
Dual-Purpose Utilization of Sri Lankan Apatite for Rare Earth Recovery Integrated into Sustainable Nitrophosphate Fertilizer Manufacturing

 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Characterization
2.3. Methods
2.3.1. Acidolysis of Rock Phosphate
2.3.2. Cooling Crystallization
2.3.3. Partial Neutralization
2.3.4. Nitrophosphate Fertilizer Synthesis
2.3.5. Selective Dissolution of REE
2.3.6. Sodium Rare Earth Double Sulfate Precipitation
3. Results
3.1. Chemical Characterization of Eppawala Rock Phosphate
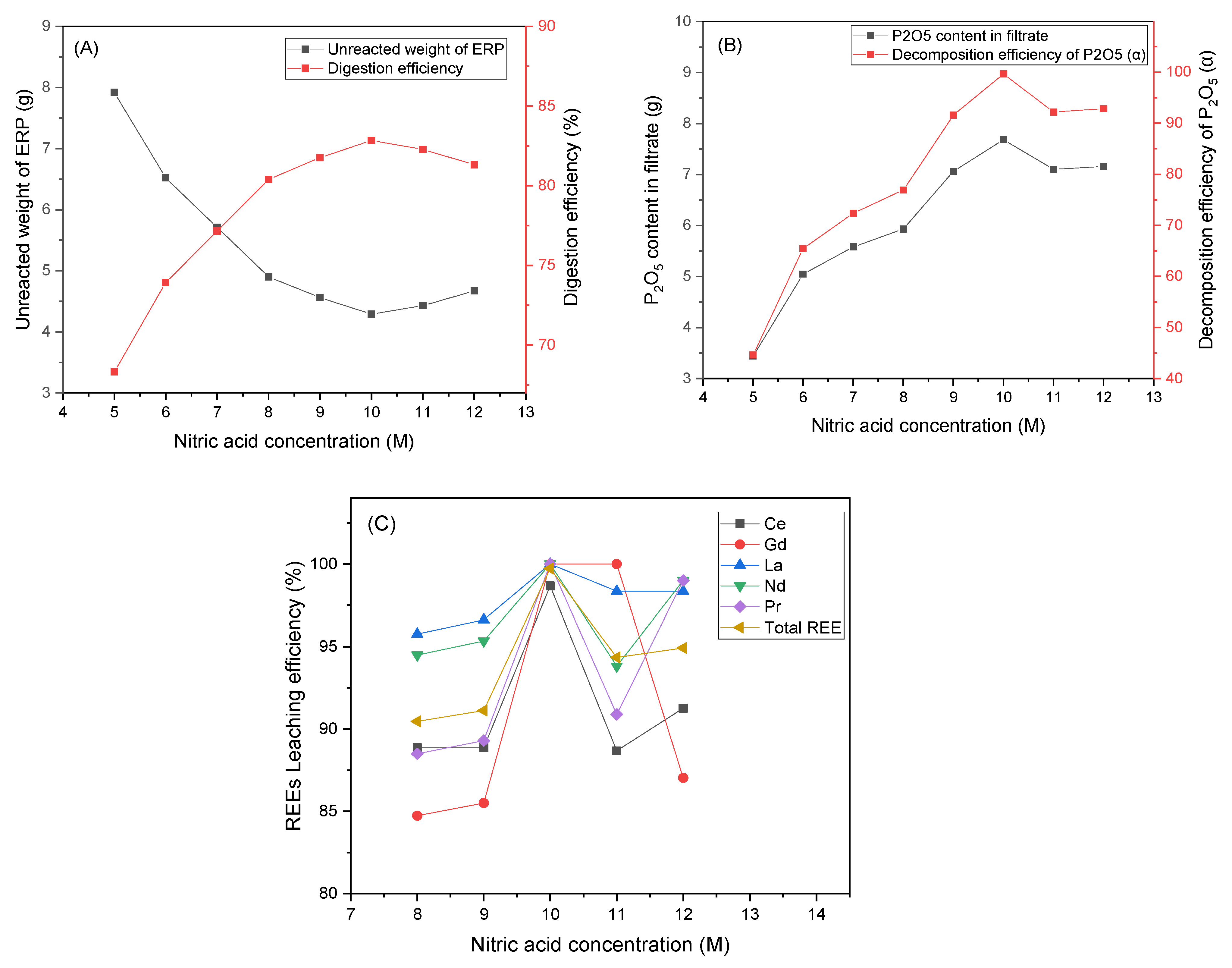
3.2. Effect of Nitric Acid Concentration on Leaching
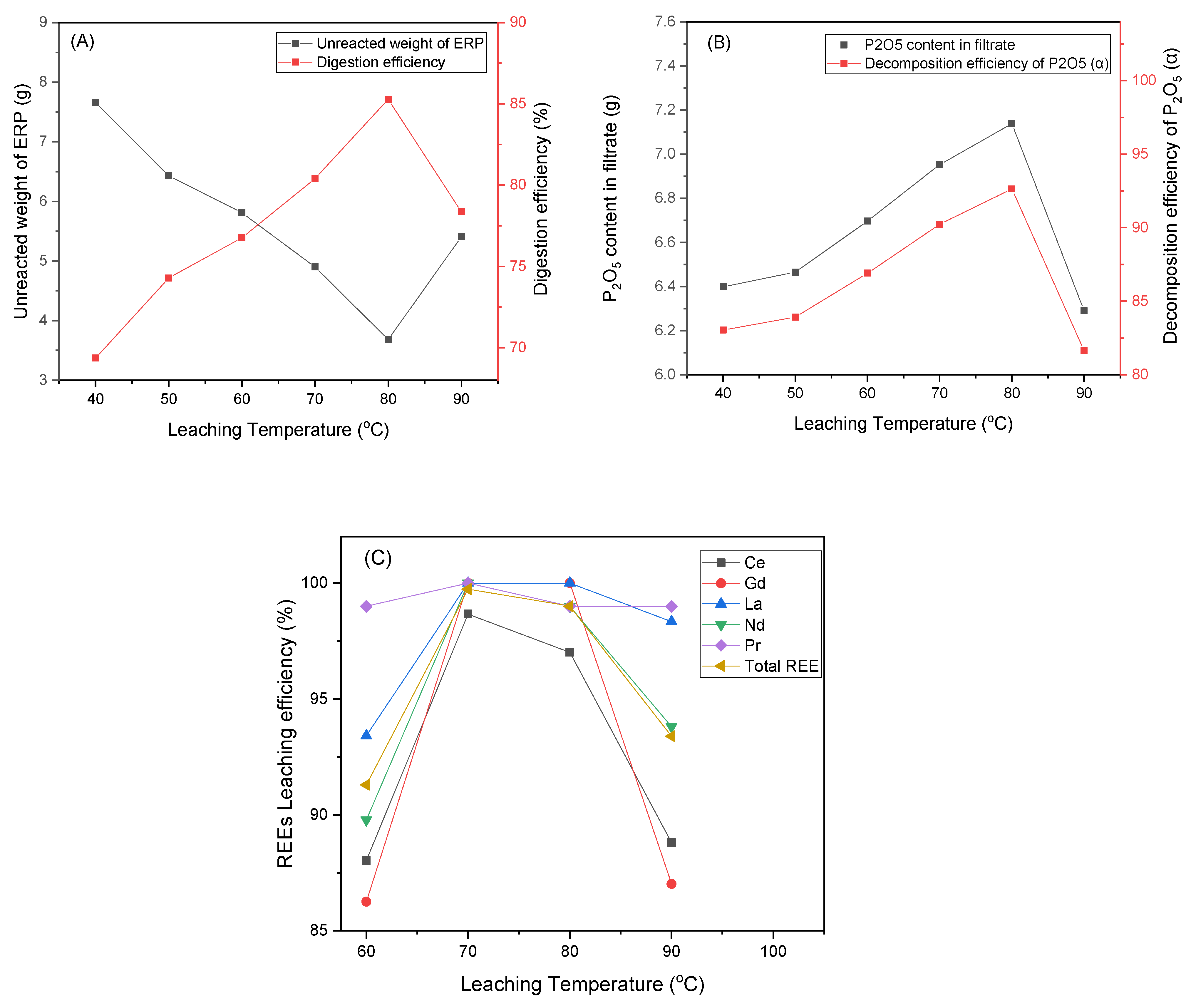
3.3. Effect of Leaching Temperature
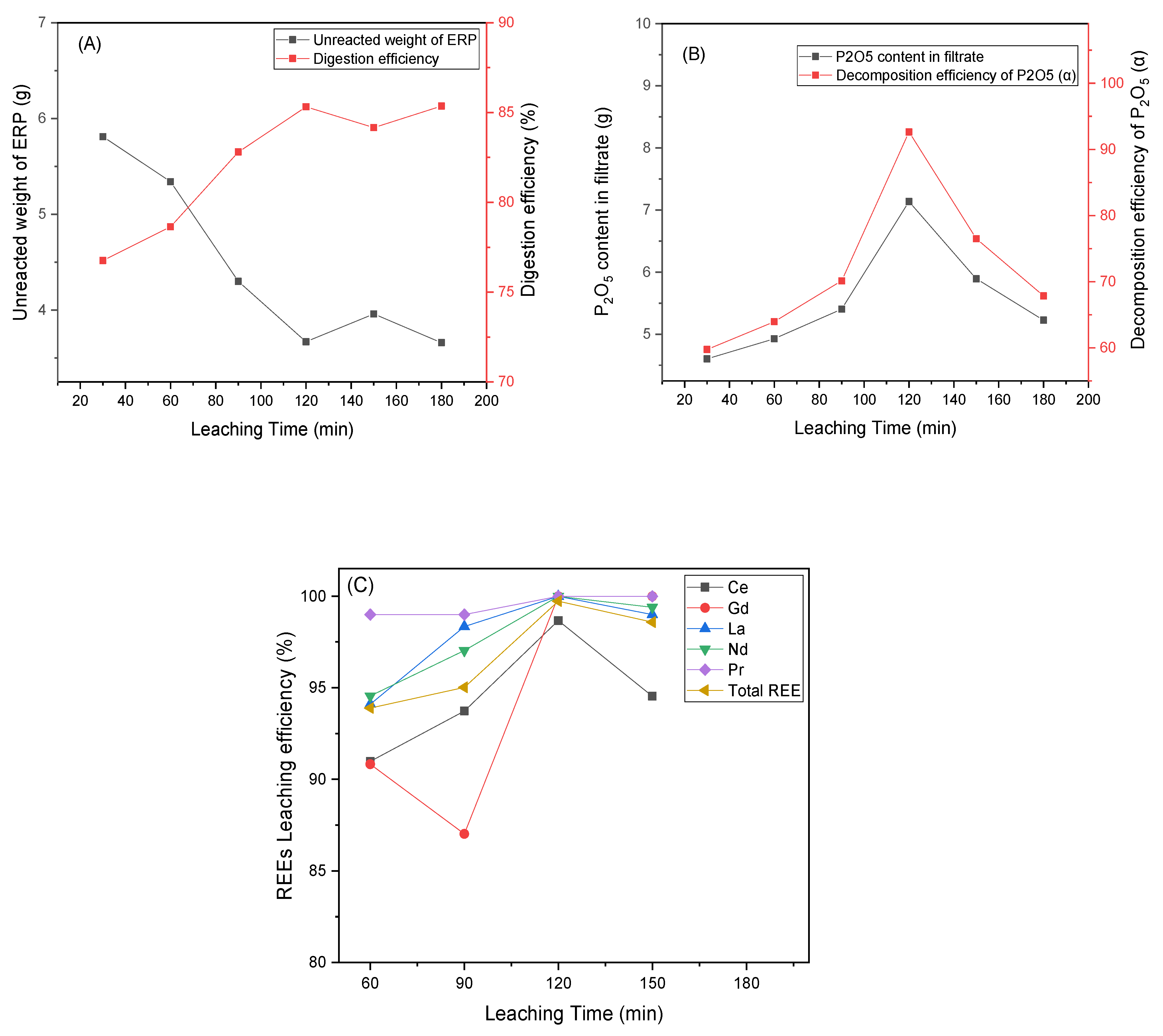
3.4. Effect of Leaching Time
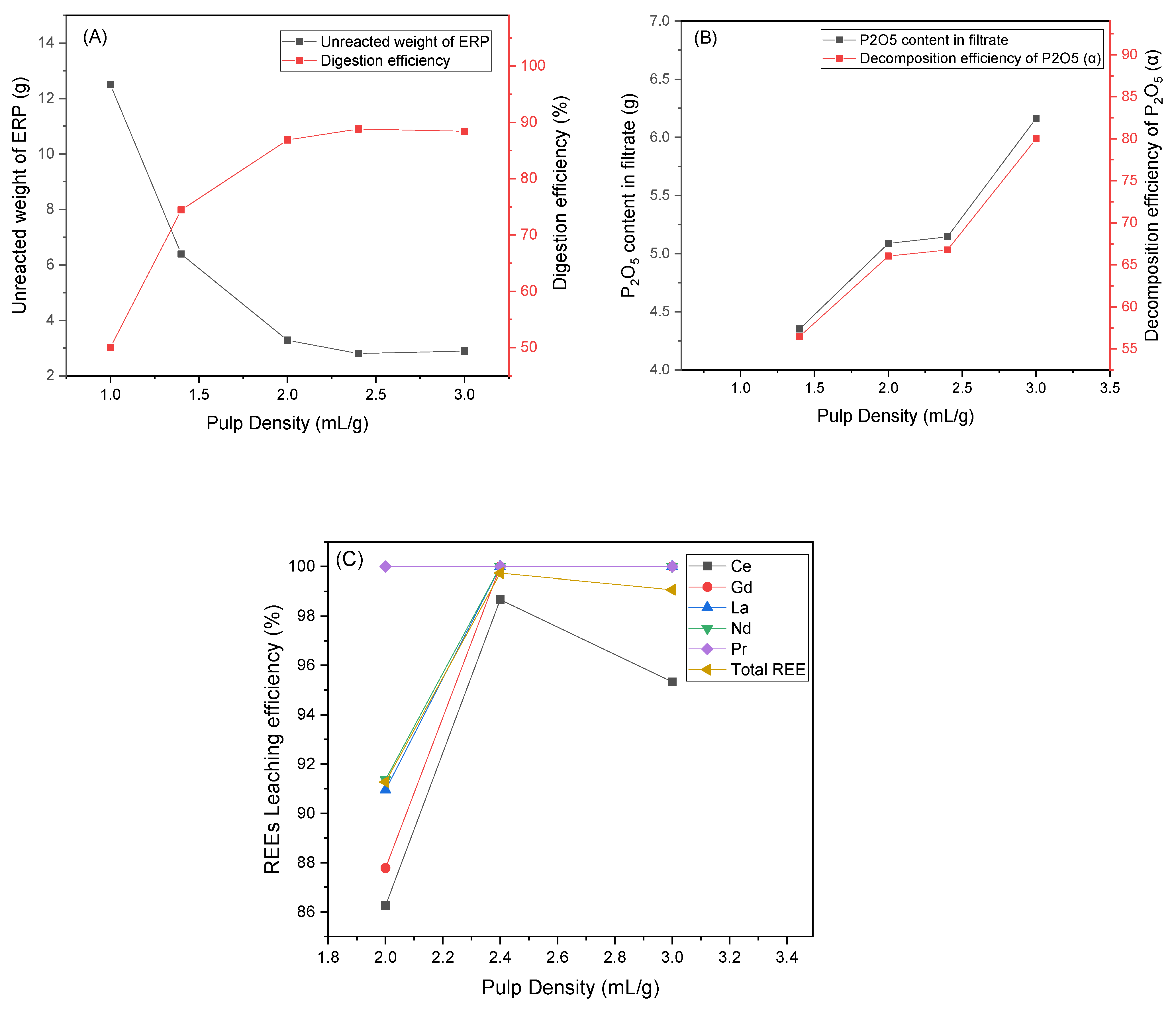
3.5. Effect of Pulp Density
3.6. Selective Recovery of REEs
3.7. Nitrophosphate Synthesis
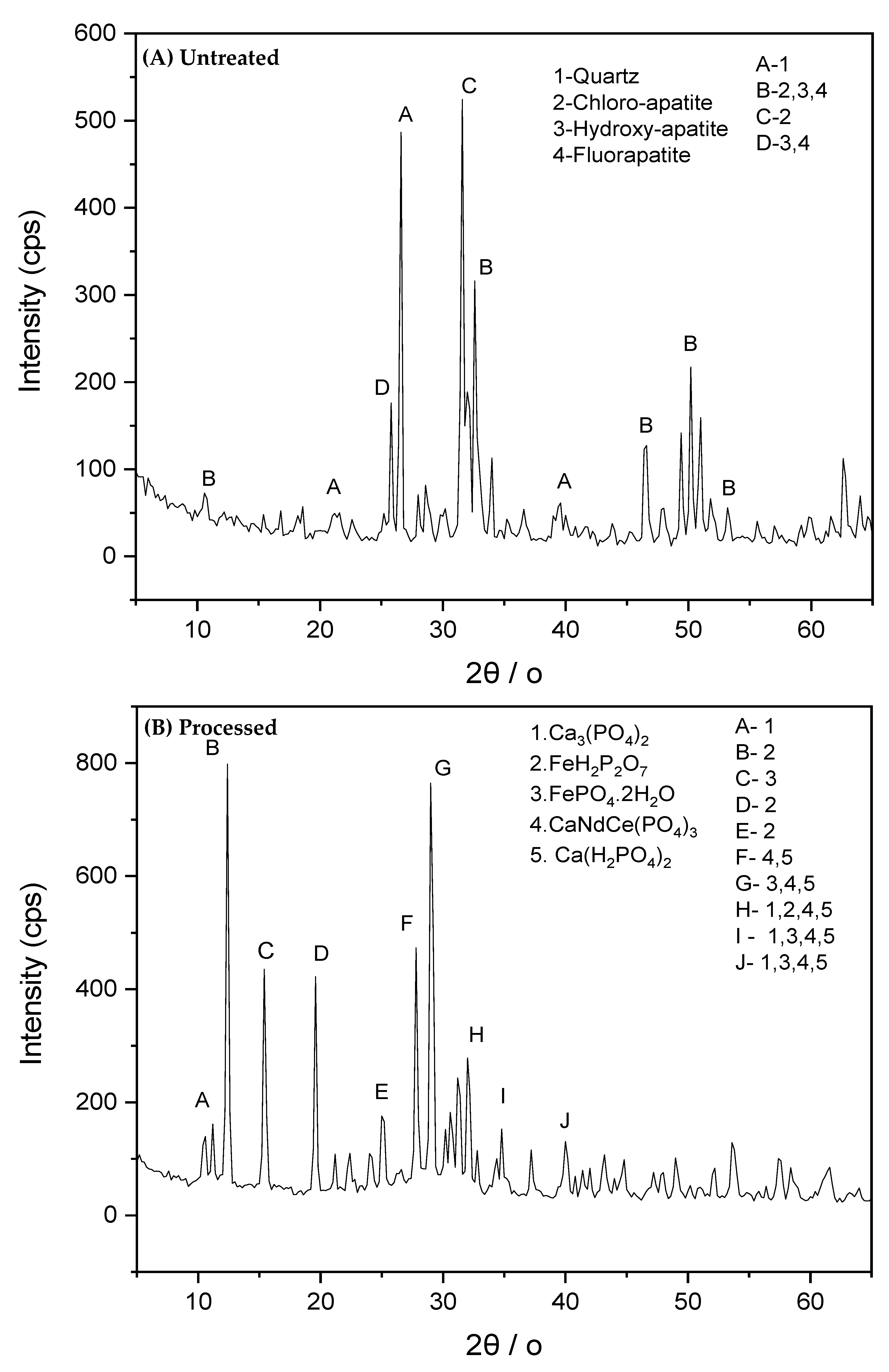
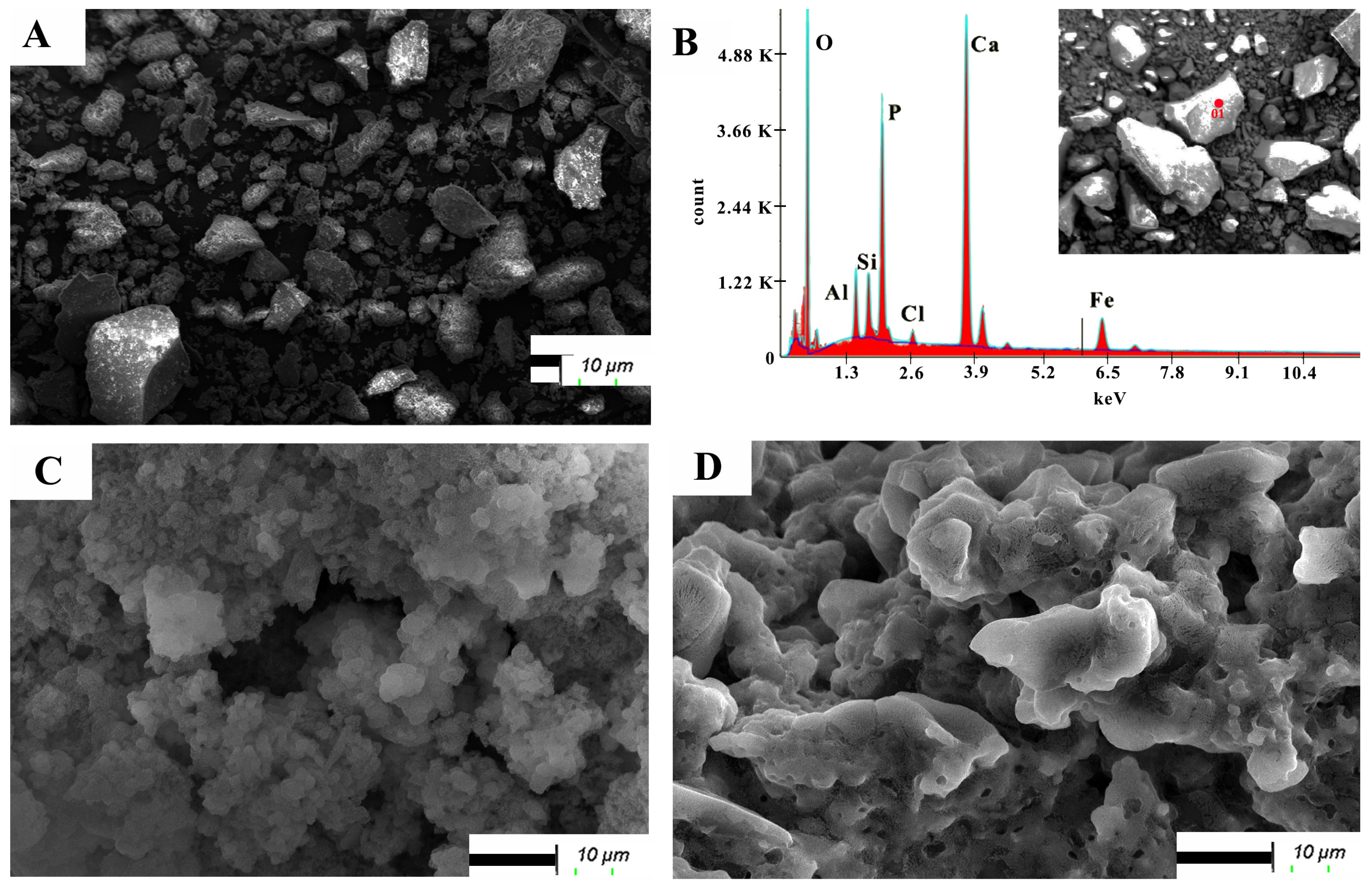
3.8. Evolution of Crystalline Phases and Surface Composition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Drobniak, A.; Mastalerz, M. Rare Earth Elements: A brief overview. Indiana J. Earth Sci. 2022, 4, 1–7. [Google Scholar] [CrossRef]
- Suli, L.M.; Ibrahim, W.H.W.; Aziz, B.A.; Deraman, M.R.; Ismail, N.A. A Review of Rare Earth Mineral Processing Technology. Chem. Eng. Res. Bull. 2017, 19, 20. [Google Scholar] [CrossRef]
- Du, X.; Graedel, T.E. Global in-use stocks of the rare earth elements: A first estimate. Environ. Sci. Technol. 2011, 45, 4096–4101. [Google Scholar] [CrossRef]
- Balaram, V. Rare earth elements: A review of applications, occurrence, exploration, analysis, recycling, and environmental impact. Geosci. Front. 2019, 10, 1285–1303. [Google Scholar] [CrossRef]
- Rupasinghe, M.S.; Dissanayake, C.B. The rare-earth element abundance in the sedimentary gem deposits of Sri Lanka. Lithos 1984, 17, 329–342. [Google Scholar] [CrossRef]
- Lin, P.; Yang, X.; Werner, J.M.; Honaker, R.Q. Application of Eh-pH Diagrams on Acid Leaching Systems for the Recovery of REEs from Bastnaesite, Monazite and Xenotime. Metals 2021, 11, 734. [Google Scholar] [CrossRef]
- Vinoj, P.K.L.; Kapilarathna, M.W.C.S.; Samarakoon, S.M.P.S.; Dushyantha, N.P. Investigation of Rare Earth Element Potential in Granitic Rocks of Sri Lanka Special Reference to Thonigala Granite. In Proceedings of the International Symposium on Earth Resources Management & Environment, Jakarta, Indonesia, 25–26 September 2021; pp. 128–134. [Google Scholar]
- Dostal, J. Rare earth element deposits of alkaline igneous rocks. Resources 2017, 6, 34. [Google Scholar] [CrossRef]
- Dissanayake, C.B.; Chandrajith, R.; Tobschall, H.J. The geology, mineralogy and rare element geochemistry of the gem deposits of Sri Lanka. Bull. Geol. Soc. Finl. 2000, 72, 5–20. [Google Scholar] [CrossRef]
- Toama, H.Z. World Phosphate Industry. Iraqi Bull. Geol. Min. Spec. Issue 2017, 7, 5–23. [Google Scholar]
- Guelfi, D.; Nunes, A.P.P.; Sarkis, L.F.; Oliveira, D.P. Innovative Phosphate Fertilizer Technologies to Improve Phosphorus Use Efficiency in Agriculture. Sustainability 2022, 14, 4266. [Google Scholar] [CrossRef]
- Khan, M.; Ahmad, S.; Sharif, M.; Billah, M.; Aslam, M. Formulation of single super phosphate fertilizer from rock phosphate of Hazara, Pakistan. Soil Environ. 2012, 31, 96–99. [Google Scholar]
- Reijnders, L. Phosphorus resources, their depletion and conservation, a review. Resour. conserv. recycl. 2014, 93, 32–49. [Google Scholar] [CrossRef]
- Hughes, J.M.; Rakovan, J. The crystal structure of apatite, Ca5(PO4)3(F,OH,Cl). Phosphates Geochem. Geobiol. Mater. Importance 2019, 48, 1–12. [Google Scholar] [CrossRef]
- Cheremisina, O.; Sergeev, V.V.; Fedorov, A. Rare Earth Metal Extraction from Apatite Ores. Metallurgist 2019, 63, 300–307. [Google Scholar] [CrossRef]
- Gamage, S. Determination of Rare Earth Element Contents in the Pulmoddai-Based Monazite. Int. J. Adv. Res. 2018, 6, 1229–1236. [Google Scholar] [CrossRef]
- Manthilake, M.A.G.M.; Sawada, Y.; Sakai, S. Genesis and evolution of Eppawala carbonatites, Sri Lanka. J. Asian Earth Sci. 2008, 32, 66–75. [Google Scholar] [CrossRef]
- Batapola, N.M.; Dushyantha, N.P.; Premasiri, H.M.R.; Abeysinghe, A.M.K.B.; Rohitha, L.P.S.; Ratnayake, N.P.; Dissanayake, D.M.D.O.K.; Ilankoon, I.M.S.K.; Dharmaratne, P.G.R. A comparison of global rare earth element (REE) resources and their mineralogy with REE prospects in Sri Lanka. J. Asian Earth Sci. 2020, 200, 104475. [Google Scholar] [CrossRef]
- Dushyantha, N.; Batapola, N.M.; Ratnayake, N.P. Leaching Potential of Rare Earth Elements (REES) from Eppawala Phosphate Deposit, Sri Lanka for Sustainable Critical Metals Recovery. In Proceedings of the 39th Technical Session of Geological Society of Sri Lanka (GSSL), Kandy, Sri Lanka, 24 February 2023; p. 6. [Google Scholar]
- Ribeiro, G.P.; Dantas, S.C.; Ribeiro, E.J.; Hori, C.E. Production of Single Superphosphates Using Igneous Phosphate Rocks with High Iron Oxide Concentrations. Ind. Eng. Chem. Res. 2023, 62, 12963–12973. [Google Scholar] [CrossRef]
- Udawatte, C.P.; Panagoda, P.V.A.; Wickramasinghe, W.M.A.D.B.; Wijewardena, J.D.H.; Sirisena, D.N.; Emitiyagoda, S.; Bandara, H.R.U.D. Use of single superphosphate fertiliser produced using eppawala rock phosphate as a source of phosphorous for rice cultivation. J. Natl. Sci. Found. Sri Lanka 2020, 48, 131–142. [Google Scholar] [CrossRef]
- Idi, A.A.B.; Ousmane, M.I.C.; Sahirou, B.M.; Manzola, A.S. Manufacture of simple superphosphate from Tahoua rock phosphate. World J. Adv. Res. Rev. 2023, 18, 1139–1148. [Google Scholar] [CrossRef]
- Nicola Doebelin, R.K. Profex: A graphical user interface for the Rietveld refinement program BGMN. J. Appl. Crystallogr. 2015, 48, 1573–1580. [Google Scholar] [CrossRef]
- Sri Lanka Standards Institution. Methods of Test for Fertilizers and Soil Conditioners Part 2: Determination of Moisture and Ash (First Revision). 2023. Available online: https://slsi.lk/web/wp-content/uploads/2023/04/DSLS-645-2.pdf (accessed on 22 April 2025).
- Vindiola, A.G., Jr. Laboratory Safety. In Official Methods of Analysis of AOAC International, Latimer, G.W., Ed.; 22nd ed.; AOAC Publications: New York, NY, USA, 2023. [Google Scholar]
- Sri Lanka Standards Institution (SLS). Methods of Test for Fertilizers and Soil Conditioners Part 1: Determination of Nitrogen (Second Revision). 2023. Available online: https://slsi.lk/web/wp-content/uploads/2023/04/SLS-645-1.pdf (accessed on 20 April 2025).
- Sri Lanka Standards Institution (SLS). Methods of Test for Fertilizers and Soil Con Ditioners Part 5: Determination of Phosphorous; Sri Lanka Standards Institution (SLS): Colombo, Sri Lanka, 1985. [Google Scholar]
- Sri Lanka Standards Institution (SLS). Triple Super-Phosphate (Fertilizer Grade); Sri Lanka Standards Institution (SLS): Colombo, Sri Lanka, 2014. [Google Scholar]
- Zuo, R.F.; Du, G.X.; Yang, W.G.; Liao, L.B.; Li, Z. Mineralogical and chemical characteristics of a powder and purified quartz from Yunnan Province. Open Geosci. 2016, 8, 606–611. [Google Scholar] [CrossRef]
- García-Tuñón, E.; Couceiro, R.; Franco, J.; Saiz, E.; Guitián, F. Synthesis and characterisation of large chlorapatite single-crystals with controlled morphology and surface roughness. J. Mater. Sci. Mater. Med. 2012, 23, 2471–2482. [Google Scholar] [CrossRef] [PubMed]
- Kostov-Kytin, V.V.; Dyulgerova, E.; Ilieva, R.; Petkova, V. Powder X-ray diffraction studies of hydroxyapatite and β-TCP mixtures processed by high energy dry milling. Ceram. Int. 2018, 44, 8664–8671. [Google Scholar] [CrossRef]
- Loy, C.W.; Matori, K.A.; Zainuddin, N.; Whitten, A.E.; Rehm, C.; de Campo, L.; Sokolova, A.; Schmid, S. Crystallographic characterization of fluorapatite glass-ceramics synthesized from industrial waste. Powder Diffr. 2017, 32, S61–S65. [Google Scholar] [CrossRef]
- Dahanayake, K.; Subasinghe, S.M.N.D. Mineralogical, chemical and solubility variations in the Eppawala phosphate deposit of Sri Lanka—A case for selective mining for fertilizers. Fertil. Res. 1991, 28, 233–238. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Z.; You, Y.; Zheng, X.; Wen, J. Synthesis of different structured FePO4 for the enhanced conversion of methyl cellulose to 5-hydroxymethylfurfural. RSC Adv. 2017, 7, 51281–51289. [Google Scholar] [CrossRef]
- Pralong, V.; Baies, R.; Calgnaert, V.; Raveau, B. Structure and physical properties of new iron hydrogenophosphates K2Fe(HP2O7)(H2PO4)2, LiH3Fe2(P2O7)2, and FeH2P2O7. Inorg. Chem. 2009, 48, 6835–6844. [Google Scholar] [CrossRef]
- Ardanova, L.I.; Get’Man, E.I.; Loboda, S.N.; Prisedsky, V.V.; Tkachenko, T.V.; Marchenko, V.I.; Antonovich, V.P.; Chivireva, N.A.; Chebishev, K.A.; Lyashenko, A.S. Isomorphous substitutions of rare earth elements for calcium in synthetic hydroxyapatites. Inorg. Chem. 2010, 49, 10687–10693. [Google Scholar] [CrossRef]
- Alemrajabi, M. Recovery of Rare Earth Elements from an Apatite Concentrate. Ph.D. Thesis, KTH Royal Institute of Technology, Stockholm, Sweden, 2018. Available online: https://www.diva-portal.org/smash/get/diva2%3A1263554/FULLTEXT01.pdf (accessed on 15 January 2024).
- Roy, R.N. Use of Phosphate Rocks for Sustainable Agriculture. Zapata, F., Ed.; FAO Land and Water Development Division and the International Atomic Energy Agency: Rome, Italy, 2004. [Google Scholar]
- Kim, R.; Cho, H.; Han, K.N.; Kim, K.; Mun, M. Optimization of Acid Leaching of Rare-Earth Elements from Mongolian Apatite-Based Ore. Minerals 2016, 6, 63. [Google Scholar] [CrossRef]
- Alemrajabi, M.; Rasmuson, Å.C.; Korkmaz, K.; Forsberg, K. Upgrading of a rare earth phosphate concentrate within the nitrophosphate process. J. Clean. Prod. 2018, 198, 551–563. [Google Scholar] [CrossRef]
- Stone, K.; Bandara, A.M.T.S.; Senanayake, G.; Jayasekera, S. Processing of rare earth phosphate concentrates: A comparative study of pre- leaching with perchloric, hydrochloric, nitric and phosphoric acids and deportment of minor/major elements. Hydrometallurgy 2016, 163, 137–147. [Google Scholar] [CrossRef]
- Meor, Y.M.S. Rate of rare earths leaching in HCI, H2SO4 AND HNO3. Adv. Mater. Res. 2013, 795, 1–4. [Google Scholar] [CrossRef]
- Premasiri, H.M.R. Leaching Potential of Rare Earth Elements (REES) from Eppawala Phosphate Deposit, Sri Lanka for Sustainable Critical Metals Recovery. In Proceedings of the Geological Society of Sri Lanka (GSSL), Kandy, Sri Lanka, 24 February 2023; pp. 1–2. [Google Scholar]
- Wu, S.; Zhao, L.; Wang, L.; Huang, X.; Dong, J.; Feng, Z.; Cui, D.; Zhang, L. feng Dissolution behaviors of rare earth elements in phosphoric acid solutions. Trans. Nonferrous Met. Soc. China (Engl. Ed.) 2018, 28, 2375–2382. [Google Scholar] [CrossRef]
- Li, H.; Guo, F.; Zhang, Z.; Li, D.; Wang, Z. A new hydrometallurgical process for extracting rare earths from apatite using solvent extraction with P350. J. Alloys Compd. 2006, 412, 995–998. [Google Scholar] [CrossRef]
- Habashi, F. The Recovery of the Lanthanides from Phosphate Rock. J. Chem. Technol. Biotechnol. 1985, 4, 5–14. [Google Scholar] [CrossRef]
- Liu, X.; Byrne, R.H. Rare earth and yttrium phosphate solubilities in aqueous solution. Geochim. Cosmochim. Acta 1997, 61, 1625–1633. [Google Scholar] [CrossRef]
- Sigtryggsson, C.; Hamnér, K.; Kirchmann, H. Laboratory studies on dissolution of nitrogen fertilizers by humidity and precipitation. Agric. Environ. Lett. 2020, 5, e20016. [Google Scholar] [CrossRef]
- P, D. CRC Handbook of Chemistry and Physics: Editor-in-chief D.R. Lide; CRC Press, Boca Raton, FL, USA, 71st edn, 1990–1991, pp. 2324, price $117.00 (USA $99.50). J. Mol. Struct. 1992, 268, 320. [Google Scholar] [CrossRef]
- Beaumont, A.B.; Mooney, R.A. Hygroscopicity and Cakiness of Fertilizer Materials. Ind. Eng. Chem. 1925, 17, 635–636. [Google Scholar] [CrossRef]
- Han, K.N. Characteristics of precipitation of rare earth elements with various precipitants. Minerals 2020, 10, 178. [Google Scholar] [CrossRef]
- Zhang, T.; Lu, Y.; Luo, G. Effects of temperature and phosphoric acid addition on the solubility of iron phosphate dihydrate in aqueous solutions. Chin. J. Chem. Eng. 2017, 25, 211–215. [Google Scholar] [CrossRef]
- Cetiner, Z.S.; Wood, S.A.; Gammons, C.H. The aqueous geochemistry of the rare earth elements. Part XIV. The solubility of rare earth element phosphates from 23 to 150 °C. Chem. Geol. 2005, 217, 147–169. [Google Scholar] [CrossRef]
- Cui, H.; Zhong, R.; Xie, Y.; Yuan, X.; Liu, W.; Brugger, J.; Yu, C. Forming sulfate-and REE-rich fluids in the presence of quartz. Geology 2020, 48, 145–148. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | SiO2 | Al2O3 | BaO | CaO | Fe2O3 | K2O | MgO | MnO | Na2O | P2O5 |
|---|---|---|---|---|---|---|---|---|---|---|
| (w/w) % | 15.29 | 5.64 | 0.21 | 39.51 | 6.80 | 0.24 | 0.54 | 0.25 | 1.32 | 25.88 |
| Rare Earth Elements | Ce | Dy | Gd | La | Nd | Pr | Sm | Y |
|---|---|---|---|---|---|---|---|---|
| ppm (mg/kg) | 1124.9 | 1.5 | 41.4 | 482.7 | 477.7 | 138.1 | 72.3 | 90.0 |
| REE | Ce | Gd | Dy | La | Nd | Pr | Sm | Y |
|---|---|---|---|---|---|---|---|---|
| Leaching Efficiency (%) | 95.4 | 89.3 | 99.9 | 98.1 | 100 | 92.3 | 100 | 100 |
| Co-precipitation (σ) % | 1.6 | 1.6 | 1.8 | 0.5 | 1.8 | 0.5 | 1.2 | 2.0 |
| REE | Ce | Dy | Gd | La | Nd | Pr | Sm | Y |
|---|---|---|---|---|---|---|---|---|
| Extraction efficiency % | 98.8 | 92.5 | 98.5 | 95.8 | 99.5 | 100 | 98.2 | 97.9 |
| Selective dissolution efficiency % | 99.8 | 99.9 | 99.9 | 99.8 | 99.8 | 99.9 | 100.0 | 99.9 |
| Elements | Na | Ca | Fe | Al | Mn | Mg | REEs |
| % | 68.4 | 4.9 | 2.2 | 2.2 | <0.1 | <0.1 | 22.2 |
| REE recovery efficiency (ε) from (NaREE(SO4)2·xH2O) as a percentage of TREEs | |||||||
| REE | Ce | Dy | Gd | La | Nd | Pr | Sm |
| TREE % | 31.2 | 59.5 | 19.6 | 24.9 | 34.9 | 52.5 | 29.6 |
| Parameter | Results (% by Mass) | Method |
|---|---|---|
| Moisture | 0.60 | SLS 645 Part 2: 2023 [24] |
| Total nitrogen content as N, present by dry basis | 18.20 | AOAC 892.01 [25] SLS 645 Part 1: 2023 [26] |
| Total phosphate content as P2O5, percent by dry basis | 13.90 | SLS 645 Part 5: 1985 [27] |
| Free phosphoric acid as P2O5, percent by dry basis | 0.10 | SLS 812: 2014: Appendix B [28] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bandara, D.B.H.I.; Prasad, A.; Dulanjana, K.D.A.; Samarasekere, P.W. Dual-Purpose Utilization of Sri Lankan Apatite for Rare Earth Recovery Integrated into Sustainable Nitrophosphate Fertilizer Manufacturing. Sustainability 2025, 17, 6353. https://doi.org/10.3390/su17146353
Bandara DBHI, Prasad A, Dulanjana KDA, Samarasekere PW. Dual-Purpose Utilization of Sri Lankan Apatite for Rare Earth Recovery Integrated into Sustainable Nitrophosphate Fertilizer Manufacturing. Sustainability. 2025; 17(14):6353. https://doi.org/10.3390/su17146353
Chicago/Turabian StyleBandara, D. B. Hashini Indrachapa, Avantha Prasad, K. D. Anushka Dulanjana, and Pradeep Wishwanath Samarasekere. 2025. "Dual-Purpose Utilization of Sri Lankan Apatite for Rare Earth Recovery Integrated into Sustainable Nitrophosphate Fertilizer Manufacturing" Sustainability 17, no. 14: 6353. https://doi.org/10.3390/su17146353
APA StyleBandara, D. B. H. I., Prasad, A., Dulanjana, K. D. A., & Samarasekere, P. W. (2025). Dual-Purpose Utilization of Sri Lankan Apatite for Rare Earth Recovery Integrated into Sustainable Nitrophosphate Fertilizer Manufacturing. Sustainability, 17(14), 6353. https://doi.org/10.3390/su17146353