Abstract
In wastewater treatment plants, the stability of the related microbiota is pivotal for a steady and appropriate operation in biological wastewater treatment (BWT). The interactions between phages, which are high in amounts and diversity, and their particular hosts are significant due to their specificity in this microbial community. As drivers of diversity, phages are capable of changing the microbial structure by affecting host cells. This study aimed to enhance the stability of the functional microbiota, a primary concern of BWT, by evaluating the influence of bioaugmentation with bacteriophage cocktails. The sequencing data revealed significant alterations in the entire microbiome, including microeukaryotes, induced by the bacteriophages. These alterations led to a reduction in opportunistic microorganisms while preserving the beneficial ones. The genus Proteobacteria was found unaffected by three antibiotics in the bioreactor due to its antibiotic-resistant members, leading to its further growth, while the antibiotic-sensitive genus decreased in quantity. Furthermore, the phages notably influenced the metabolic pathway such as nitrogen, carbohydrate, and amino acid metabolisms by eliminating opportunistic microbes and providing improved growth conditions to bacterial species that are essential for effective reactor performance and wastewater treatment.
1. Introduction
In recent years, water quality has notably decreased due to the increment in both population and industrial development. Wastewater treatment plants (WWTP) receive tons of wastewater coming from various sources such as hospitals, slaughterhouses, livestock farming, pharmaceutical facilities, and household sewage. In conditions where the treatment is not efficacious, effluent water may still have pollutants resulting in adverse effects on both human health and aquatic environments through being released from the treatment process. Therefore, the appropriate treatment of wastewater is of critical importance in terms of disposal. Various treatment methods such as land application, chemical treatment, and discharge in sewers are in use, however, there are constraints to their utilization since unfavorable odor and contamination of aquatic environments issue in the exploitation of the techniques of land application and discharge in sewers. In addition, it is not recommended to use a chemical treatment, as chemicals used in the process raise the treatment cost, and the challenge in the disposal of remaining sludge-containing chemicals makes the process cumbersome and costly [1]. On the contrary, the BWT approach provides a substantial remedy with its eco-friendly and cost-effective characteristic in removing pollutants from wastewater. In BWT, diverse functional populations of bacteria that are responsible for removing organic matter, nitrogen, and phosphorus from the wastewater play a critical role in the effective treatment of wastewater. Heterotrophic bacteria take a substantial role in organic matter removal, ammonia-oxidizing bacteria (AOB) in nitrification, denitrifying bacteria in nitrogen removal, and phosphate-accumulating organisms (PAOs) in phosphorus removal. Appropriate pollutant removal can be achieved through collaborative interactions among these functional groups [2]. In this process, bacterial strains adjust their population density and behaviors via a cell-to-cell communication pathway named quorum sensing (QS), which is also responsible for the formation of bacterial biofilms. In addition to the formation of biofilm, bacteria through QS also regulate the removal of pollutants, and pathogenesis control, and they determine the composition and behavior of the biofilm community. Therefore, the manipulation of the bacterial community in BWT is of critical interest, as it has a substantial role in obtaining effective treatment and optimal removal [3].
In recent years, alternative treatment systems have attracted a lot of attention in order to treat wastewater with a high quality, due to the limited area and strict discharge issues in conventional systems. Membrane bioreactor (MBR) is a promising strategy that has been frequently implemented in the treatment of both domestic and industrial wastewater in recent years, and it is an innovative technology for the treatment and reuse of treated water by combining the activated sludge process with the membrane filtration technique. In this system, pollutants with a molecular weight greater than the molecular weight cut-off of the membranes are trapped via the membrane’s sieving effect. This brings pollutants into contact with the degrading microorganisms that exist inside the MBR, allowing for their full degradation [4]. The usage of MBR technology ensures augmented treatment efficacy, specifically if the wastewater contains effluents from diverse sources when compared to the conventional treatment approaches, including activated sludge system, and physical or chemical treatment. MBR requiring relatively low-maintenance needs offers several advantages that make it a more appealing technology. Firstly, it provides a consistent treatment quality for the wastewater, regardless of the sludge conditions in the bioreactor. MBR is also beneficial in a lower production of sludge while facilitating high removal rates of nutrients, organic compounds, and pollutants. Another advantage of MBR usage is its physical footprint, which occupies a smaller area compared to conventional treatment systems, resulting in space-saving. Additionally, the combination of BWT with membrane filtration ensures an advanced capability to retain microorganisms, which have recently turned into a major concern in WWTP [5].
Microorganisms have a pivotal role in the efficiency of the BWT process, however, characteristics of both bio-sludge and wastewater influence the effectiveness of microorganisms. Therefore, optimization of the interactions between microbial strains and modifications in the microbial community can have a substantial impact on a sustainable process. In this regard, bacteriophages or simply phages have the potential to enhance aerobic BWT processes by targeting specific bacterial strains. The control of the microbial community through selective targeting with phages can provide solutions to the challenges seen in aerobic BWT and can be helpful in the optimization of the microbial community dynamics, resulting in improved efficiency in wastewater treatment [6]. Since phage treatment with single species can face bacterial resistance development, the usage of specific phage mixtures, known as phage cocktails, offers improved treatment in various processes. In addition, single-phage species exhibit limited effectiveness in the mitigation of biofilm formation, while phage cocktails containing diverse phage species increase the host range and alleviate the phage resistance by bacterial pathogens [7]. The significance of phage treatment’s adaptability lies in its achievement of effective implementation within both aerobic and anaerobic BWT process, as well as across various systems without necessitating alterations to the pre-existing operational dynamics of the systems [6].
Microbial community analyses have a substantial part to understand and regulate the internal dynamics of the microbial communities in BWT. Although 16S rRNA sequencing is a widely used approach in the identification and characterization of microbial communities, it has various limitations, especially in providing high-resolution data about genetic diversity and community dynamics. Therefore, researchers have focused on whole genome shotgun (WGS) metagenomic sequencing to obtain comprehensive data about microbial communities, and to eliminate the limitations of 16S rRNA sequencing [8]. Even though WGS is a relatively more expensive sequencing method, it provides a deeper understanding of microbial community, reaching beyond simple taxonomic identification. Through WGS, it is possible to explore the entire genetic makeup, metabolic pathways, and microbial interactions of the community [9].
In this study, shotgun metagenomic sequencing was used to investigate the impact of the bioaugmentation of phage cocktails on the functional microbiota, which is of great importance in the BWT process, with the addition of three common antibiotics (erythromycin, tetracycline, and sulfamethoxazole) that anthropogenically affect aquatic spots. The pyophage cocktail used in the current study was acquired from the Eliava Institute located in Tbilisi, Georgia. The pyophage cocktail has proven effective against five bacterial species, namely Staphylococcus sp., E. coli, Streptococcus sp., Proteus sp., and Pseudomonas sp. The results indicated that phages altered the whole microbiome including microeukaryotes, and substantially affected the metabolic pathways related to carbohydrates and amino acid metabolisms. This study reveals the potential of phages as promising tools to enhance the functional capabilities of microbial communities in WWTP systems.
2. Materials and Methods
2.1. Operation of Aerobic Membrane Bioreactor
The previous study which investigated the effect of pyophage cocktail on biofilm formation through 16S sequencing provided comprehensive details regarding the setup and operation of the bioreactors [10]. Three aerobic membrane bioreactors were used in a continuous feeding mode in the experimental setup to examine the bioaugmentation of wastewater treatment via phage cocktail. Pharmaceutical wastewater conditions were simulated, and erythromycin, tetracycline, and sulfamethoxazole antibiotics were included in these bioreactors, according to the previous research study [10]. A submerged hollow-fiber membrane module with a surface area of 0.125 m2 was provided for each bioreactor. An air diffuser was used at the base of the bioreactors to ensure membrane surface-cleaning and aeration, and the volume of the aeration tank with dimensions of R × H = 7.25 cm × 34 cm was 5 L. A hollow-fiber membrane module with an average pore size of 0.04 μm was used to perform ultrafiltration. To measure the pressure, the transmembrane pressure (TMP) sensor was attached to a vacuum line, and values were recorded on a computer automatically. Three different sets of bioreactors were prepared: the control (C1) reactor included only aerobic sludge, the control with aerobic sludge included antibiotics (C2) and the BP bioreactor included a pyophage cocktail in addition to aerobic sludge and antibiotics [10]. All bioreactors were run under the same environmental conditions to eliminate the alterations that could result from external factors. Samples were collected twice daily from the influent, effluent, and reactor sludge, and were analysed to define different parameters such as chemical oxygen demand (COD), total suspended solids (TSS), total solids (TS), antibiotic removal rate, and the occurrence of antibiotic transformation products. When the TMP value exceeded 25 kPa, the membrane module was removed from the reactors. Samples obtained from the bioreactor sludge were extracted and cleaned through OASIS HLB cartridges (6 mL, 200 mg, Waters, Milford, MA, USA) to prepare the samples for LC/MS (liquid chromatography/mass spectrometry) analysis.
2.2. Microbial Community Analysis Using Shotgun Metagenomic Sequencing
Shotgun metagenomic sequencing is an efficient approach to studying biodiversity and functional characteristics of microbial communities. To detect the alterations in aerobic microbial communities and the persistence of antibiotic resistance genes (ARGs) in the presence of antibiotics and pyophage cocktail, Illumina sequencing was employed and the preparation for the sequencing was realized at Gentera Biotechnology Co., Ltd. (Istanbul, Turkey). Sludge from the BP reactor was obtained as the sample for DNA extraction, and whole genome DNA extraction was realized through Genomic DNA Isolation Kit (Norgen Biotek. Corp., Thorol, ON, Canada) according to instructions of the manufacturer. Quality control was conducted at each phase during the library construction. An amount of 1% of agarose gel electrophoresis was employed in DNA purification and integration, and the measurement of DNA concentration was realized with Qubit 2.0 fluorometer quantitation with a Qubit dsDNA Assay Kit (Life Technologies, CA, USA). In library construction, each sample was prepared with 1 μg DNA, and NEBNext® UltraTM DNA Library Prep Kit for Illumina (MA, USA) was used to construct libraries. After samples were tagged with specific index codes, CovarisTM S220 Sonicator (Covaris, Inc., Woburn, MA, USA) was employed in the physical fractionation of each sample to 350 bp. Following obtaining the fragments as A-tailed and end-polished, fragments were ligated with PCR-amplification for Illumina sequencing. The purification of samples was performed through the AMPure XP system, and the Agilent 2100 Bioanalyzer and Q-PCR were applied to provide sufficient enrichment. Afterwards, the cBot Cluster Genereation System was employed for the sequencing following the manufacturer’s instructions. Subsequently, paired-end reads were generated from library preparations by using Illumina NextSeq 500/HiSeq platforms. Quality control and host filtering were applied to the obtained raw data to acquire clean data, which was later to be used in metagenome assembly. Bases with a value below 38 and reads surpassing the 40 bp threshold were subjected to trimming during the data pre-processing. In addition, reads with excessive ambiguous nucleotides (N) were discarded, while reads that overlapped significantly with adapters were trimmed. The Soap 2.21 software was employed to reduce potential contamination from the host DNA.
Following the quality control, MEGAHIT was used in the metagenome assembly as the early step [11]. Scaftigs, meaning uninterrupted sequences within scaffolds, were obtained by trimming at the time the “N” symbol occurred. Scaftigs with a length below 500 bp acquired from mixed reads and individual samples were cut. The unutilized reads formed by the combination of each sample were analyzed to identify the low-abundance species. MetaGeneMark was used to perform gene prediction, using scaftigs assembled from mixed and individual samples. Afterwards, the predicted genes were assembled for the dereplication, which subsequently resulted in the construction of an exhaustive gene catalog. The Clean Data obtained in the previous steps were subjected to ORF prediction through MetaGeneMark [12]. The SoapAligner tool that mapped the clean data to the catalog was used to perform the calculation of the gene abundance by using the total quantity of mapped reads and gene length [13]. This approach allowed for the determination of gene-abundance values (Figure 1).
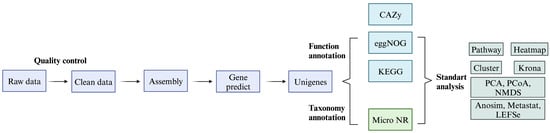
Figure 1.
Workflow of metagenome analysis.
The MEGAN software (version 6.21.14) was used in the taxonomical annotation of metagenomic homologs and made a comparison with the NR database from NCBI (NCBI: version 2016-11-05) [14]. For the function determination of coding sequences and relative abundances of different types of functions in the metagenome, similarity research was conducted in databases such as KEGG, eggnog, and CAZy. Analysis involving NMDS, Anosim, and PCA clustering was performed based on taxonomic and functional abundance tables. In addition, LEfSe and Metastats were employed for statistical analysis when comparing the functional contents of different groups. In addition, the identification of ARGs abundance and distribution in species, the dataset was compared to the Comprehensive Antibiotic Resistance Database (CARD). In this comparison, BLASTp, which is a bioinformatics tool that compares protein sequences, was utilized and the e-value threshold in the comparison was ≤1 × 10−5, providing the determination of the statistical significance of matches between the dataset and the CARD database [15].
3. Results and Discussion
3.1. Interactions between Bacteriophage and Indigenous Prokaryotic Microbial Diversity in Aerobic Membrane Bioreactor
In WWTPs, phages and bacteria are critical microbial groups in terms of microbial dynamics, the process of material circulation, and energy metabolism. Phages are viruses that target and infect specific bacterial strains, and they substitute a large abundance when compared to bacteria, with an approximate ratio of 10:1. Bacteriophage displays synergistic interactions with bacteria in communities, indicating their impact on bacterial communities. They can infect bacterial strains at a rate of approximately 1023 infections per second, and this ability has the potential to decrease target populations, especially antibiotic-resistant ones [16]. Previous studies have shown that pathogenic bacteria commonly found in municipal and industrial wastewater include Staphylococcus spp., E. coli, Streptococcus spp., Proteus spp., and Pseudomonas spp., as shown in [17,18]. As expected, with the addition of the cocktail containing the phages specific to these pathogenic strains to the reactor, these strains were hardly detected in the reactor. On the other hand, previous studies also reveal that Proteobacteria and Bacteroidetes are the most dominant bacterial phyla, while commonly found genera involve Aeromonas, Acinetobacter, Pseudomonas, Bacillus, etc., in different WWTPs [19]. Illumina sequencing provided comprehensive data about the microbial community structure, and indicated that the exposure to the combination of pyophage cocktail and antibiotics resulted in substantial alterations in the microbial community structure of the reactor sludge (Figure 2).

Figure 2.
The abundance of the predominant ten taxa in the sludge of the BP reactor. (a) At the phylum level, Proteobacteria were found to be the most predominant phylum. (b) At the genus level, Runella and Dechloromonas were the two most dominant genera.
At the phylum level, Proteobacteria accounted for 62% of the bacterial population, showing an increase due to the tolerance, while the second most abundant phylum Bacteroidetes has a ratio of 23% in the BP reactor with antimicrobials, similar to the previous study [20]. On the other hand, Firmicutes is known as the third predominant phylum in WWTPs [21]. However, with the exposure of the pyophage cocktail along with antibiotics, the abundance of Firmicutes had a low rate, accounting for only 5% of the total bacterial population in the BP reactor. Previous studies have revealed that members belonging to the Firmicutes phylum can harbor antibiotic-resistance genes [22,23]. Considering this information, it is feasible to suggest that the reduction seen in the abundance of Firmicutes may be attributed to the effect of the pyophage cocktail.
Furthermore, Nitrospirae was found as the third predominant phylum in the bacterial population of the BP reactor sludge, constituting 4% of the entire bacterial population. Nitrospirae is a predominant phylum of nitrite-oxidizing bacteria (NOB) that has a substantial role in biological nutrient removal in wastewater systems, especially with low levels of dissolved oxygen and substrate availability [24]. Compared to the relative abundance of Nitrospirae in normally operating wastewater treatment systems, the rate of Nitrospirae detected in the BP reactor in the current study is higher, and it is appropriate to conclude that this increase is due to the pyophage cocktail [25,26]. Considering the lack of success in the nitrification process is generally linked to the low concentration of nitrifying bacteria in activated sludge systems [27], this increment in Nitrospirae demonstrates that the pyophage cocktail has the potential to improve the performance of MBR.
The development of the bacterial community within the BP reactor in the presence of the pyophage cocktail and antibiotics are also can be observed at the genus level. Previous studies have indicated that the genera of Dechloromonas and Nitrospira were one of the core microbial groups in most WWTPs and could show resistant characteristics when faced with antibacterial agents [28]. In the current study, Runella (5%), Dechloromonas (5%), Nitrospira (4%), Haliangium (4%), and Haliscomenobacter (3%) were identified as the most prevalent genera within the bacterial community. The importance of these five predominant genera, despite the existence of antimicrobial agents, points to possible resistance and their ability to develop in a community in which antibiotic-sensitive ones could have decreased, and this observation aligns with previous studies that have revealed similar findings [10,20]. Another genus found and notable in the BP reactor is Zoogloea (2%), and this group of bacteria is typically found in activated sludge and involved in the degradation of organic and inorganic compounds in wastewater [29].
3.2. Interactions between Bacteriophage and Indigenous Eukaryotic Microbial Diversity in Aerobic Membrane Bioreactor
We investigated eukaryotic microorganisms in bioreactors and found that these microorganisms constitute only a small percentage of the whole community. Phyla of Ascomycota (10%), Chytridiomycota (2%), and Basidiomycota (1%) were observed as the core fungal groups, while microalgal phylum Chlorophyta (2%) and diatom group Thalassiosira (1%) were the other notable eukaryotic microorganisms. A significant amount of microorganism assemblage was evaluated as unclassified, which could be the result of the lack of reference sequence data. Ascomycota and Chytridiomycota are able to secrete cellulase, thus, these fungal groups can be used in the treatment of cellulose-containing wastewater due to their cellulose-degrading abilities. However, the relative abundance of Ascomycota with a ratio of 10% in eukaryotes, is of notable importance (Figure 3). In a study conducted on aerobic sludge, the high abundance of Ascomycota detected in the sludge was correlated with the addition of rich cellulose in the membrane reactor [30]. Based on this study, the high abundance of Ascomycota phylum with a ratio of 10% in the current study may be cellulose-containing plants that account for as much as 32% of the eukaryotic organisms in the reactor. On the other hand, the genus Aspergillus, which constitutes 17% of Ascomycota in the BP reactor, is a filamentous fungus and is widely employed as a bioremediation agent for wastewater contaminated with heavy metal, petroleum, and other pollutants [31].

Figure 3.
The relative abundance of Ascomycota in the BP reactor. Ascomycota accounts for a significant 10% of all eukaryotes in the BP reactor.
In the BP reactor antimicrobials, the existence of the Chlorophyta group (2%) in eukaryotic microorganisms, is noteworthy. The microalgal group Chlorophyta has been used in various streams, including industrial and municipal wastewater treatments with the purpose of nutrient removal. This utilization can be attributed to the substantial adaptation capability of microalgae in specific conditions [32]. Even though a significant challenge in the cultivation of microalgae in untreated wastewater is the presence of other microorganisms that restrict biomass productivity, Chlorophyta in the BP reactor has shown the ability to thrive in the existence of the combination of pyophage cocktail and antibiotics. Thus, if the BP reactor operates with long-term exposure to these antimicrobials, the microalgal group Chlorophyta may have improved growth opportunities. Therefore, the application of a pyophage cocktail with antibiotics would effectively hamper the development of microbes and this would result in the increased capacity and potential of the wastewater treatment systems for nutrient removal. These findings suggest that the pyophage cocktail positively influences the WWTP process by enhancing the development of microorganisms that are beneficial in bioremediation. By targeting specific pathogens or opportunistic microorganisms, the pyophage cocktail may help create a more suitable environment for these species to thrive, ultimately improving the treatment efficiency and overall performance of the system. Indeed, it is also vital to consider the potential synergistic relationship between antibiotics and the pyophage cocktail in the current study.
3.3. Interactions between Bacteriophage and the Metabolic Pathway
To gain further insights into the effects of bacteriophage on the metabolic pathway of the microbial community from the proteomic level, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway classification in the BP reactor was also carried out. Predominantly seen metabolic pathways are amino acid metabolism, carbohydrate metabolism, energy metabolism, cofactors and vitamins metabolism, nucleotide metabolism, lipid metabolism, xenobiotics biodegradation, and metabolism and biosynthesis of other seconder metabolites (Figure 4). The core genera in nitrogen metabolism were observed as Nitrosomonas (0.2%) and Nitrospira (4%), which are known as potential nitrifying bacterial groups, and they have been identified as predominant ammonia-oxidizing bacteria (AOB) in different activated sludge systems, indicating their tolerance to environmental alterations [33]. Therefore, the application of the combination of a pyophage cocktail and antibiotics did not appear to affect the existence of these genera. The group of these two is known for their contribution to the reactor performance by enhancing nitrification. Specifically, the genus of comammox Nitrospira has a substantial role in the nitrification process, which is the pivotal step in the nitrogen cycle. The process of nitrification briefly occurs with the conversion of ammonia (NH3) to nitrate (NO3−) via two sequential oxidation reactions. While it was considered at first nitrification was realized by two different bacterial groups comprising ammonia-oxidizing bacteria (AOB) and nitrite-oxidizing bacteria (NOB). With the discovery of comammox Nitrospira, this understanding was totally changed. The unique feature of this genus is its capability in performing both ammonia oxidation and nitrite oxidation without the need for additional bacteria to realize the second step. In other words, the members of Nitrospira can perform the whole nitrification independently, increasing the efficiency of the process. Their abundance in diverse environments such as soil, fresh water and WWT systems, make them a key player in nitrogen metabolism [34]. In fact, the metabolic capabilities of the genus Nitrospira reach beyond nitrite oxidation, covering various abilities including the growth on formate and hydrogen under aerobic conditions, and the reduction of nitrate to nitrite when it is in anoxic condition [35].

Figure 4.
KEGG pathway classification in the BP reactor.
The BP reactor exhibited the existence of Dechloromonas (5%), Zoogloea (2%), and Accumulibacter (2%) genera, constituting a total of 9% of the bacterial population, well known for denitrification potentials. Especially, species in Zoogloea genus have been identified as denitrifying aerobic heterotrophic bacterial strains in traditionally operating activated sludge systems, providing nitrate reduction even in high levels of oxygen [36]. Haliangium ochraceum (1%) species have been associated with both carbohydrate and amino acid metabolisms. This group of bacteria displays genes encoding key enzymes that took part in the production of polyketide and peptide metabolites, including polyketide synthases and nonribosomal peptide synthases. They also have the ability to decompose macromolecules such as starch, DNA, casein, gelatine, and chitin [37].
Phosphorus removal of cofactor metabolism in activated sludge systems can be attributed to the activity of phosphorus-accumulating organisms (PAOs). The genus Accumulibacter is also recognized as a PAO genus [38], thus the existence of Accumulibacter (2%) in the BP reactor indicates this genus contribution to phosphorus removal as a PAO. Photosynthetic Rhodobacterales constituting 3% of the bacterial genome in the BP reactor have been previously described in energy and vitamin metabolisms [39]. In addition, the genus of Sorangium (0.9%) has a substantial role in the synthesis of approximately half of the secondary metabolites [40]. These metabolic pathways are widely encountered in wastewater treatment systems, and the genera and species above mentioned are one of the key players in the pathways. Despite the presence of a phage cocktail, the maintenance of these metabolic pathways suggests that the usage of phages for pathogen removal and the enrichment of WWTP does not have an adverse impact on bacterial metabolic pathways, which are crucial in reactor systems. Furthermore, the phage cocktail may have provided suitable environments for bacteria that are responsible for these pathways, by eliminating opportunistic pathogens from the BP reactor, resulting in the persistence of the pathways.
3.4. Interactions between Bacteriophage and Occurrence of Antibiotic Resistance Genes in Aerobic Membrane Bioreactor
WWTPs serve as prominent hotspots for the dissemination of resistance genes and the proliferation of resistant bacteria. This kind of contaminated environment harbors a diverse array of ARGs and a great amount of bacteria, fostering microbial interactions. Furthermore, WWTPs are subject to various stresses such as microplastic and heavy metal, which further enhance the selection and horizontal gene transfer (HGT). With the analysis of antibiotic resistance genes (ARGs) alongside microbial community structures in the bioreactor, we obtained insight into the correlations among ARGs. The presence of antibiotics led to an enrichment of gram-negative Proteobacteria in the BP reactor, which exhibited the highest levels of antibiotic resistance profiles. The considerable presence and wide range of Proteobacteria in the BP reactor could potentially be attributed to antibiotic-resistant species of Proteobacteria encompassing numerous significant human pathogens, including Klebsiella spp., Salmonella spp., Acinetobacter spp., and various others.
The increase in Proteobacteria was also observed in a previous study that indicated the addition of antibiotics into the influent caused a remarkable change in the microbial community composition, and an increase in the abundance of ARGs in the membrane reactor [41]. Different resistance genes occur through different mechanisms. Figure 5 illustrates different phyla containing ARGs in the BP reactor. Since Proteobacteria is the predominant genus, the most dominant resistant mechanism used by bacteria to acquire resistance genes is RND-type Drug Efflux Pumps, while mtrA has been identified as the common resistance gene (Figure 6b). RND antibiotic efflux pumps are a kind of efflux pump present in Gram-negative bacterial phylum such as Proteobacteria, and they significantly contribute to this group of bacteria developing resistance against a wide range of antibiotics. The mtrA gene, also known as mtrR, is a crucial resistant gene in various Gram-negative bacteria, and this gene encodes a transcriptional repressor protein that is responsible for the MtrCDE efflux pump system expression. This pump system plays a substantial role in bacterial resistance to different antimicrobials. The MtrA gene ensures that bacterial cells actively release antibiotics by upregulating the MtrCDE efflux pump expression, resulting in a decrease in intracellular concentrations of antibiotics and making them less efficient [42]. Although the existence of Staphylococcus aureus substitutes a very small amount in the whole bacterial genome (0.003%), the resistance gene (arlR) belonging to this species was observed as one of the most prominent resistance genes in the BP reactor. ArlR gene is part of a two-component regulatory system named arlRS and this system has been strongly associated with several gene regulations that play a role in antimicrobial resistance mechanisms [43]. Interestingly, lytic phage application did not appear to substantially improve the ARGs abundance; nevertheless, they were in high abundance in the BP reactor. In further investigations through phage bioaugmentation, it would be beneficiary to incorporate supplementary agents for the bioaugmentation, that can mitigate ARG formation, particularly by targeting Proteobacteria, in the aerobic membrane bioreactor.
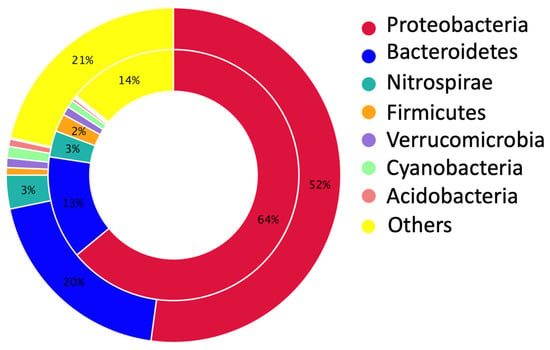
Figure 5.
The inner circle indicates the abundance of diverse phyla containing ARGs, while the outer ring represents the rate of ARGs across diverse phyla within this group.
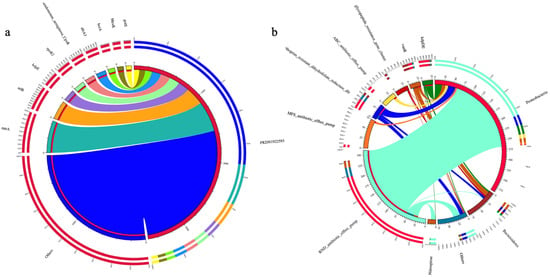
Figure 6.
(a) Overview Circle chart of resistance genes, (b) the relationship between resistance mechanism and phyla.
3.5. The Potential Use of Bacteriophages in the Aerobic Membrane Reactor
Except for the function of phages in removing membrane contamination by targeting pathogens, phages display various capabilities in aerobic WWTPs in which microorganisms have a game-changer role and augment the treatment efficiency. Filamentous bacterial species such as Gordonia and Nocordia generally result in bulking and foaming issues in the activated sludge. Chemical approaches have been used to tackle this problem, however, this application can also have adverse results by hampering the development of bacteria that are key players in the process [44]. As an alternative approach, studies revealed the potential of phage treatment in controlling bulking and foaming in sludge systems. Four bacteriophages were isolated and mixed to reduce the Gordonia abundance, and the phage cocktail provided a successful reduction in comparison with the control [45]. Furthermore, the polyvalent phage isolated from a WWTP succeeded in the removal of different Gordonia and Nocardia species [46]. Excessive sludge-forming caused by filamentous bacteria and EPS secreted by Zoogloea and Thauerea genera is the other problem to tackle in activated sludge systems [47]. The high abundance of EPS prevents the dehydration of sludge through retaining water and subsequently results in the reduction of the sedimentation capacity and biofilm formation. The success of phages in disrupting these biofilm formations has been demonstrated by various studies [48].
The PN/AMX is a useful method in nitrogen removal and generation of low sludge in WWTP. Nevertheless, the competition between NOB (Nitrobacter and Nitrospiraceae) and anammox bacteria poses a difficulty. A study using N. multiformis indicated that two pyophages were induced under specific conditions, such as acidic pH, and the increment of these viral particles led to a decrease in nitrification activity through the reduction in N. multiformis abundance [49]. Phage-mediated disruption has also achieved successful reductions in nitrification through the removal of certain bacterial species in different research studies [10,50,51].
4. Conclusions
The current study investigated the impact of combining a phage cocktail with three antibiotics frequently present in wastewater on the microbial community, in a bioreactor using shotgun metagenomic analyses. The phage cocktail used in this study was specific to Staphylococcus sp., E. coli, Streptococcus sp., Proteus sp., and Pseudomonas sp. Pathogens, which are normally and frequently found in wastewater. The results indicate that the phage treatment was successful with no detection of these species in the BP reactor. This is of importance, since if untreated appropriately, these pathogens could potentially be released into aquatic environments. Antibiotics used in addition to the pyophage cocktail provided synergy with the phage treatment, being effective in removing opportunistic bacterial species that are outside the host range of the phage cocktail and generally found in wastewater but barely detected in the BP reactor. This combination supported bacteria such as NOB, which are responsible for effective reactor performance and wastewater treatment. The impact of the phage cocktail on metabolic pathways was also examined, and it was observed that the phage application did not have adverse effects on these metabolic pathways. In fact, the pyophage cocktail may have even supported bacterial groups involved in pathways, by removing opportunistic species from the reactor. However, more comprehensive knowledge about the effects of phages on microbial community dynamics will require further investigation under long-term exposure.
Author Contributions
Conceptualization, S.A.; methodology, S.A.; software, S.A.; validation, S.A.; resources, S.A.; writing—original draft preparation, Ş.R. & S.A.; writing—review and editing, Ş.R. & S.A.; project administration, S.A.; funding acquisition, S.A. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by the Scientific Research Projects Coordination Unit of Istanbul University. Grant Number: FYL-2023-39728.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data will be available on request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Aziz, A.; Basheer, F.; Sengar, A.; Irfanullah; Khan, S.U.; Farooqi, I.H. Biological wastewater treatment (anaerobic-aerobic) technologies for safe discharge of treated slaughterhouse and meat processing wastewater. Sci. Total Environ. 2019, 686, 681–708. [Google Scholar] [CrossRef]
- Liu, R.; Li, Z.; Han, G.; Cun, S.; Yang, M.; Liu, X. Bacteriophage ecology in biological wastewater treatment systems. Appl. Microbiol. Biotechnol. 2021, 105, 5299–5307. [Google Scholar] [CrossRef] [PubMed]
- Maddela, N.R.; Sheng, B.; Yuan, S.; Zhou, Z.; Villamar-Torres, R.; Meng, F. Roles of quorum sensing in biological wastewater treatment: A critical review. Chemosphere 2019, 221, 616–629. [Google Scholar] [CrossRef] [PubMed]
- Goswami, L.; Kumar, R.V.; Borah, S.N.; Manikandan, N.A.; Pakshirajan, K.; Pugazhenthi, G. Membrane bioreactor and integrated membrane bioreactor systems for micropollutant removal from wastewater: A review. J. Water Process. Eng. 2018, 26, 314–328. [Google Scholar] [CrossRef]
- Lebleu, N.; Roques, C.; Aimar, P.; Causserand, C. Effects of membrane alterations on bacterial retention. J. Membr. Sci. 2009, 348, 56–65. [Google Scholar] [CrossRef]
- Reisoglu, Ş.; Aydin, S. Bacteriophages as a promising approach for the biocontrol of antibiotic resistant pathogens and the reconstruction of microbial interaction networks in wastewater treatment systems: A review. Sci. Total Environ. 2023, 890, 164291. [Google Scholar] [CrossRef]
- Zhang, B.; Yu, P.; Wang, Z.; Alvarez, P.J.J. Hormetic Promotion of Biofilm Growth by Polyvalent Bacteriophages at Low Concentrations. Environ. Sci. Technol. 2020, 54, 12358–12365. [Google Scholar] [CrossRef]
- Lazarevic, V.; Whiteson, K.; Gaïa, N.; Gizard, Y.; Hernandez, D.; Farinelli, L.; Østerås, M.; François, P.; Schrenzel, J. Analysis of the salivary microbiome using culture-independent techniques. J. Clin. Bioinform. 2012, 2, 4. [Google Scholar] [CrossRef]
- Hasan, N.A.; Young, B.A.; Minard-Smith, A.T.; Saeed, K.; Li, H. Microbial Community Profiling of Human Saliva Using Shotgun Metagenomic Sequencing. PLoS ONE 2014, 9, 97699. [Google Scholar] [CrossRef]
- Aydin, S.; Can, K. Pyophage cocktail for the biocontrol of membrane fouling and its effect in aerobic microbial biofilm community during the treatment of antibiotics. Bioresour. Technol. 2020, 318, 123965. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Lomsadze, A.; Borodovsky, M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010, 38, e132. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Fang, L.; Xu, X. Using SOAPaligner for Short Reads Alignment. Curr. Protoc. Bioinform. 2013, 44, 11. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.J.; Tappu, R. MEGAN Community Edition-Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2019, 48, D517–D525. [Google Scholar] [CrossRef]
- Worley-Morse, T.O.; Gunsch, C.K. Modeling phage induced bacterial disinfection rates and the resulting design implications. Water Res. 2015, 68, 627–636. [Google Scholar] [CrossRef]
- Kumaraswamy, R.; Amha, Y.M.; Anwar, M.Z.; Henschel, A.; Rodríguez, J.; Ahmad, F. Molecular Analysis for Screening Human Bacterial Pathogens in Municipal Wastewater Treatment and Reuse. Environ. Sci. Technol. 2014, 48, 11610–11619. [Google Scholar] [CrossRef]
- Yasir, M. Analysis of Microbial Communities and Pathogen Detection in Domestic Sewage Using Metagenomic Sequencing. Diversity 2020, 13, 6. [Google Scholar] [CrossRef]
- Zhao, R.; Feng, J.; Liu, J.; Fu, W.; Li, X.; Li, B. Deciphering of microbial community and antibiotic resistance genes in activated sludge reactors under high selective pressure of different antibiotics. Water Res. 2018, 151, 388–402. [Google Scholar] [CrossRef]
- Du, B.; Wang, Q.; Yang, Q.; Wang, R.; Yuan, W.; Yan, L. Responses of bacterial and bacteriophage communities to long-term exposure to antimicrobial agents in wastewater treatment systems. J. Hazard. Mater. 2021, 414, 125486. [Google Scholar] [CrossRef]
- Aydin, S.; Can, K.; Çalışkan, M.; Balcazar, J.L. Bacteriophage cocktail as a promising bio-enhancer for methanogenic activities in anaerobic membrane bioreactors. Sci. Total Environ. 2022, 832, 154716. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Deng, D.; Hu, C.; Lou, L.; Luo, L.; He, J.; Tian, D.; Xiao, Y.; He, Y.; Zhang, S.; et al. More effective removal of antibiotic resistance genes from excess sludge by microwave integrated fenton treatment. Int. Biodeterior. Biodegrad. 2020, 149, 104920. [Google Scholar] [CrossRef]
- Schwarz, S.; Shen, J.; Wendlandt, S.; Feßler, A.T.; Wang, Y.; Kadlec, K.; Wu, C.M. Plasmid-Mediated Antimicrobial Resistance in Staphylococci and Other Firmicutes. Microbiol. Spectr. 2014, 421–444. [Google Scholar] [CrossRef] [PubMed]
- Geng, M.; You, S.; Guo, H.; Ma, F.; Xiao, X.; Zhang, J.; Ma, X. Response of aerobic granular sludge to loading shock: Performance and proteomic study. Chem. Eng. J. 2022, 444, 136458. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, L.; Xiang, F.; Zhao, L.; Qiao, Z. Activated sludge microbial community and treatment performance of wastewater treatment plants in industrial and municipal zones. Int. J. Environ. Res. Public Health 2020, 17, 436. [Google Scholar] [CrossRef]
- Xiao, X.; Guo, H.; Ma, F.; Zhang, J.; Ma, X.; You, S. New insights into mycelial pellets for aerobic sludge granulation in membrane bioreactor: Bio-functional interactions among metazoans, microbial communities and protein expression. Water Res. 2023, 228, 119361. [Google Scholar] [CrossRef]
- Wanner, J.; Ruzickova, I.; Krhutkova, O.; Pribyl, M. Activated sludge population dynamics and wastewater treatment plant design and operation. Water Sci. Technol. 2000, 41, 217–225. [Google Scholar] [CrossRef]
- Wu, L.; Ning, D.; Zhang, B.; Li, Y.; Zhang, P.; Shan, X.; Zhang, Q.; Brown, M.R.; Li, Z.; Van Nostrand, J.D.; et al. Global diversity and biogeography of bacterial communities in wastewater treatment plants. Nat. Microbiol. 2019, 4, 1183–1195. [Google Scholar] [CrossRef]
- Wang, X.; Hu, M.; Xia, Y.; Wen, X.; Ding, K. Pyrosequencing Analysis of Bacterial Diversity in 14 Wastewater Treatment Systems in China. Appl. Environ. Microbiol. 2012, 78, 7042–7047. [Google Scholar] [CrossRef]
- Chen, C.; Gan, Z.; Xu, R.; Meng, F. Cellulose-induced shifts in microbial communities and microbial interactions in an anoxic/aerobic membrane bioreactor. J. Water Process. Eng. 2021, 42, 102106. [Google Scholar] [CrossRef]
- El-Bondkly, A.M.A.; El-Gendy, M.M.A.A. Bioremoval of some heavy metals from aqueous solutions by two different indigenous fungi Aspergillus sp. AHM69 and Penicillium sp. AHM96 isolated from petroleum refining wastewater. Heliyon 2022, 8, e09854. [Google Scholar] [CrossRef]
- Abinandan, S.; Shanthakumar, S. Challenges and opportunities in application of microalgae (Chlorophyta) for wastewater treatment: A review. Renew. Sustain. Energy Rev. 2015, 52, 123–132. [Google Scholar] [CrossRef]
- Geets, J.; Boon, N.; Verstraete, W. Strategies of aerobic ammonia-oxidizing bacteria for coping with nutrient and oxygen fluctuations. FEMS Microbiol. Ecol. 2006, 58, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Latocheski, E.C.; da Rocha, M.C.V.; Braga, M.C.B. Nitrospira in wastewater treatment: Applications, opportunities and research gaps Graphical abstract. Rev. Environ. Sci. Biotechnol. 2022, 21, 905–930. [Google Scholar] [CrossRef]
- Koch, H.; van Kessel, M.A.H.J.; Lücker, S. Complete nitrification: Insights into the ecophysiology of comammox Nitrospira. Appl. Microbiol. Biotechnol. 2019, 103, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Straub, K.L.; Schönhuber, W.A.; Buchholz-Cleven, B.E.E.; Schink, B. Diversity of Ferrous Iron-Oxidizing, Nitrate-Reducing Bacteria and their Involvement in Oxygen-Independent Iron Cycling. Geomicrobiol. J. 2004, 21, 371–378. [Google Scholar] [CrossRef]
- Ivanova, N.; Daum, C.; Lang, E.; Abt, B.; Kopitz, M.; Saunders, E.; Lapidus, A.; Lucas, S.; Del Rio, T.G.; Nolan, M.; et al. Complete genome sequence of Haliangium ochraceum type strain (SMP-2T). Stand. Genom. Sci. 2010, 2, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Mielczarek, A.T.; Nguyen, H.T.T.; Nielsen, J.L.; Nielsen, P.H. Population dynamics of bacteria involved in enhanced biological phosphorus removal in Danish wastewater treatment plants. Water Res. 2013, 47, 1529–1544. [Google Scholar] [CrossRef]
- Sasaki, K.; Watanabe, M.; Suda, Y.; Ishizuka, A.; Noparatnaraporn, N. Applications of photosynthetic bacteria for medical fields. J. Biosci. Bioeng. 2005, 100, 481–488. [Google Scholar] [CrossRef]
- Chen, H.; Wang, M.; Chang, S. Disentangling Community Structure of Ecological System in Activated Sludge: Core Communities, Functionality, and Functional Redundancy. Microb. Ecol. 2020, 80, 296–308. [Google Scholar] [CrossRef]
- Wen, Q.; Yang, L.; Zhao, Y.; Huang, L.; Chen, Z. Insight into effects of antibiotics on reactor performance and evolutions of antibiotic resistance genes and microbial community in a membrane reactor. Chemosphere 2018, 197, 420–429. [Google Scholar] [CrossRef]
- Rouquette, C.; Harmon, J.B.; Shafer, W.M. Induction of the mtrCDE-encoded ef¯ux pump system of Neisseria gonorrhoeae requires MtrA, an AraC-like protein. Mol. Microbiol. 1999, 33, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Otarigho, B.; O Falade, M. Open access Analysis of antibiotics resistant genes in different strains of Staphylococcus aureus. Bioinformation 2018, 14, 113–122. [Google Scholar] [CrossRef]
- Collivignarelli, M.C.; Baldi, M.; Abbà, A.; Caccamo, F.M.; Miino, M.C.; Rada, E.C.; Torretta, V. Foams in Wastewater Treatment Plants: From Causes to Control Methods. Appl. Sci. 2020, 10, 2716. [Google Scholar] [CrossRef]
- Liu, M.; Gill, J.J.; Young, R.; Summer, E.J. Bacteriophages of wastewater foaming-associated filamentous Gordonia reduce host levels in raw activated sludge. Sci. Rep. 2015, 5, 13754. [Google Scholar] [CrossRef] [PubMed]
- Petrovski, S.; Seviour, R.J.; Tillett, D. Prevention of Gordonia and Nocardia Stabilized Foam Formation by Using Bacteriophage GTE7. Appl. Environ. Microbiol. 2011, 77, 7864–7867. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Ge, Y.; Yuan, S.; Yu, Y.; Shi, Z.; Zhou, S.; Deng, J. Enhanced dewaterability of waste activated sludge by a combined use of permanganate and peroxymonosulfate. RSC Adv. 2019, 9, 27593–27601. [Google Scholar] [CrossRef]
- Sheng, G.-P.; Yu, H.-Q.; Li, X.-Y. Extracellular polymeric substances (EPS) of microbial aggregates in biological wastewater treatment systems: A review. Biotechnol. Adv. 2010, 28, 882–894. [Google Scholar] [CrossRef]
- Choi, J.; Kotay, S.M.; Goel, R. Various physico-chemical stress factors cause prophage induction in Nitrosospira multiformis 25196- an ammonia oxidizing bacteria. Water Res. 2010, 44, 4550–4558. [Google Scholar] [CrossRef]
- Liao, Y.; Zhang, J.; Wang, M.; Wu, Y.; Wang, S.; Pan, Y.; Cao, G. Nitrogen removal from wastewater for heterotrophic nitrification-aerobic denitrification bacterium with the combination of bacteriophage DEY7 and Fe nanoparticles. Biochem. Eng. J. 2023, 191, 108805. [Google Scholar] [CrossRef]
- Liu, R.; Qi, R.; Wang, J.; Zhang, Y.; Liu, X.; Rossetti, S.; Tandoi, V.; Yang, M. Phage-host associations in a full-scale activated sludge plant during sludge bulking. Appl. Microbiol. Biotechnol. 2017, 101, 6495–6504. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).