Energy-Dense Diets and Mineral Metabolism in the Context of Chronic Kidney Disease–Metabolic Bone Disease (CKD-MBD)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
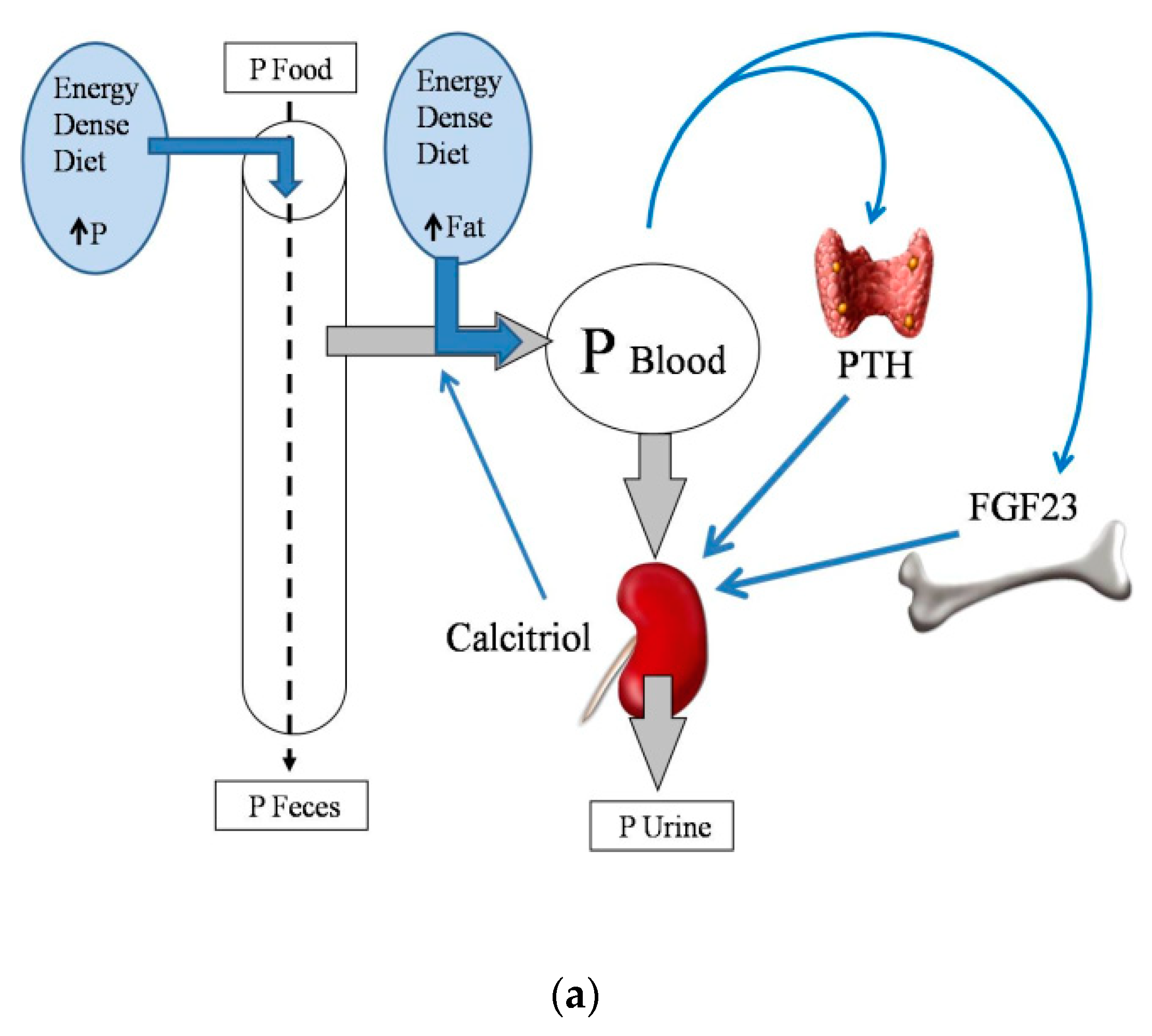
2. Energy-Dense Diets and Mineral Metabolism
2.1. Energy-Dense Diets and Calcium
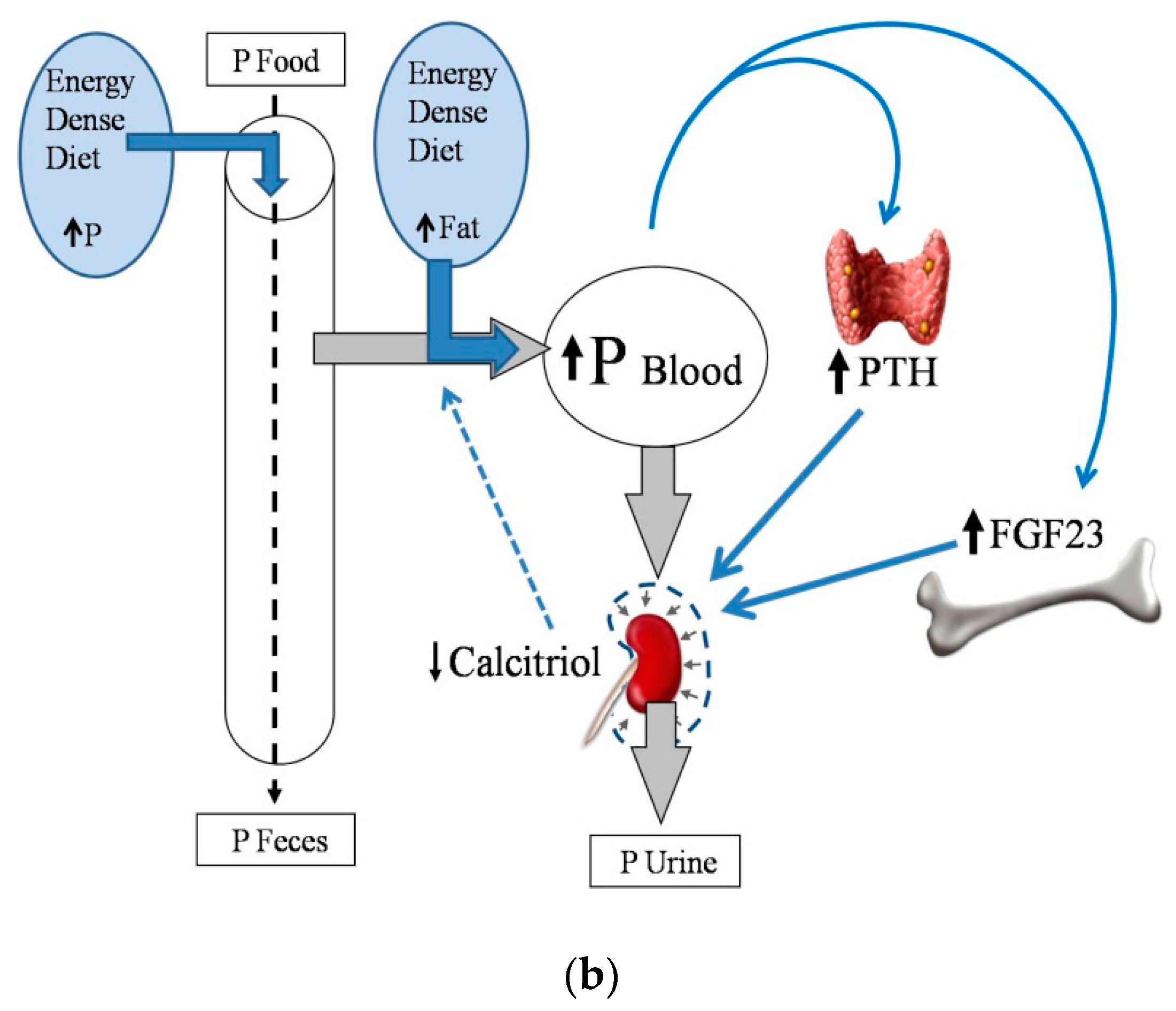
2.2. Energy-Dense Diets and Phosphorus
2.3. Energy-Dense Diets and Magnesium
2.4. Energy-Dense Diets and PTH
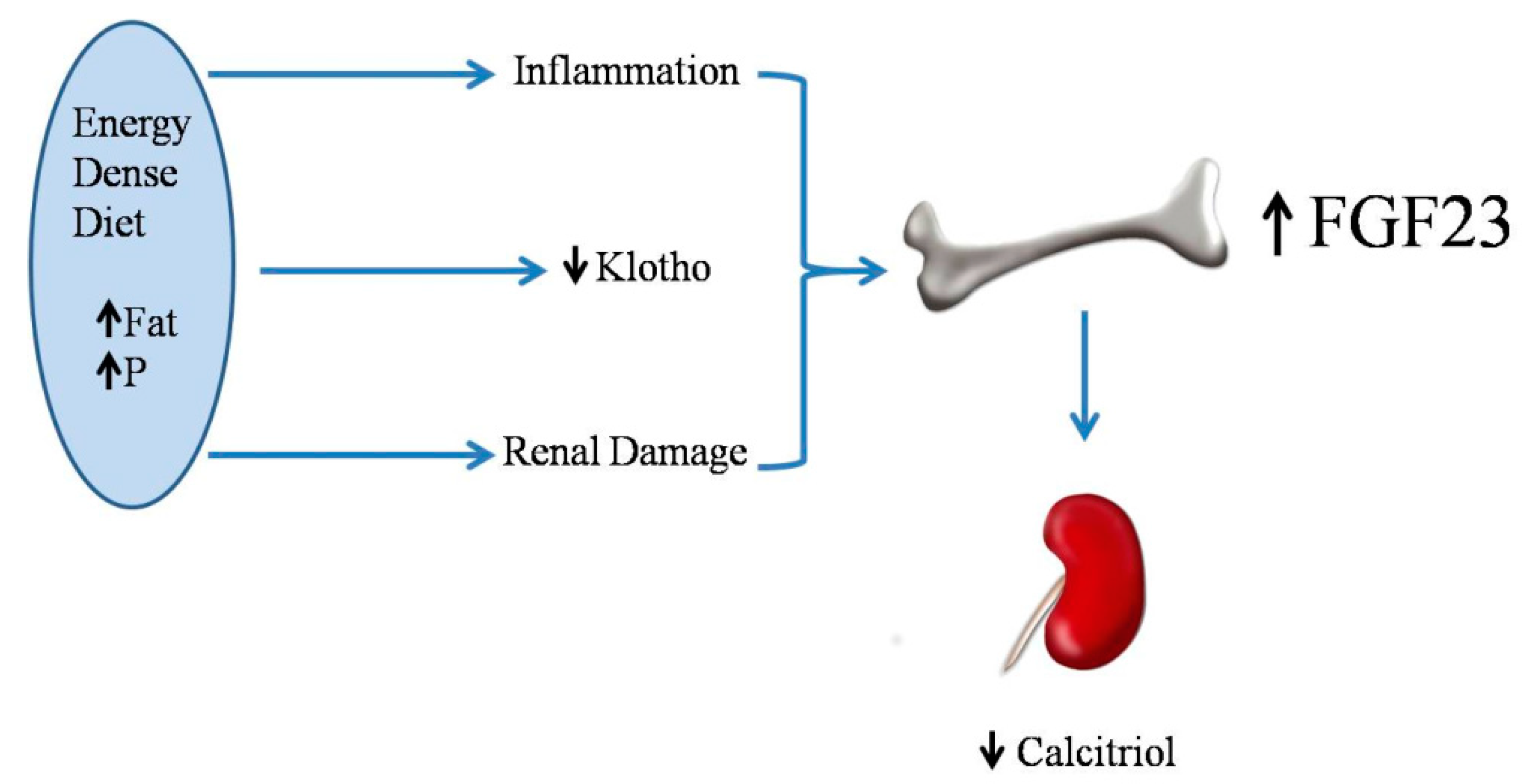
2.5. Energy-Dense Diets and FGF23/Klotho
2.6. Energy-Dense Diets and Vitamin D
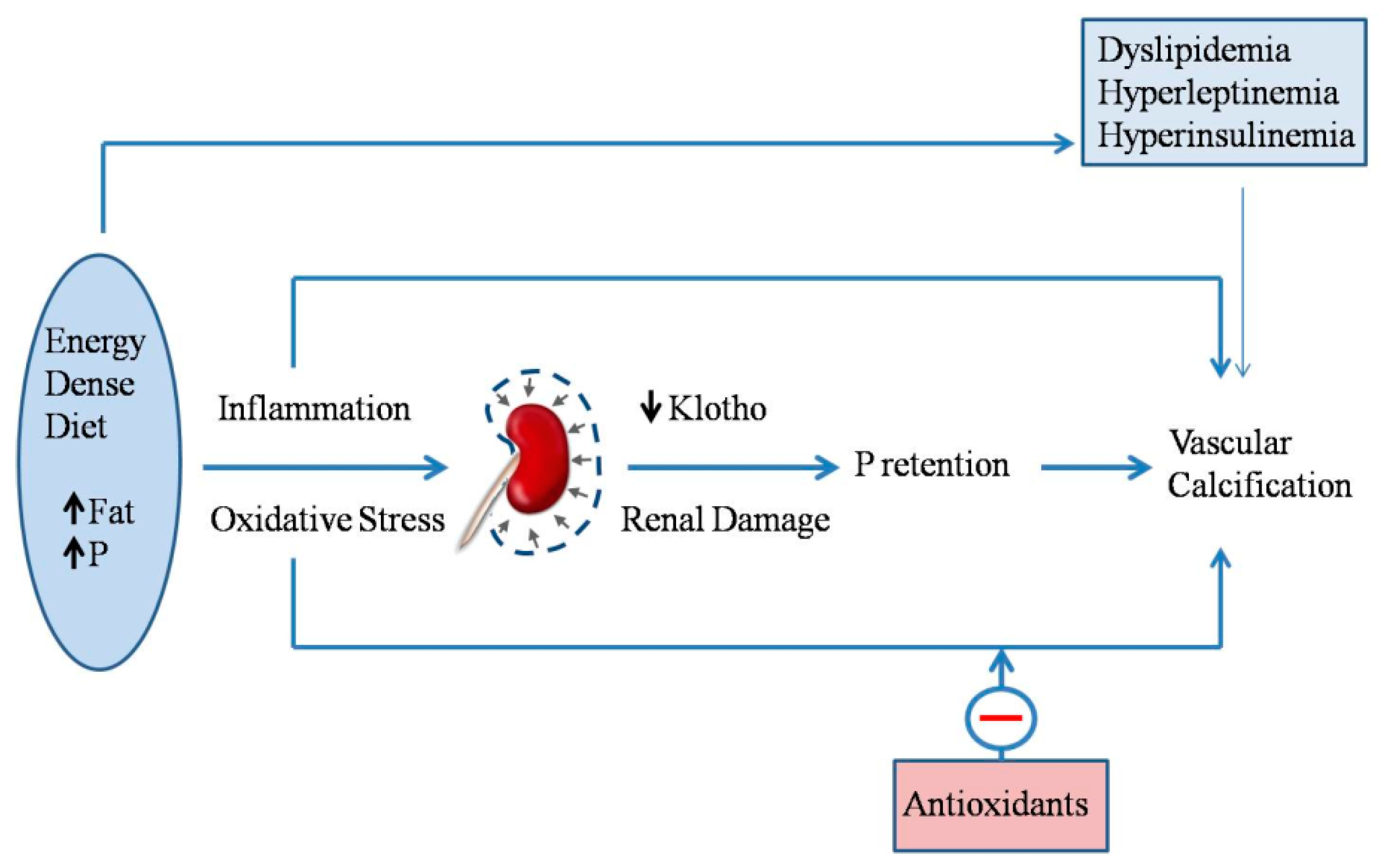
2.7. Energy-Dense Diets and Vascular Calcification
3. Conclusions
4. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Zafar, U.; Khaliq, S.; Ahmad, H.U.; Manzoor, S.; Lone, K.P. Metabolic syndrome: An update on diagnostic criteria, pathogenesis, and genetic links. Hormones (Athens) 2018, 17, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Weinsier, R.L.; Hunter, G.R.; Heini, A.F.; Goran, M.I.; Sell, S.M. The etiology of obesity: Relative contribution of metabolic factors, diet, and physical activity. Am. J. Med. 1998, 105, 145–150. [Google Scholar] [CrossRef]
- Ting, S.M.S.; Nair, H.; Ching, I.; Taheri, S.; Dasgupta, I. Overweight, obesity and chronic kidney disease. Nephron Clin. Pract. 2009, 112, c121–c127. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, F.; Del Vecchio, L.; Aicardi, V. Nutritional issues with incremental dialysis: The role of low-protein diets. Semin. Dial. 2017, 30, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Ku, E.; Kopple, J.D.; Johansen, K.L.; McCulloch, C.E.; Go, A.S.; Xie, D.; Lin, F.; Hamm, L.L.; He, J.; Kusek, J.W.; et al. Longitudinal weight change during CKD progression and its association with subsequent mortality. Am. J. Kidney Dis. 2018, 71, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Cheung, W.; Yu, P.X.; Little, B.M.; Cone, R.D.; Marks, D.L.; Mak, R.H. Role of leptin and melanocortin signaling in uremia-associated cachexia. J. Clin. Investig. 2005, 115, 1659–1665. [Google Scholar] [CrossRef] [PubMed]
- Mills, S.R.; Wilcox, C.R.; Ibrahim, K.; Roberts, H.C. Can fortified foods and snacks increase the energy and protein intake of hospitalised older patients? A systematic review. J. Hum. Nutr. Diet. 2018, 31, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Kalantar-Zadeh, K.; Rhee, C.M.; Chou, J.; Ahmadi, S.F.; Park, J.; Chen, J.L.T.; Amin, AN. The obesity paradox in kidney disease: How to reconcile it with obesity management. Kidney Int. Rep. 2017, 2, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, S.F.; Zahmatkesh, G.; Ahmadi, E.; Streja, E.; Rhee, C.M.; Gillen, D.L.; De Nicola, L.; Minutolo, R.; Ricardo, A.C.; Kovesdy, C.P.; et al. Association of body mass index with clinical outcomes in non-dialysis-dependent chronic kidney disease: A systematic review and meta-analysis. Cardiorenal Med. 2015, 6, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, M.; López, I.; Muñoz, J.; Aguilera-Tejero, E.; Almaden, Y. FGF23 and mineral metabolism, implications in CKD-MBD. Nefrologia 2012, 32, 275–278. [Google Scholar] [CrossRef] [PubMed]
- Villa-Bellosta, R.; Millan, A.; Sorribas, V. Role of calcium-phosphate deposition in vascular smooth muscle cell calcification. Am. J. Physiol. Cell. Physiol. 2011, 300, C210–C220. [Google Scholar] [CrossRef] [PubMed]
- Ketteler, M.; Block, G.A.; Evenepoel, P.; Fukagawa, M.; Herzog, C.A.; McCann, L.; Moe, S.M.; Shroff, R.; Tonelli, M.A.; Toussaint, N.D.; et al. Executive summary of the 2017 KDIGO Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD) Guideline Update: What’s changed and why it matters. Kidney Int. 2017, 92, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.J.; Gregoire, B.R.; Gao, H. High-fat diet decreases cancellous bone mass but has no effect on cortical bone mass in the tibia in mice. Bone 2009, 44, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Ionova-Martin, S.S.; Do, S.H.; Barth, H.D.; Szadkowska, M.; Porter, A.E.; Ager, J.W., 3rd; Ager, J.W., Jr.; Alliston, T.; Vaisse, C.; Ritchie, R.O. Reduced size-independent mechanical properties of cortical bone in high-fat diet-induced obesity. Bone 2010, 46, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Martins, J.; Castro, J.H.; Sainz Rueda, N.A.; Dos Reis, L.M.; Jorgetti, V.; Affonso Moysés, R.M.; Caramori, J.T. Renal osteodystrophy in the obesity era: Is metabolic syndrome relevant? PLoS ONE 2017, 12, e0180387. [Google Scholar] [CrossRef] [PubMed]
- Gacs, G.; Barltrop, D. Significance of Ca-soap formation for calcium absorption in the rat. Gut 1977, 18, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Agnew, J.E.; Holdsword, C.D. The effect of fat on calcium absorption from a mixed meal in normal subjects, patients with malabsorptive disease, and patients with partial gastrectomy. Gut 1971, 12, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Sarathy, S.; Sullivan, C.; Leon, J.B.; Sehgal, A.R. Fast food, phosphorus-containing additives, and the renal diet. J. Ren. Nutr. 2008, 18, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Calvo, M.S. Hidden sources of phosphorus in the typical American diet: Does it matter in nephrology? Semin. Dial. 2003, 16, 186–188. [Google Scholar] [CrossRef] [PubMed]
- Frommelt, L.; Bielohuby, M.; Stoehr, B.J.; Menhofer, D.; Bidlingmaier, M.; Kienzle, E. Effects of low-carbohydrate, high-fat diets on apparent digestibility of minerals and trace elements in rats. Nutrition 2014, 30, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Raya, A.I.; Rios, R.; Pineda, C.; Rodriguez-Ortiz, M.E.; Diez, E.; Almaden, Y.; Muñoz-Castañeda, J.; Rodriguez, M.; Aguilera-Tejero, E.; Lopez, I. Energy-dense diets increase FGF23, lead to phosphorus retention and promote vascular calcifications in rats. Sci. Rep. 2016, 6, 36881. [Google Scholar] [CrossRef] [PubMed]
- Simental-Mendia, L.E.; Sahebkar, A.; Rodriguez-Moran, M.; Guerrero-Romero, F. A systematic review and meta-analysis of randomized controlled trials on the effects of magnesium supplementation on insulin sensitivity and glucose control. Pharmacol. Res. 2016, 111, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Toprak, O.; Kurt, H.; Sari, Y.; Sarkis, C.; Us, H. Magnesium replacement improves the metabolic profile in obese and pre-diabetic patients with mild-to-moderate chronic kidney disease: A 3 month, randomised, double-blind, placebo-controlled study. Kidney Blood Press. Res. 2017, 42, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Sales, C.H.; Santos, A.R.; Cintra, D.E.; Colli, C. Magnesium-deficient high-fat diet: Effects on adiposity, lipid profile and insulin sensitivity in growing rats. Clin. Nutr. 2014, 33, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Bertinato, J.; Lavergne, C.; Rahimi, S.; Rachid, H.; Vu, N.A.; Plouffe, L.J.; Swist, E. Moderately low magnesium intake impairs growth of lean body mass in obese-prone an obese-resistant rats fed a high-energy diet. Nutrients 2016, 8, 253. [Google Scholar] [CrossRef] [PubMed]
- Kurstjens, S.; van Diepen, J.A.; Overmars-Bos, C.; Alkema, W.; Bindels, R.J.M.; Ashcroft, F.M.; Tack, C.J.J.; Hoenderop, J.G.J.; de Baaij, J.H.F. Magnesium deficiency prevents high-fat-diet-induced obesity in mice. Diabetologia 2018, 61, 2030–2042. [Google Scholar] [CrossRef] [PubMed]
- Van Laecke, S.; Nagler, E.V.; Verbeke, F.; van Biesen, W.; Vanholder, R. Hypomagenesemia and the risk of death and GFR decline in chronic kidney disease. Am. J. Med. 2013, 126, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Tocados, J.M.; Peralta-Ramirez, A.; Rodríguez-Ortiz, M.E.; Raya, A.I.; Lopez, I.; Pineda, C.; Herencia, C.; Montes de Oca, A.; Vergara, N.; Steppan, S.; et al. Dietary magnesium supplementation prevents and reverses vascular and soft tissue calcifications in uremic rats. Kidney Int. 2017, 92, 1084–1099. [Google Scholar] [CrossRef] [PubMed]
- Sage, A.P.; Lu, J.; Atti, E.; Tetradis, S.; Ascenzi, M.G.; Adams, D.J.; Demer, L.L.; Tintut, Y. Hyperlipidemia induces resistance to PTH bone anabolism in mice via oxidized lipids. J. Bone Miner. Res. 2011, 26, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Lopez, I.; Pineda, C.; Raya, A.I.; Rodriguez-Ortiz, M.E.; Diaz-Tocados, J.M.; Rios, R.; Rodriguez, J.M.; Aguilera-Tejero, E.; Almaden, Y. Leptin directly stimulates parathyroid hormone secretion. Endocrine 2017, 56, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Marsell, R.; Mirza, M.A.; Mallmin, H.; Karlsson, M.; Mellström, D.; Orwoll, E.; Ohlsson, C.; Jonsson, K.B.; Ljunggren, O.; Larsson, T.E. Relation between fibroblast growth factor-23, body weight and bone mineral density in elderly men. Osteoporos. Int. 2009, 20, 1167–1173. [Google Scholar] [CrossRef] [PubMed]
- Di Giuseppe, R.; Kühn, T.; Hirche, F.; Buijsse, B.; Dierkes, J.; Fritsche, A.; Kaaks, R.; Boeing, H.; Stangl, G.I.; Weikert, C. Potential predictors of plasma fibroblast growth factor 23 concentrations: Cross-sectional analysis in the EPIC-Germany study. PLoS ONE 2015, 10, e0133580. [Google Scholar] [CrossRef] [PubMed]
- Rios, R.; Pineda, C.; Lopez, I.; Muñoz-Castañeda, J.; Rodriguez, M.; Aguilera-Tejero, E.; Raya, A.I. Phosphorus restriction does not prevent the increase in fibroblast growth factor 23 elicited by high fat diet. PLoS ONE 2018, 13, e0198481. [Google Scholar] [CrossRef] [PubMed]
- Sastre, C.; Rubio-Navarro, A.; Buendia, I.; Gómez-Guerrero, C.; Blanco, C.; Mas, S.; Egido, J.; Blanco-Colio, L.M.; Ortiz, A.; Moreno, J.A. Hyperlipidemia-associated renal damage decreases Klotho expression in kidneys from ApoE knockout mice. PLoS ONE 2013, 8, e83713. [Google Scholar] [CrossRef] [PubMed]
- Kanbay, M.; Vervoelt, M.; Cozzolino, M.; Siriopol, D.; Covic, A.; Goldsmith, D.; Solak, Y. Novel faces of fibroblast growth factor 23 (FGF23): Iron deficiency, inflammation, insulin resistance, left ventricular hypertrophy, proteinuria and acute kidney injury. Calcif. Tissue Int. 2017, 100, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Poret, J.M.; Souza-Smith, F.; Marcell, S.J.; Gaudet, D.A.; Tzeng, T.H.; Braymer, H.D.; Harrison-Bernard, L.M.; Primeaux, S.D. High fat diet consumption differentially affects adipose tissue inflammation and adipocyte size in obesity-prone and obesity-resistant rats. Int. J. Obes. (Lond.) 2018, 42, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Wickman, C.; Kramer, H. Obesity and kidney disease. Potential mechanisms. Semin. Nephrol. 2013, 33, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Glosse, P.; Fajol, A.; Hirche, F.; Feger, M.; Voelkl, J.; Lang, F.; Stangl, G.I.; Föller, M. A high-fat diet stimulates fibroblast growth factor 23 formation in mice through TNFα upregulation. Nutr. Diabetes 2018, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, K.; Maeda, T.; Kawane, T.; Matsunuma, A.; Horiuchi, N. Leptin stimulates fibroblast growth factor 23 expression in bone and suppresses renal 1alpha,25-dihydroxyvitamin D3 synthesis in leptin-deficient mice. J. Bone Miner. Res. 2010, 25, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- Saini, R.K.; Kaneko, I.; Jurutka, P.W.; Forster, R.; Hsieh, A.; Hsieh, J.C.; Haussler, M.R.; Whitfield, G.K. 1,25-dihydroxyvitamin D(3) regulation of fibroblast growth factor-23 expression in bone cells: Evidence for primary and secondary mechanisms modulated by leptin and interleukin-6. Calcif. Tissue Int. 2013, 92, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, J.M.; Pastor, J.; Sun, K.; Park, S.K.; Bobulescu, I.A.; Chen, C.T.; Moe, O.W.; Scherer, P.E. Adiponectin alters renal calcium and phosphate excretion through regulation of klotho expression. Kidney Int. 2017, 91, 324–337. [Google Scholar] [CrossRef] [PubMed]
- Scialla, J.J.; Xie, H.; Rahman, M.; Anderson, A.H.; Isakova, T.; Ojo, A.; Zhang, X.; Nessel, L.; Hamano, T.; Grunwald, J.E.; et al. Fibroblast growth factor-23 and cardiovascular events in CKD. J. Am. Soc. Nephrol. 2014, 25, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Kuro-O, M. Klotho and endocrine fibroblast growth factors: Marker of chronic kidney disease progression and cardiovascular complications? Nephrol. Dial. Transplant 2018. [Google Scholar] [CrossRef] [PubMed]
- Vimaleswaran, K.S.; Berry, D.J.; Lu, C.; Tikkanen, E.; Pilz, S.; Hiraki, L.T.; Cooper, J.D.; Dastani, Z.; Li, R.; Houston, D.K.; et al. Causal relationship between obesity and vitamin D status: Bi-directional Mendelian randomization analysis of multiple cohorts. PLos Med. 2013, 10, e1001383. [Google Scholar] [CrossRef] [PubMed]
- Peralta-Ramírez, A.; Montes de Oca, A.; Raya, A.I.; Pineda, C.; López, I.; Guerrero, F.; Diez, E.; Muñoz-Castañeda, J.R.; Martinez, J.; Almaden, Y.; et al. Vitamin E protection of obesity-enhanced vascular calcification in uremic rats. Am. J. Physiol. Renal Physiol. 2014, 306, F422–F429. [Google Scholar] [CrossRef] [PubMed]
- Dawson-Hughes, B.; Harris, S.S.; Lichtenstein, A.H.; Dolnikowski, G.; Palermo, N.J.; Rasmussen, H. Dietary fat increases vitamin D-3 absorption. J. Acad. Nutr. Diet. 2015, 115, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Holick, M.F. Vitamin D deficiency. N. Engl. J. Med. 2007, 357, 266–281. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Hasegawa, H.; Yamazaki, Y.; Muto, T.; Hino, R.; Takeuchi, Y.; Fujita, T.; Nakahara, K.; Fukumoto, S.; Yamashita, T. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J. Bone Miner. Res. 2004, 19, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Singh, J.; Kumar, J. Vitamin D and cardiovascular disease in chronic kidney disease. Pediatr. Nephrol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, L.; Lundwall, K.; Moshfegh, A.; Jacobson, S.H.; Lundahl, J.; Spaak, J. Vitamin D receptor activation reduces inflammatory cytokines and plasma MicroRNAs in moderate chronic kidney disease—A randomized trial. BMC Nephrol. 2017, 18, 161. [Google Scholar] [CrossRef] [PubMed]
- Vila Cuenca, M.; Ferrantelli, E.; Meinster, E.; Pouw, S.M.; Kovačević, I.; de Menezes, R.X.; Niessen, H.W.; Beelen, R.H.J.; Hordijk, P.L.; Vervloet, M.G. Vitamin D attenuates endothelial dysfunction in uremic rats and maintains human endothelial stability. J. Am. Heart. Assoc. 2018, 7, e008776. [Google Scholar] [CrossRef] [PubMed]
- van Schoor, N.M.; Visser, M.; Pluijm, S.M.; Kuchuk, N.; Smit, J.H.; Lips, P. Vitamin D deficiency as a risk factor for osteoporotic fractures. Bone 2008, 42, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Rios, R.; Raya, A.I.; Pineda, C.; Rodriguez, M.; Lopez, I.; Aguilera-Tejero, E. Vitamin E protects against extraskeletal calcification in uremic rats fed high fat diets. BMC Nephrol. 2017, 18, 374. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, M.; Brancaccio, D.; Gallieni, M.; Slatopolsky, E. Pathogenesis of vascular calcification in chronic kidney disease. Kidney Int. 2005, 68, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Giachelli, C.M. The emerging role of phosphate in vascular calcification. Kidney Int. 2009, 75, 890–897. [Google Scholar] [CrossRef] [PubMed]
- McCabe, K.M.; Zelt, J.G.; Kaufmann, M.; Laverty, K.; Ward, E.; Barron, H.; Jones, G.; Adams, M.A.; Holden, R.M. Calcitriol accelerates vascular calcification irrespective of vitamin K status in a rat model of chronic kidney disease with hyperphosphatemia and secondary hyperparathyroidism. J. Pharmacol. Exp. Ther. 2018, 366, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Zeadin, M.G.; Butcher, M.K.; Shaughnessy, S.G.; Werstuck, G.H. Leptin promotes osteoblast differentiation and mineralization of primary cultures of vascular smooth muscle cells by inhibiting glycogen synthase kinase (GSK)-3β. Biochem. Biophys. Res. Commun. 2012, 425, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Iribarren, C.; Husson, G.; Go, A.S.; Lo, J.C.; Fair, J.M.; Rubin, G.D.; Hlatky, M.A.; Fortmann, S.P. Plasma leptin levels and coronary artery calcification in older adults. J. Clin. Endocrinol. MeTable 2007, 92, 729–732. [Google Scholar] [CrossRef] [PubMed]
- Parhami, F.; Tintut, Y.; Ballard, A.; Fogelman, A.M.; Demer, L.L. Leptin enhances the calcification of vascular cells: Artery wall as a target of leptin. Circ. Res. 2001, 88, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.K.; Fortmann, S.P.; Fair, J.M.; Iribarren, C.; Rubin, G.D.; Varady, A.; Go, A.S.; Quertermous, T.; Hlatky, M.A. Insulin resistance independently predicts the progression of coronary artery calcification. Am. Heart J. 2009, 157, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Olesen, P.; Nguyen, K.; Wogensen, L.; Ledet, T.; Rasmussen, L.M. Calcification of human vascular smooth muscle cells: Associations with osteoprotegerin expression and acceleration by high-dose insulin. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1058–H1064. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.; Sorribas, V.; Sharma, G.; Levi, M.; Draznin, B. Insulin attenuates vascular smooth muscle calcification but increases vascular smooth muscle cell phosphate transport. Atherosclerosis 2007, 195, e65–e75. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.Q.; Zhu, J.H.; Wang, H.W.; Liang, Q.H.; Xie, H.; Wu, X.P.; Zhou, H.; Cui, R.R.; Sheng, Z.F.; Zhou, H.D.; et al. RANKL is a downstream mediator for insulin-induced osteoblastic differentiation of vascular smooth muscle cells. PLoS ONE 2011, 6, e29037. [Google Scholar] [CrossRef] [PubMed]
- Towler, D.A.; Bidder, M.; Latifi, T.; Coleman, T.; Semenkovich, C.F. Diet-induced diabetes activates an osteogenic gene regulatory program in the aortas of low density lipoprotein receptor-deficient mice. J. Biol. Chem. 1998, 273, 30427–30434. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Oshima, M.; Watanabe, Y.; Miyake, H. Arterial location-specific calcification at the carotid artery and aortic arch for chronic kidney disease, diabetes mellitus, hypertension, and dyslipidemia. Calcif. Tissue Int. 2014, 95, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Amann, K. Media calcification and intima calcification are distinct entities in chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2008, 3, 1599–1605. [Google Scholar] [CrossRef] [PubMed]
- Skalicky, J.; Muzakova, V.; Kandar, R.; Meloun, M.; Rousar, T.; Palicka, V.; Palicka, V. Evaluation of oxidative stress and inflammation in obese adults with metabolic syndrome. Clin. Chem. Lab. Med. 2008, 46, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Ravarotto, V.; Simioni, F.; Pagnin, E.; Davis, P.A.; Calò, L.A. Oxidative stress—Chronic kidney disease—Cardiovascular disease: A vicious circle. Life Sci. 2018, 210, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Mody, N.; Parhami, F.; Sarafian, T.A.; Demer, L.L. Oxidative stress modulates osteoblastic differentiation of vascular and bone cells. Free Radic. Biol. Med. 2001, 31, 509–519. [Google Scholar] [CrossRef]
- Muteliefu, G.; Enomoto, A.; Jiang, P.; Takahashi, M.; Niwa, T. Indoxyl sulphate induces oxidative stress and the expression of osteoblast-specific proteins in vascular smooth muscle cells. Nephrol. Dial. Transplant. 2009, 24, 2051–2058. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Gurnani, P.; Nandi, A.; Kurosu, H.; Miyoshi, M.; Ogawa, Y.; Castrillon, D.H.; Rosenblatt, K.P.; et al. Regulation of oxidative stress by the anti-aging hormone klotho. J. Biol. Chem. 2005, 280, 38029–38034. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Shi, M.; Zhang, J.; Quiñones, H.; Griffith, C.; Kuro-o, M.; Moe, O.W. Klotho deficiency causes vascular calcification in chronic kidney disease. J. Am. Soc. Nephrol. 2011, 22, 124–136. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodriguez, M.; Aguilera-Tejero, E. Energy-Dense Diets and Mineral Metabolism in the Context of Chronic Kidney Disease–Metabolic Bone Disease (CKD-MBD). Nutrients 2018, 10, 1840. https://doi.org/10.3390/nu10121840
Rodriguez M, Aguilera-Tejero E. Energy-Dense Diets and Mineral Metabolism in the Context of Chronic Kidney Disease–Metabolic Bone Disease (CKD-MBD). Nutrients. 2018; 10(12):1840. https://doi.org/10.3390/nu10121840
Chicago/Turabian StyleRodriguez, Mariano, and Escolastico Aguilera-Tejero. 2018. "Energy-Dense Diets and Mineral Metabolism in the Context of Chronic Kidney Disease–Metabolic Bone Disease (CKD-MBD)" Nutrients 10, no. 12: 1840. https://doi.org/10.3390/nu10121840
APA StyleRodriguez, M., & Aguilera-Tejero, E. (2018). Energy-Dense Diets and Mineral Metabolism in the Context of Chronic Kidney Disease–Metabolic Bone Disease (CKD-MBD). Nutrients, 10(12), 1840. https://doi.org/10.3390/nu10121840