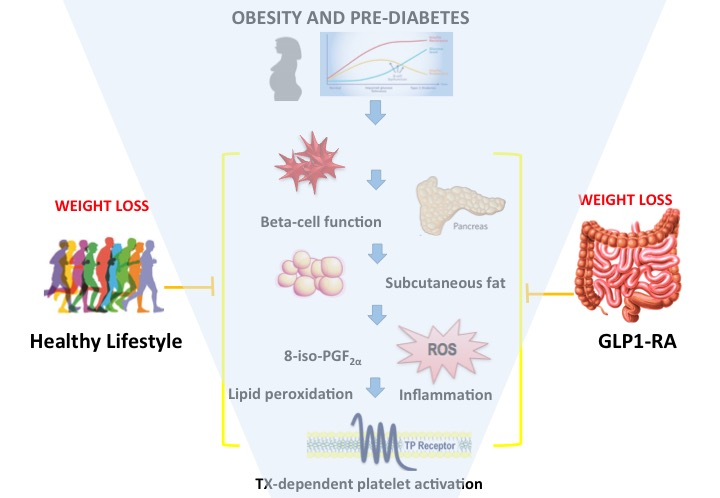
1. Introduction
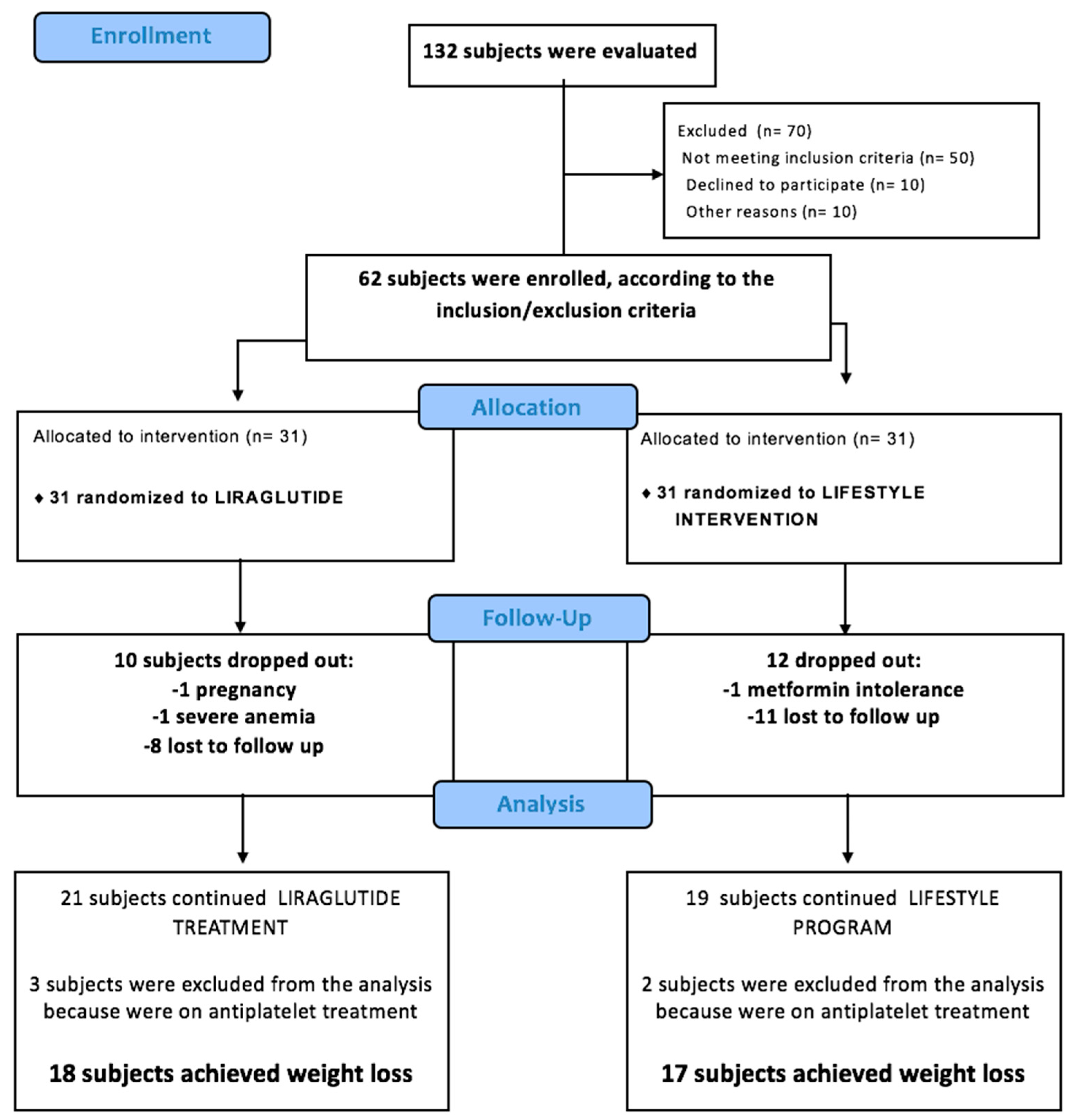
2. Materials and Methods
2.1. Subjects and Study Design
2.1.1. Lifestyle Intervention Program
2.1.2. Oral glucose tolerance test (OGTT) with Frequent Sampling
2.1.3. Magnetic Resonance Imaging (MRI) Quantification of Visceral and Subcutaneous Fat
2.2. Analytical Measurements
2.2.1. Biological Material Collection
2.2.2. Biochemical Measurements
2.2.3. Urinary Eicosanoid Assays
2.3. Statistical Analysis
3. Results
3.1. Baseline Evaluation
3.2. Effects of Liraglutide and Lifestyle Interventions
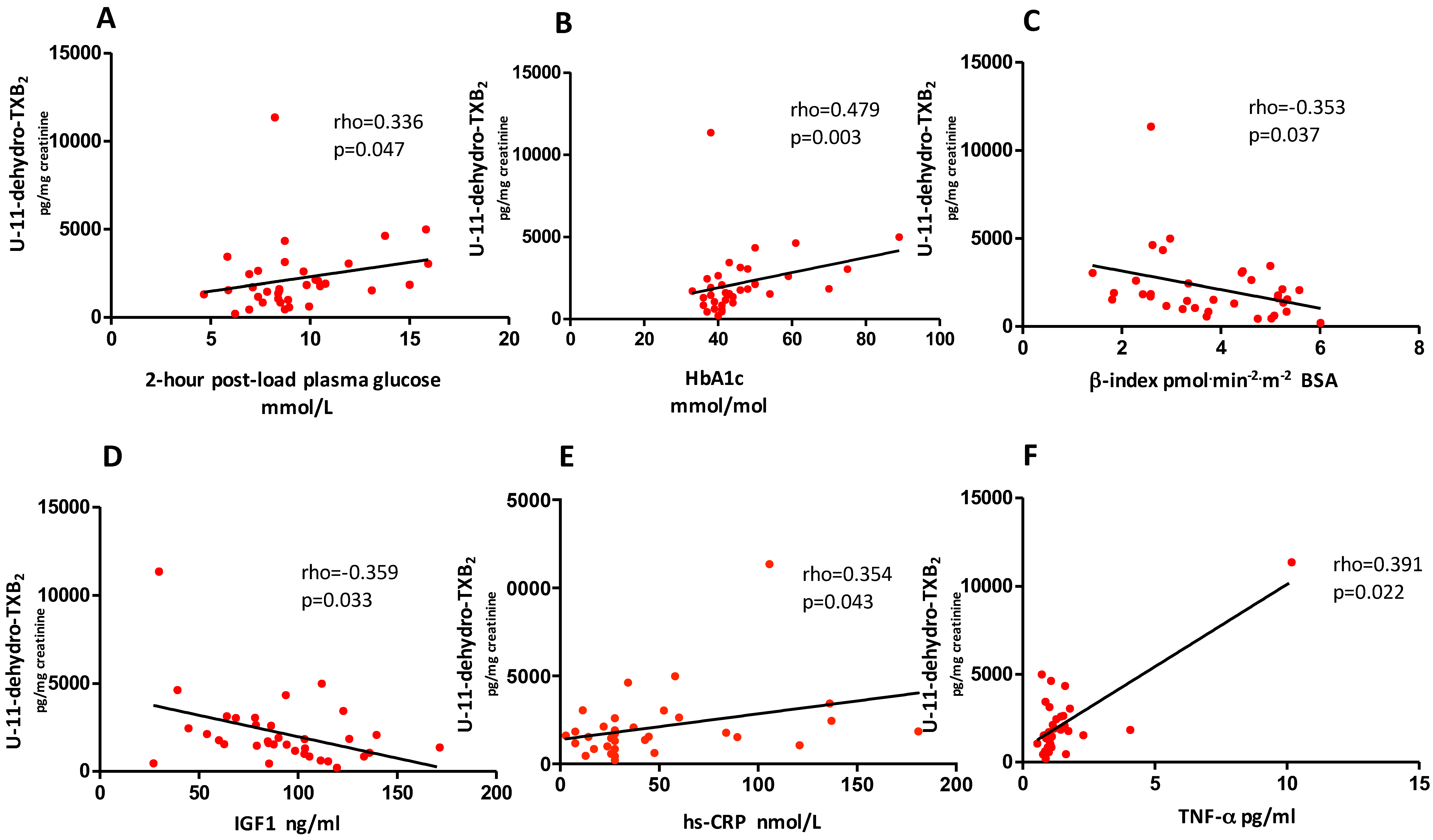
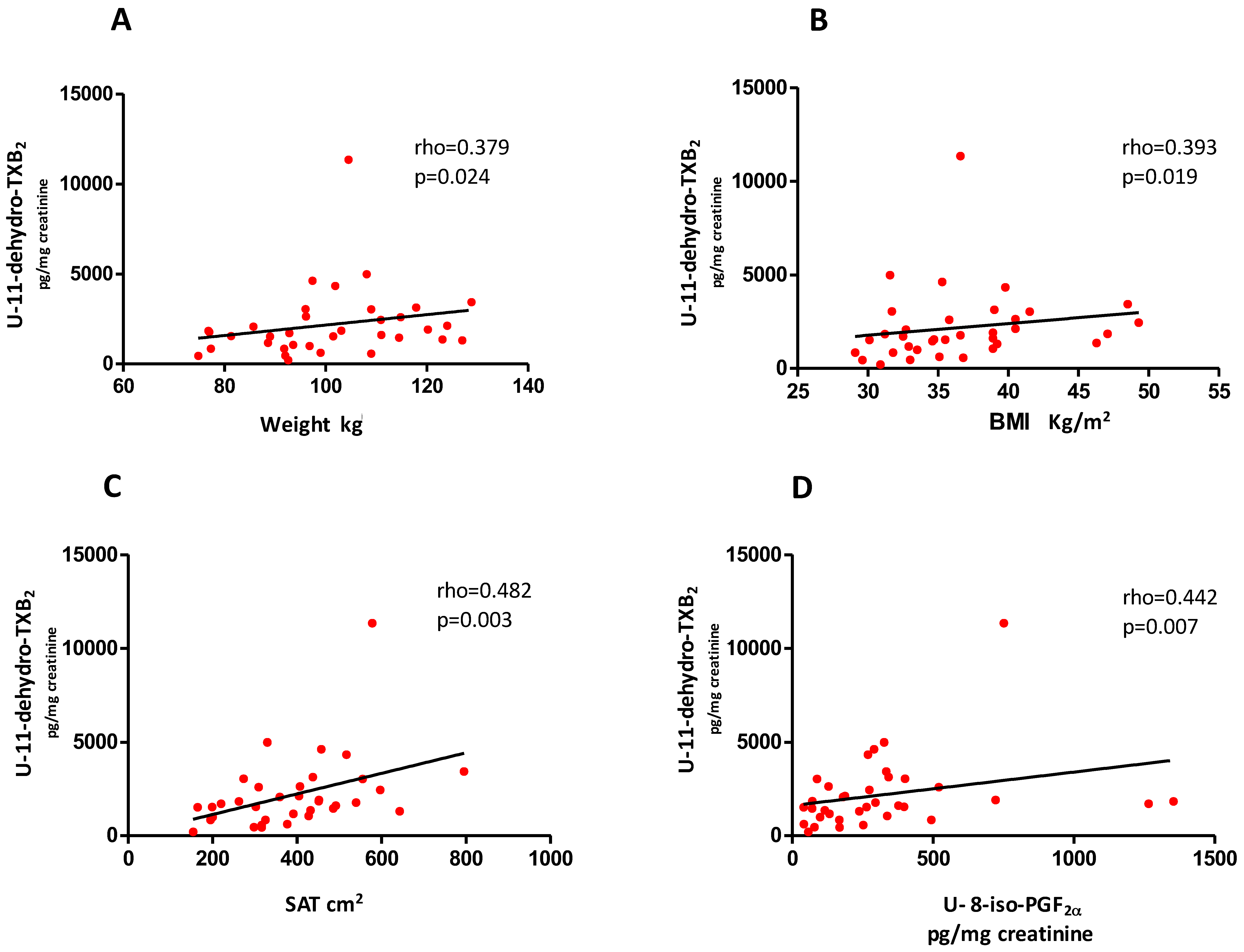
3.3. Predictors of Change in U-11-Dehydro-TXB2 after Weight Loss
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Després, J.P. Intra-abdominal obesity: An untreated risk factor for Type 2 diabetes and cardiovascular disease. J. Endocrinol. Invest. 2006, 29, 77–82. [Google Scholar] [PubMed]
- Davì, G.; Catalano, I.; Averna, M.; Notarbartolo, A.; Strano, A.; Ciabattoni, G.; Patrono, C. Thromboxane biosynthesis and platelet function in type II diabetes mellitus. N. Engl. J. Med. 1990, 322, 1769–1774. [Google Scholar] [CrossRef] [PubMed]
- Santilli, F.; Davì, G.; Consoli, A.; Cipollone, F.; Mezzetti, A.; Falco, A.; Taraborelli, T.; Devangelio, E.; Ciabattoni, G.; Basili, S.; et al. Thromboxane-dependent CD40 ligand release in type 2 diabetes mellitus. J. Am. Coll. Cardiol. 2006, 47, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Davì, G.; Patrono, C. Platelet activation and atherothrombosis. N. Engl. J. Med. 2007, 357, 2482–2494. [Google Scholar] [CrossRef] [PubMed]
- Davì, G.; Guagnano, MT.; Ciabattoni, G.; Basili, S.; Falco, A.; Marinopiccoli, M.; Nutini, M.; Sensi, S.; Patrono, C. Platelet activation in obese women: Role of inflammation and oxidant stress. JAMA 2002, 288, 2008–2014. [Google Scholar] [CrossRef] [PubMed]
- Santilli, F.; Formoso, G.; Sbraccia, P.; Averna, M.; Miccoli, R.; Di Fulvio, P.; Ganci, A.; Pulizzi, N.; Lattanzio, S.; Ciabattoni, G.; et al. Postprandial hyperglycemia is a determinant of platelet activation in early type 2 diabetes mellitus. J. Thromb. Haemost. 2010, 8, 828–837. [Google Scholar] [CrossRef]
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998, 352, 837–853. [Google Scholar] [CrossRef]
- Holman, R.R.; Paul, S.K.; Bethel, M.A.; Matthews, D.R.; Neil, H.A. 10-year follow-up of intensive glucose control in type 2 diabetes. N. Engl, J. Med. 2008, 359, 1577–1589. [Google Scholar] [CrossRef]
- Gerstein, H.C.; Miller, M.E.; Byington, R.P.; Goff, D.C.Jr.; Bigger, J.T.; Buse, J.B.; Cushman, W.C.; Genuth, S.; Ismail-Beigi, F.; Grimm, R.H.Jr.; et al. Action to Control Cardiovascular Risk in Diabetes Study Group. Effects of intensive glucose lowering in type 2 diabetes. N. Engl. J. Med. 2008, 358, 2545–2559. [Google Scholar] [CrossRef]
- Patel, A.; MacMahon, S.; Chalmers, J.; Neal, B.; Billot, L.; Woodward, M.; Marre, M.; Cooper, M.; Glasziou, P.; Grobbee, D.; et al. ADVANCE Collaborative Group. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 2008, 358, 2560–2572. [Google Scholar] [CrossRef]
- Duckworth, W.; Abraira, C.; Moritz, T.; Reda, D.; Emanuele, N.; Reaven, P.D.; Zieve, F.J.; Marks, J.; Davis, S.N.; Hayward, R.; et al. VADT Investigators. Glucose control and vascular complications in veterans with type 2 diabetes. N. Engl, J. Med. 2009, 360, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Stefan, N.; Fritsche, A.; Schick, F.; Häring, H.U. Phenotypes of prediabetes and stratification of cardiometabolic risk. Lancet Diabetes Endocrinol. 2016, 4, 789–798. [Google Scholar] [CrossRef]
- Monami, M.; Dicembrini, I.; Marchionni, N.; Rotella, C.M.; Mannucci, E. Effects of glucagon-like peptide-1 receptor agonists on body weight: A meta-analysis. Exp. Diabetes Res. 2012, 2012, 672658. [Google Scholar] [CrossRef] [PubMed]
- Orskov, C.; Wettergren, A.; Holst, J.J. Secretion of the incretin hormones glucagon-like peptide-1 and gastric inhibitory polypeptide correlates with insulin secretion in normal man throughout the day. Scand. J. Gastroenterol. 1996, 31, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Cameron-Vendrig, A.; Reheman, A.; Siraj, M.A.; Xu, X.R.; Wang, Y.; Lei, X.; Afroze, T.; Shikatani, E.; El-Mounayri, O.; Noyan, H.; et al. Glucagon-Like Peptide 1 Receptor Activation Attenuates Platelet Aggregation and Thrombosis. Diabetes 2016, 65, 1714–1723. [Google Scholar] [CrossRef] [PubMed]
- Barale, C.; Buracco, S.; Cavalot, F.; Frascaroli, C.; Guerrasio, A.; Russo, I. Glucagon-like peptide 1-related peptides increase nitric oxide effects to reduce platelet activation. Thromb. Haemost. 2017, 117, 1115–1128. [Google Scholar] [CrossRef] [PubMed]
- Santilli, F.; Simeone, P.; Guagnano, M.T.; Leo, M.; Maccarone, M.T.; Di Castelnuovo, A.; Sborgia, C.; Bonadonna, R.C.; Angelucci, E.; Federico, V.; et al. Effects of Liraglutide on weight loss, fat distribution and beta-cell function in obese subjects with prediabetes or early type 2 diabetes mellitus. Diabetes Care 2017, 40, 1556–1564. [Google Scholar] [CrossRef]
- American Diabetes Association. Classification and diagnosis of diabetes. Diabetes Care 2015, 38, S8–S16. [Google Scholar] [CrossRef]
- Knowler, W.C.; Barrett-Connor, E.; Fowler, S.E.; Hamman, R.F.; Lachin, J.M.; Walker, E.A.; Nathan, D.M. Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N. Engl. J. Med. 2002, 346, 393–403. [Google Scholar] [CrossRef]
- Basili, S.; Pacini, G.; Guagnano, M.T.; Manigrasso, M.R.; Santilli, F.; Pettinella, C.; Ciabattoni, G.; Patrono, C.; Davì, G. Insulin resistance as a determinant of platelet activation in obese women. J. Am. Coll. Cardiol. 2006, 48, 2531–2538. [Google Scholar] [CrossRef]
- Ross, R.; Léger, L.; Morris, D.; de Guise, J.; Guardo, R. Quantification of adipose tissue by MRI: Relationship with anthropometric variables. J. Appl. Physiol. 1992, 72, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Cretti, A.; Lehtovirta, M.; Bonora, E.; Brunato, B.; Zenti, M.G.; Tosi, F.; Caputo, M.; Caruso, B.; Groop, L.C.; Muggeo, M.; et al. Assessment of beta-cell function during the ora glucose tolerance test by a minimal model of insulin secretion. Eur. J. Clin. Invest. 2001, 31, 405–416. [Google Scholar] [CrossRef]
- Matsuda, M.; DeFronzo, R.A. Insulin sensitivity indices obtained from oral glucose tolerance testing: Comparison with the euglycemic insulin clamp. Diabetes Care 1999, 22, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Khuu, H.M.; Robinson, C.A.; Goolsby, K.; Hardy, R.W.; Konrad, R.J. Evaluation of a fully automated high-performance liquid chromatography assay for hemoglobin A1c. Arch. Pathol. Lab. Med. 1999, 123, 763–767. [Google Scholar] [PubMed]
- Matthews, D.R.; Hosker, J.P.; Rudenski, A.S.; Naylor, B.A.; Treacher, D.F.; Turner, R.C. Homeostasis model assessment: Insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985, 28, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Cianfarani, S.; Inzaghi, E.; Alisi, A.; Germani, D.; Puglianiello, A.; Nobili, V. Insulin-like growth factor-I and -II levels are associated with the progression of nonalcoholic fatty liver disease in obese children. J. Pediatr. 2014, 165, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Davì, G.; Averna, M.; Catalano, I.; Barbagallo, C.; Ganci, A.; Notarbartolo, A.; Ciabattoni, G.; Patrono, C. Increased thromboxane biosynthesis in type IIa hypercholesterolemia. Circulation 1992, 85, 1792–1798. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ciabattoni, G.; Créminon, C.; Lawson, J.; Fitzgerald, G.A.; Patrono, C.; Maclouf, J. Immunological characterization of urinary 8-epi-prostaglandin F2 alpha excretion in man. J. Pharmacol. Exp. Ther. 1995, 275, 94–100. [Google Scholar]
- Brownlee, M. The Pathobiology of Diabetic Complications. A Unifying Mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef]
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; et al. LEADER Steering Committee; LEADER Trial Investigators. Liraglutide and cardiovascular outcomes in type 2 diabetes. N. Engl J. Med. 2016, 375, 311–322. [Google Scholar] [CrossRef]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jódar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. SUSTAIN-6 Investigators. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 2016, 375, 1834–1844. [Google Scholar] [CrossRef] [PubMed]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. EMPAREG OUTCOME Investigators. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Ghani, M.; DeFronzo, R.A.; Del Prato, S.; Chilton, R.; Singh, R.; Ryder, R.E.J. Cardiovascular Disease and Type 2 Diabetes: Has the Dawn of a New Era Arrived? Diabetes Care 2017, 40, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Defronzo, R.A. From the triumvirate to the ominous octet: A new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 2009, 58, 773–795. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E. Definition of intervention points in prediabetes. Lancet Diabetes Endocrinol. 2014, 2, 667–675. [Google Scholar] [CrossRef]
- Abdul-Ghani, M.A.; Lyssenko, V.; Tuomi, T.; DeFronzo, R.A.; Groop, L. Fasting versus postload plasma glucose concentration and the risk for future type 2 diabetes: Results from the Botnia Study. Diabetes Care 2009, 32, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Faerch, K.; Witte, D.R.; Tabák, A.G.; Perreault, L.; Herder, C.; Brunner, E.J.; Kivimäki, M.; Vistisen, D. Trajectories of cardiometabolic risk factors before diagnosis of three subtypes of type 2 diabetes: A post-hoc analysis of the longitudinal Whitehall II cohort study. Lancet Diabetes Endocrinol. 2013, 1, 43–51. [Google Scholar] [CrossRef]
- Nauck, M.A.; Vardarli, I.; Deacon, C.F.; Holst, J.J.; Meier, J.J. Secretion of glucagon-like peptide-1 (GLP-1) in type 2 diabetes: What is up, what is down? Diabetologia 2011, 54, 10–18. [Google Scholar] [CrossRef]
- Halvorsen, B.; Santilli, F.; Scholz, H.; Sahraoui, A.; Gulseth, H.L.; Wium, C.; Lattanzio, S.; Formoso, G.; Di Fulvio, P.; et al. LIGHT/TNFSF14 is increased in patients with type 2 diabetes mellitus and promotes islet cell dysfunction and endothelial cell inflammation in vitro. Diabetologia 2016, 59, 2134–2144. [Google Scholar] [CrossRef]
- Davì, G.; Santilli, F.; Vazzana, N. Thromboxane receptors antagonists and/or synthase inhibitors. Handb. Exp. Pharmacol. 2012, 210, 261–286. [Google Scholar] [CrossRef]
- Shuster, A.; Patlas, M.; Pinthus, J.H.; Mourtzakis, M. The clinical importance of visceral adiposity: A critical review of methods for visceral adipose tissue analysis. Br. J. Radiol. 2012, 85, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Spoto, B.; Di Betta, E.; Mattace-Raso, F.; Sijbrands, E.; Vilardi, A.; Parlongo, R.M.; Pizzini, P.; Pisano, A.; Vermi, W.; Testa, A.; et al. Pro- and anti-inflammatory cytokine gene expression in subcutaneous and visceral fat in severe obesity. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 1137–1143. [Google Scholar] [CrossRef] [PubMed]
- Schlecht, I.; Fischer, B.; Behrens, G.; Leitzmann, M.F. Relations of Visceral and Abdominal Subcutaneous Adipose Tissue, Body Mass Index, and Waist Circumference to Serum Concentrations of Parameters of Chronic Inflammation. Obes. Facts 2016, 9, 144–157. [Google Scholar] [CrossRef] [PubMed]
- Fox, C.S.; Massaro, J.M.; Hoffmann, U.; Pou, K.M.; Maurovich-Horvat, P.; Liu, C.Y.; Vasan, R.S.; Murabito, J.M.; Meigs, J.B.; Cupples, L.A.; et al. Abdominal visceral and subcutaneous adipose tissue compartments: Association with metabolic risk factors in the Framingham Heart Study. Circulation 2007, 116, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Abraham, T.M.; Pedley, A.; Massaro, J.M.; Hoffmann, U.; Fox, C.S. Association between visceral and subcutaneous adipose depots and incident cardiovascular disease risk factors. Circulation 2015, 132, 1639–1647. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Pedley, A.; Hoffmann, U.; Massaro, J.M.; Fox, C.S. Association of Changes in Abdominal Fat Quantity and Quality With Incident Cardiovascular Disease Risk Factors. J. Am. Coll. Cardiol. 2016, 68, 1509–1521. [Google Scholar] [CrossRef] [PubMed]
- Mohamed-Ali, V.; Goodrick, S.; Rawesh, A.; Katz, D.R.; Miles, J.M.; Yudkin, J.S.; Klein, S.; Coppack, S.W. Subcutaneous adipose tissue releases interleukin-6, but not tumor necrosis factor-alpha, in vivo. J. Clin. Endocrinol. Metab. 1997, 82, 4196–4200. [Google Scholar] [CrossRef]
- Matsubara, Y.; Murata, M.; Ikeda, Y. Culture of megakaryocytes and platelets from subcutaneous adipose tissue and a preadipocyte cell line. Methods. Mol. Biol. 2012, 788, 249–258. [Google Scholar] [CrossRef]
- Chon, S.; Gautier, J.F. An Update on the Effect of Incretin-Based Therapies on β-Cell Function and Mass. Diabetes Metab. J. 2016, 40, 99–114. [Google Scholar] [CrossRef]
- Retnakaran, R.; Kramer, C.K.; Choi, H.; Swaminathan, B.; Zinman, B. Liraglutide and the preservation of pancreatic β-cell function in early type 2 diabetes: The LIBRA trial. Diabetes Care 2014, 37, 3270–3278. [Google Scholar] [CrossRef]
- Jia, G.; Aroor, A.R.; Sowers, J.R. Glucagon-Like Peptide 1 Receptor Activation and Platelet Function: Beyond Glycemic Control. Diabetes 2016, 65, 1487–1489. [Google Scholar] [CrossRef] [PubMed]
- Lattanzio, S.; Santilli, F.; Liani, R.; Vazzana, N.; Ueland, T.; Di Fulvio, P.; Formoso, G.; Consoli, A.; Aukrust, P.; Davì, G. Circulating dickkopf-1 in diabetes mellitus: Association with platelet activation and effects of improved metabolic control and low-dose aspirin. J. Am. Heart Assoc. 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Santilli, F.; Liani, R.; Di Fulvio, P.; Formoso, G.; Simeone, P.; Tripaldi, R.; Ueland, T.; Aukrust, P.; Davì, G. Increased circulating resistin is associated with insulin resistance, oxidative stress and platelet activation in type 2 diabetes mellitus. Thromb. Haemost. 2016, 116, 1089–1099. [Google Scholar] [CrossRef] [PubMed]
- Simeone, P.; Boccatonda, A.; Liani, R.; Santilli, F. Significance of urinary 11-dehydro-thromboxane b2 in age-related diseases: Focus on atherothrombosis. Ageing Res. Rev. 2018, 48, 51–78. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association. Standards of Medical Care in Diabetes—2018. Diabetes Care 2018, 41, S4–S6. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, L.V.; Flint, A.; Olsen, A.K.; Ingwersen, S.H. Liraglutide in Type 2 Diabetes Mellitus: Clinical Pharmacokinetics and Pharmacodynamics. Clin. Pharmacokinet. 2016, 55, 657–672. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Pre-Liraglutide (n = 18) | Pre-Lifestyle (n = 17) | p-value * |
|---|---|---|---|
| Age (years) | 55.5 (48.0–64.0) | 53.0 (51.0–55.0) | 0.753 |
| Gender (male), n (%) | 11 (61) | 8 (47) | 0.505 |
| BMI (kg/m2) | 36.6 (34.6–39.0) | 33.5 (31.2–39.8) | 0.222 |
| Type 2 diabetes, n (%) | 8 (44) | 7 (41) | - |
| IGT/IFG, n (%) | 10 (59) | 10 (56) | - |
| Waist (cm) | 114.5 (112.0–127.0) | 105.0 (100.0–117.0) | 0.021 |
| Systolic BP (mmHg) | 146.0 (130.0–154.0) | 137 (123–144) | 0.120 |
| Diastolic BP (mmHg) | 84.0 (79.0–88.0) | 80 (72–84) | 0.124 |
| Smoke, n (%) | 4 (22) | 0 (0) | 0.153 |
| Hypertension, n (%) | 15 (83) | 9 (53) | 0.075 |
| Dyslipidemia, n (%) | 7 (39) | 8 (47) | 0.738 |
| CVD, n (%) | 1 (5.6) | 3 (17.6) | 0.337 |
| Previous MI, or revascularization, n (%) | 0 (0) | 0 (0) | - |
| Previous TIA/stroke, o revascularization, n (%) | 0 (0) | 1 (5.9) | 0.485 |
| PAD, n (%) | 1 (5.6) | 0 (0) | - |
| Carotid stenosis, n (%) | 1 (5.6) | 4 (23) | 0.177 |
| Microvascular disease, n (%) | 0 (0) | 1 (5.9) | 0.485 |
| Total cholesterol (mmol/L) | 4.3 (3.6–4.8) | 4.4 (4.0–4.6) | 0.632 |
| High density lipoprotein (HDL) cholesterol (mmol/L) | 1.1 (1.0–1.4) | 1.1 (1.0–1.3) | 0.934 |
| Triglycerides (mmol/L) | 1.4 (0.9–2.3) | 1.1 (0.8–1.3) | 0.116 |
| Amylase (U/L) | 60.5 (55.0–71.0) | 64.0 (52.0–75.0) | 0.973 |
| Lipase (U/L) | 106.0 (76.0–118.0) | 118.0 (71.0–156.0) | 0.241 |
| Fasting plasma glucose (mmol/L) | 5.1 (4.9–5.9) | 5.3 (5.1–5.7) | 0.791 |
| 1-h post load plasma glucose (mmol/L) | 10.6 (9.3–11.2) | 10.2 (8.7–11.3) | 0.428 |
| 2-h post load plasma glucose (mmol/L) | 8.7 (8.2–10.5) | 8.5 (6.9–10.3) | 0.338 |
| Glycated hemoglobin (HbA1c) (%) | 5.9 (5.6–6.4) | 6.1 (5.8–6.5) | 0.596 |
| HbA1c (mmol/mol) | 41 (38–46) | 43 (40–48) | 0.596 |
| Fasting plasma insulin (µU/mL) | 13.3 (9.5–21.0) | 10.8 (8.7–16.5) | 0.541 |
| 1-h post load plasma insulin (µU/mL) | 53.7 (29.2–105.8) | 78.7 (54.6–95.6) | 0.447 |
| 2-h post load plasma insulin (µU/mL) | 76.9 (44.3–101.9) | 75.3 (57.2–115.4) | 0.467 |
| Creatinine (µmol/L) | 61.6 (61.6–70.4) | 70.4 (61.6–79.2) | 0.289 |
| Total bilirubin (µmol/L) | 10 (9–15) | 12 (7–14) | 0.753 |
| hs-C-reactive protein (nmol/L) | 27.6 (25.7–58.1) | 27.6 (22.8–52.4) | 0.800 |
| Aspartate aminotransferase (AST) (U/L) | 29.0 (24.0–39.0) | 33 (27–44) | 0.427 |
| Alanine aminotransferase (ALT) (U/L) | 38.5 (36.0–45.0) | 42 (33–59) | 0.704 |
| Metformin, n (%) | 18 (100) | 17 (100) | - |
| ACE-I, n (%) | 3 (17) | 3 (18) | - |
| ARBs, n (%) | 6 (33) | 4 (23) | 0.711 |
| Diuretics, n (%) | 5 (28) | 3 (18) | 0.690 |
| B-blockers, n (%) | 6 (33) | 2 (12) | 0.228 |
| CCA, n (%) | 0 (0) | 0 (0) | - |
| Statins, n (%) | 2 (11) | 3 (18) | 0.658 |
| Fibrates, n (%) | 0 (0) | 0 (0) | - |
| Polyunsaturated fatty acid (PUFA), n (%) | 0 (0) | 0 (0) | - |
| Proton Pump Inhibitors, n (%) | 3 (17) | 2 (12) | - |
| ASA, n (%) | 0 (0) | 0 (0) | - |
| IGF-I (ng/mL) | 85.7 (64.0–111.4) | 98.5 (78.7–119.6) | 0.322 |
| Urinary-11-dehydro-thromboxane B2 (U-11-dehydro-TXB2 ) (pg/mg creatinine) | 1659.5 (1050.0–2589.0) | 1833.0 (1170.0–2636.0) | 0.947 |
| Urinary-8-iso-prostaglandin (PG)F2α (U-8-iso-PGF2α )(pg/mg creatinine) | 284.5 (115.0–377.0) | 187.0 (129.0–334.0) | 0.355 |
| SAT (cm2) | 429.2 (315.7–491.4) | 358.9 (262.0–450.6) | 0.234 |
| VAT (cm2) | 303.9 (255.3–337.6) | 253.0 (162.6–307.5) | 0.027 |
| TNF-α (pg/mL) | 1.01 (0.94–1.44) | 1.07 (0.87–1.53) | 0.958 |
| Leptin (pg/mL) | 17.02 (10.8–39.3) | 28.02 (13.27–44.73) | 0.667 |
| β-index (pmol·min−2·m−2 Body Surface Area) | 3.41 (2.58–5.08) | 4.27 (2.90–5.0) | 0.306 |
| Matsuda-index | 2.9 (2.3–4.4) | 2.8 (2.1–4.3) | 0.670 |
| Predictor Variable | Parameter Estimate | SE | P-Value | Partial R2 | Percentage of Variation Relative to 1-SD of Ln-U-11-dehydro-TXB2 for 1-SD Change in the Predictor Variable | SD |
|---|---|---|---|---|---|---|
| Ln-U-8-iso-PGF2α | 0.31 | 0.10 | 0.0088 | 25.3% | +33.2% (95% CI: 9.3% to 57.0%) | 0.83 |
| Ln-HbA1c | 2.64 | 0.69 | 0.0011 | 16.3% | +51.3% (95% CI: 23.3% to 79.7%) | 0.15 |
| Ln-TNF-α | 0.58 | 0.19 | 0.0075 | 9.8% | +39.2% (95% CI: 11.7% to 66.7%) | 0.53 |
| SAT | 0.14 | 0.06 | 0.044 | 16.4% | +25.7% (95% CI: 0.7% to 50.7%) | 1.46 |
| Variable | Pre-Treatment (n = 35) | Post-Treatment (n = 35) | p-Value * |
|---|---|---|---|
| BMI (kg/m2) | 35.5 (32.5–39.2) | 33.0 (30.2–36.2) | <0.001 |
| Waist (cm) | 112.5 (103.0–121.0) | 108.0 (99.0–117.0) | <0.001 |
| Weight (kg) | 99.0 (91.8–111.0) | 90.5 (84.3–103) | <0.001 |
| Systolic BP (mmHg) | 141.0 (127.0–150.0) | 133.0 (125.0–145.0) | 0.093 |
| Diastolic BP (mmHg) | 81.0 (77.0–86.0) | 80.0 (72.0–85.0) | 0.665 |
| Total cholesterol (mmol/L) | 4.4 (3.8–4.8) | 4.1 (3.6–4.5) | 0.011 |
| HDL cholesterol (mmol/L) | 1.1 (1.0–1.4) | 1.1 (0.9–1.3) | 0.078 |
| Triglycerides (mmol/L) | 1.3 (0.9–1.5) | 1.2 (0.9–1.7) | 0.252 |
| Amylase (U/L) | 64.0 (53.0–75.0) | 73.5 (53.0–83.0) | 0.024 |
| Lipase (U/L) | 113.0 (71.0–152.0) | 124.0 (100.0–193.0) | 0.085 |
| Fasting plasma glucose (mmol/L) | 5.3 (5.0–5.6) | 4.9 (4.6–5.2) | 0.0008 |
| 1-h post load plasma glucose (mmol/L) | 10.4 (9.0–11.3) | 9.0 (7.7–9.9) | 0.0004 |
| 2-h post load plasma glucose (mmol/L) | 8.7 (7.4–10.4) | 7.7 (5.1–10.2) | 0.0001 |
| HbA1c (%) | 6.0 (5.7–6.5) | 5.6 (5.4–6.0) | <0.0001 |
| HbA1c (mmol/mol) | 42 (39–48) | 38 (36–42) | <0.0001 |
| Fasting plasma insulin (µU/mL) | 11.5 (8.8–21.0) | 9.1 (6.4–11.9) | 0.0007 |
| 1-h post load plasma insulin (µU/mL) | 71.8 (37.1–105.8) | 65.3 (38.1–113.6) | 0.928 |
| 2-h post load plasma insulin (µU/mL) | 75.3 (50.4–107.7) | 55.9 (31.4–113.1) | 0.171 |
| Creatinine (µmol/L) | 67.8 (59.0–79.2) | 68.6 (60.7–79.2) | 0.862 |
| Total bilirubin (µmol/L) | 11 (7–15) | 10 (8–13) | 0.489 |
| hs-C-reactive protein (nmol/L) | 27.6 (23.8–58.1) 48.3 ± 7.6 * | 27.6 (12.4–43.8) 33.8 ± 5.3 * | 0.0006 |
| AST (U/L) | 32.0 (24.0–39.0) | 23.0 (20.0–28.0) | <0.0001 |
| ALT (U/L) | 40.0 (34.0–58.0) | 33.0 (27.0–41.0) | <0.0001 |
| IGF-I (ng/mL) | 90.2 (68.6–111.9) | 100.0 (75.8–128.7) | 0.035 |
| U-11-dehydro-TXB2 (pg/mg creatinine) | 1710 (1050–2636) | 1080 (619–1569) | <0.0001 |
| U-8-iso-PGF2α (pg/mg creatinine) | 264 (115–377) | 165 (95–324) | 0.035 |
| SAT (cm2) | 390.9 (297.7–485.1) | 329.1 (206.4–435.5) | <0.0001 |
| VAT (cm2) | 262.1 (180.5–336.5) | 238.0 (174.7–292.9) | <0.0001 |
| TNF-α (pg/mL) | 1.06 (0.88–1.53) | 1.14 (0.84–1.36) | 0.936 |
| Leptin (pg/mL) | 18.7 (10.8–44.7) | 12.5 (7.7–28.7) | <0.0001 |
| β-index (pmol.min−2·m−2 BSA) | 3.74 (2.62–5.08) | 4.78 (3.40–5.22) | 0.0033 |
| Matsuda-index | 2.9 (2.1–4.4) | 4.0 (3.1–5.2) | 0.0210 |
| Delta VAT | Delta SAT | Delta -index | Delta Matsuda Index | Delta leptin | Delta FPG | Delta 1-h-PPG | Delta 2-h-PPG | Delta hs-CRP | Delta HbA1c | Delta IGF-I | Delta TNF-α | Delta U-8-iso-PGF2α | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Delta U-11-dehydro-TXB2 | r = −0.199 | r = −0.110 | r = 0.150 | r = 0.124 | r = 0.380 | r = −0.07 | r = 0.010 | r = 0.097 | r = 0.099 | r = −0.136 | r = 0.069 | r = 0.561 | r = 0.357 |
| p = 0.250 | p = 0.527 | p = 0.388 | p = 0.476 | p = 0.024 | p = 0.680 | p = 0.952 | p = 0.576 | p = 0.601 | p = 0.432 | p = 0.691 | p = 0.001 | p = 0.034 |
| Predictor Variable | Standardized Parameter Estimate | SE | P-value | Partial R2 | Percentage of Variation Relative to 1-SD of Ln-U-11-Dehydro-TXB2 for 1-SD Change in the Predictor Variable | SD |
|---|---|---|---|---|---|---|
| Delta Ln-U-8-iso-PGF2α | 0.78 | 0.23 | 0.0042 | 23.1% | +88.1% (95%CI: 5.5% to 92.6%) | 0.62 |
| Delta TNF-α | 0.10 | 0.04 | 0.0294 | 6.8% | +49.1% (95%CI: 31.8% to 144.3%) | 2.63 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simeone, P.; Liani, R.; Tripaldi, R.; Di Castelnuovo, A.; Guagnano, M.T.; Tartaro, A.; Bonadonna, R.C.; Federico, V.; Cipollone, F.; Consoli, A.; et al. Thromboxane-Dependent Platelet Activation in Obese Subjects with Prediabetes or Early Type 2 Diabetes: Effects of Liraglutide- or Lifestyle Changes-Induced Weight Loss. Nutrients 2018, 10, 1872. https://doi.org/10.3390/nu10121872
Simeone P, Liani R, Tripaldi R, Di Castelnuovo A, Guagnano MT, Tartaro A, Bonadonna RC, Federico V, Cipollone F, Consoli A, et al. Thromboxane-Dependent Platelet Activation in Obese Subjects with Prediabetes or Early Type 2 Diabetes: Effects of Liraglutide- or Lifestyle Changes-Induced Weight Loss. Nutrients. 2018; 10(12):1872. https://doi.org/10.3390/nu10121872
Chicago/Turabian StyleSimeone, Paola, Rossella Liani, Romina Tripaldi, Augusto Di Castelnuovo, Maria Teresa Guagnano, Armando Tartaro, Riccardo C. Bonadonna, Virginia Federico, Francesco Cipollone, Agostino Consoli, and et al. 2018. "Thromboxane-Dependent Platelet Activation in Obese Subjects with Prediabetes or Early Type 2 Diabetes: Effects of Liraglutide- or Lifestyle Changes-Induced Weight Loss" Nutrients 10, no. 12: 1872. https://doi.org/10.3390/nu10121872
APA StyleSimeone, P., Liani, R., Tripaldi, R., Di Castelnuovo, A., Guagnano, M. T., Tartaro, A., Bonadonna, R. C., Federico, V., Cipollone, F., Consoli, A., & Santilli, F. (2018). Thromboxane-Dependent Platelet Activation in Obese Subjects with Prediabetes or Early Type 2 Diabetes: Effects of Liraglutide- or Lifestyle Changes-Induced Weight Loss. Nutrients, 10(12), 1872. https://doi.org/10.3390/nu10121872