
Chemoselective Synthesis of Mannich Adducts from 1,4-Naphthoquinones and Profile as Autophagic Inducers in Oral Squamous Cell Carcinoma

 , , , , ,
, , , , ,  ,
,  ,
,  and
and
Abstract
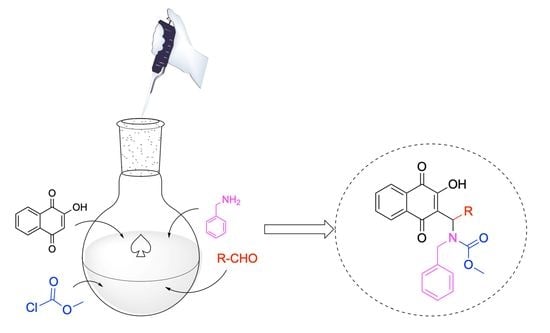
1. Introduction
2. Experimental Section
2.1. Chemistry
2.1.1. Materials and Methods
2.1.2. General Procedures for Synthesis of Naphthoquinones 5
2.1.3. General Procedures for Synthesis of Naphthoquinones 6
2.2. Biological Assays
2.3. In Silico Studies
3. Results and Discussion
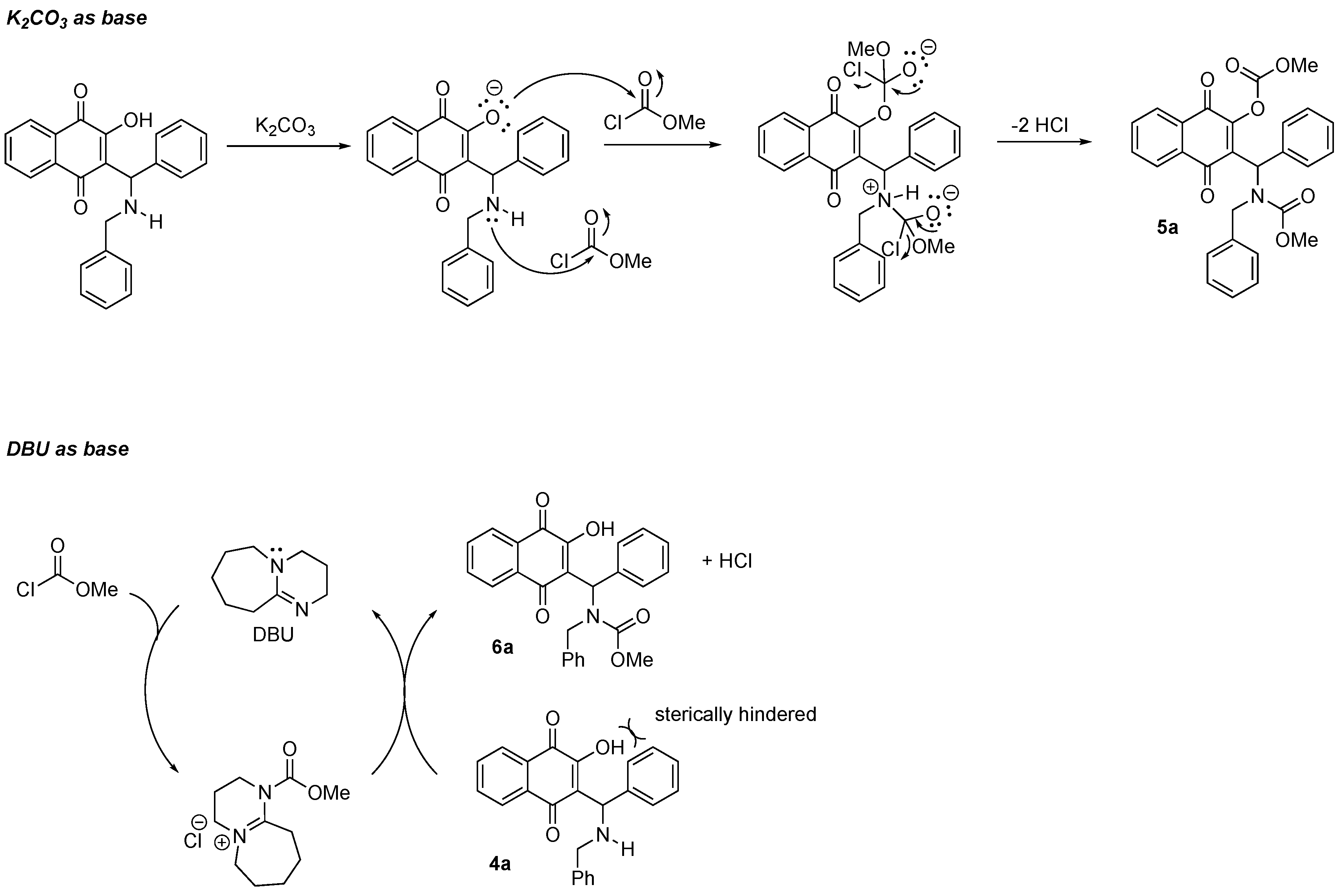
3.1. Chemistry
3.2. Biological Activity
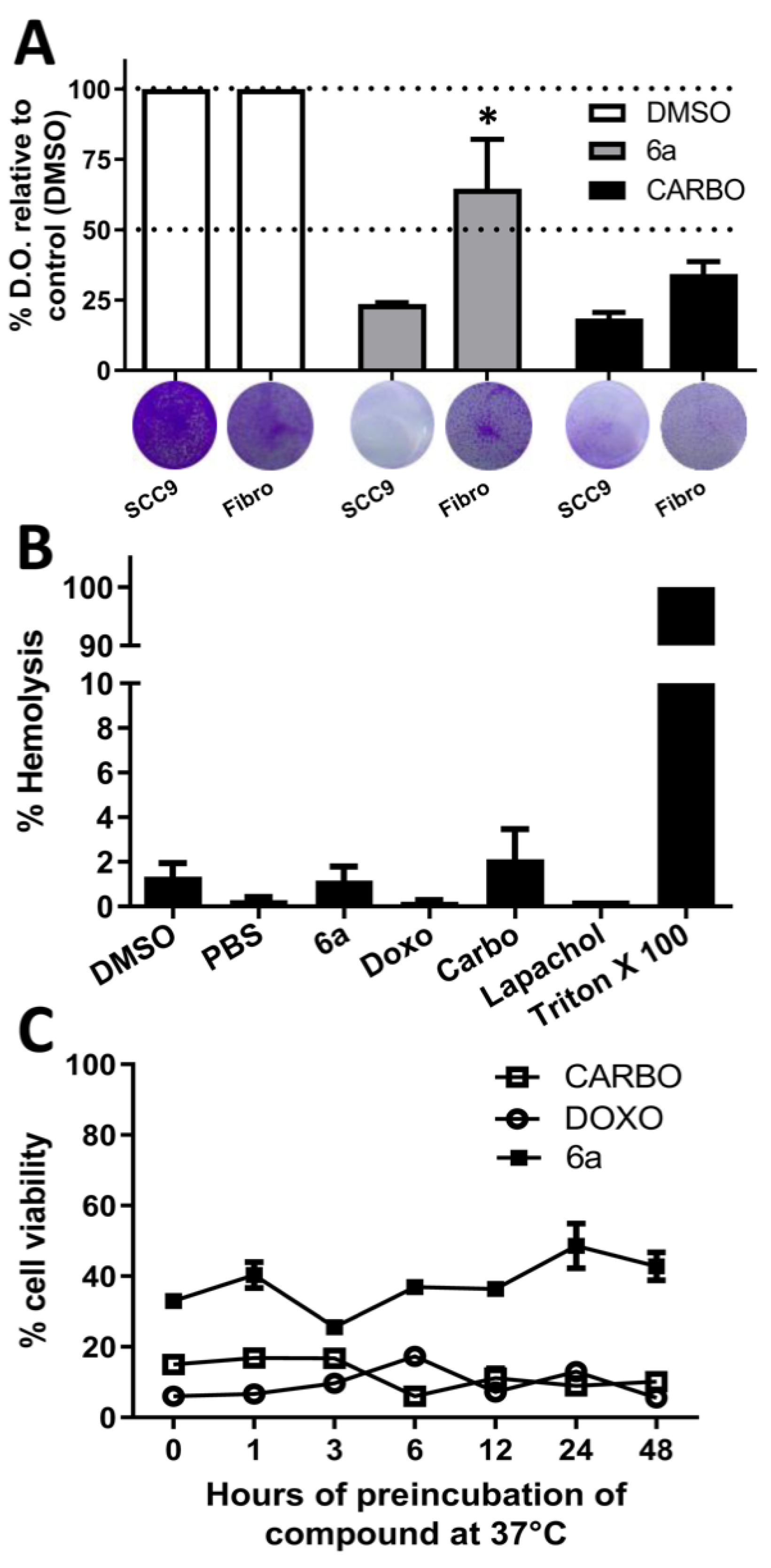
3.2.1. Cytotoxicity and Selectivity, Hemolytic Potential, and Stability of the New Compounds
3.2.2. Acute Toxicity In Vivo
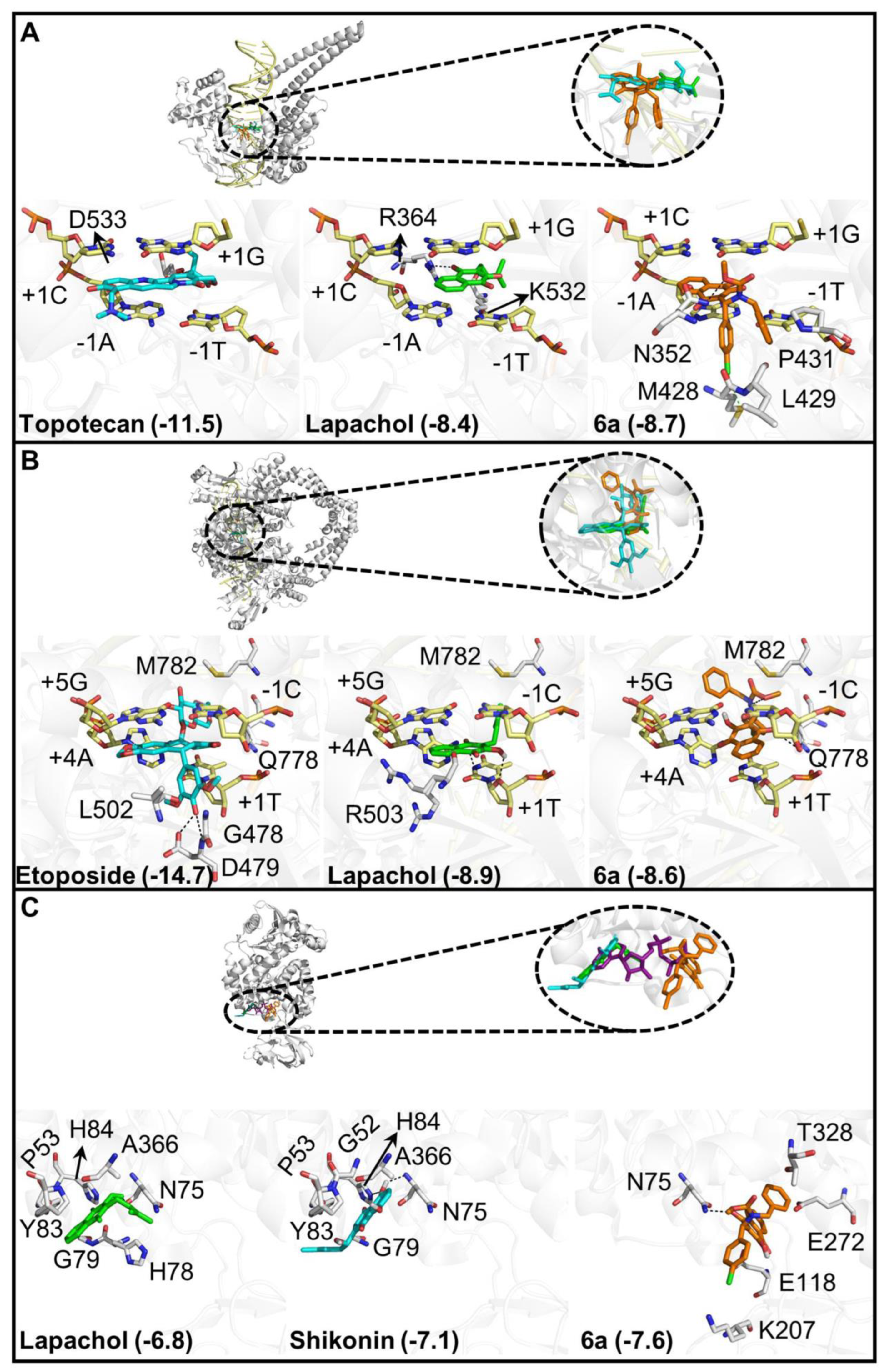
3.2.3. Molecular Docking of Compound 6a
3.2.4. Predicted Toxicity and Pharmacokinetic Properties
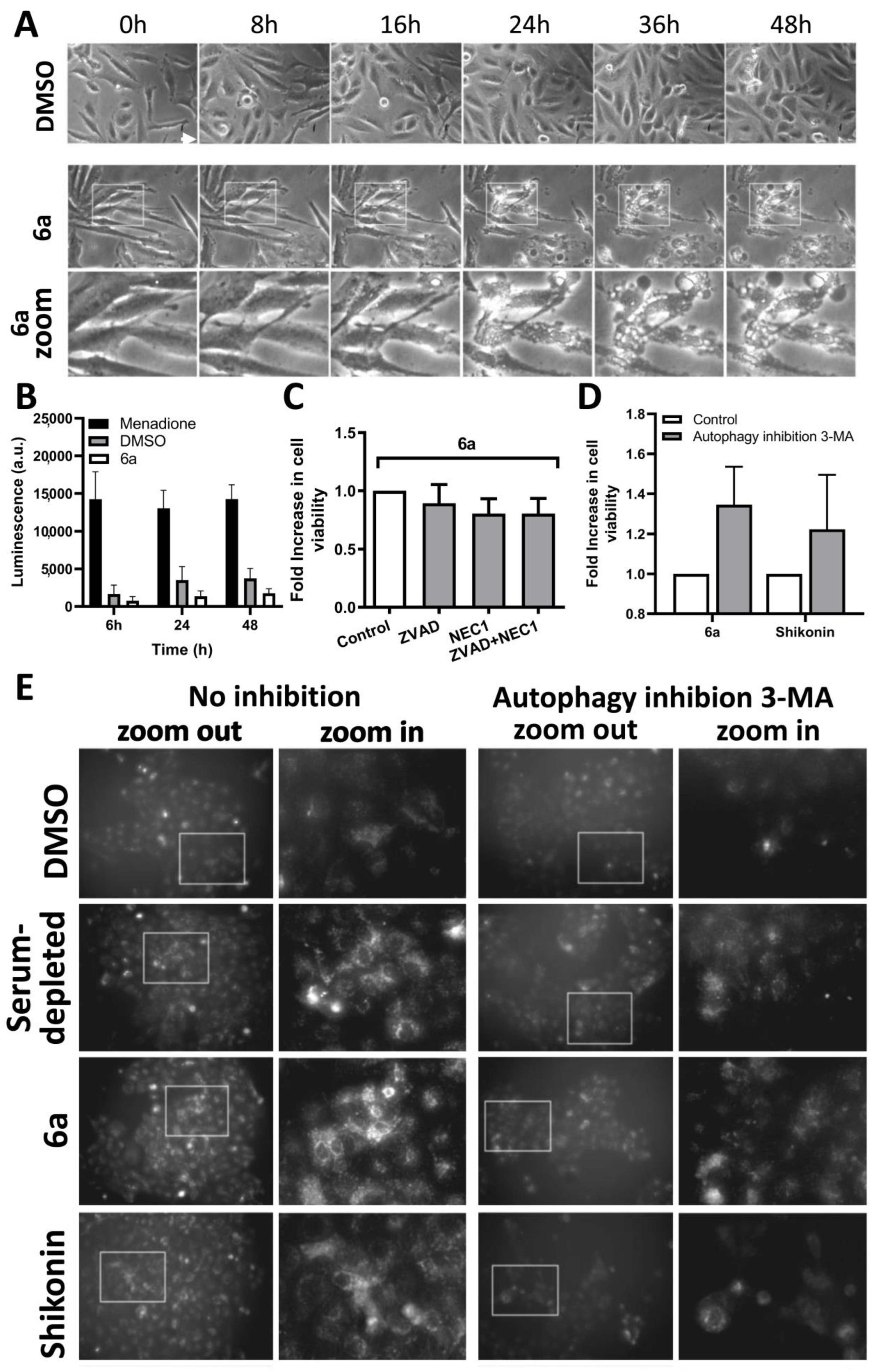
3.2.5. Investigation of the Cell Death Pathway
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Legal Aspects
References
- Chi, A.C.; Day, T.A.; Neville, B.W. Oral cavity and oropharyngeal squamous cell carcinoma-an update. CA Cancer J. Clin. 2015, 65, 401–421. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Speight, P.M.; Farthing, P.M. The pathology of oral cancer. Br. Dent. J. 2018, 225, 841–847. [Google Scholar] [CrossRef]
- Chai, A.W.Y.; Lim, K.P.; Cheong, S.C. Translational genomics and recent advances in oral squamous cell carcinoma. Semin. Cancer Biol. 2020, 61, 71–83. [Google Scholar] [CrossRef]
- Li, C.C.; Shen, Z.; Bavarian, R.; Yang, F.; Bhattacharya, A. Oral Cancer: Genetics and the Role of Precision Medicine. Surg. Oncol. Clin. N. Am. 2020, 29, 127–144. [Google Scholar] [CrossRef]
- Güneri, P.; Epstein, J.B. Late stage diagnosis of oral cancer: Components and possible solutions. Oral Oncol. 2014, 50, 1131–1136. [Google Scholar] [CrossRef]
- Aminin, D.; Polonik, S. 1,4-Naphthoquinones: Some biological properties and application. Chem. Pharm. Bull. 2020, 68, 46–57. [Google Scholar] [CrossRef]
- Pereyra, C.E.; Dantas, R.F.; Ferreira, S.B.; Gomes, L.P.; Silva, F.P., Jr. The diverse mechanisms and anticancer potential of naphthoquinones. Cancer Cell Int. 2019, 19, 207. [Google Scholar] [CrossRef]
- Biersack, B.; Ahmed, K.; Padhye, S.; Schobert, R. Recent developments concerning the application of the Mannich reaction for drug design. Expert Opin. Drug Discov. 2018, 13, 39–49. [Google Scholar] [CrossRef]
- Roman, G. Mannich bases in medicinal chemistry and drug design. Eur. J. Med. Chem. 2015, 89, 743–816. [Google Scholar]
- Machado, T.Q.; Felisberto, J.R.S.; Guimarães, E.F.; de Queiroz, G.A.; da Fonseca, A.C.C.; Ramos, Y.J.; Marques, A.M.; Moreira, D.L.; Robbs, B.K. Apoptotic effect of β-pinene on oral squamous cell carcinoma as one of the major compounds from essential oil of medicinal plant Piper rivinoides Kunth. Nat. Prod. Res. 2021, 36, 1636–1640. [Google Scholar] [CrossRef] [PubMed]
- Macedo, A.L.; da Silva, D.P.D.; Moreira, D.L.; de Queiroz, L.N.; Vasconcelos, T.R.A.; Araujo, G.F.; Kaplan, M.A.C.; Pereira, S.S.C.; de Almeida, E.C.P.; Valverde, A.L.; et al. Cytotoxicity and selectiveness of Brazilian Piper species towards oral carcinoma cells. Biomed. Pharmacother. 2019, 110, 342–352. [Google Scholar] [CrossRef]
- Parasuraman, S. Toxicological screening. J. Pharmacol. Pharmacother. 2011, 2, 74–79. [Google Scholar] [PubMed]
- Lucena, P.I.; Faget, D.V.; Pachulec, E.; Robaina, M.C.; Klumb, C.E.; Robbs, B.K.; Viola, J.P.B. NFAT2 Isoforms Differentially Regulate Gene Expression, Cell Death, and Transformation through Alternative N-Terminal Domains. Mol. Cell. Biol. 2016, 36, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Faget, D.V.; Lucena, P.I.; Robbs, B.K.; Viola, J.P.B. NFAT1 C-Terminal Domains Are Necessary but Not Sufficient for Inducing Cell Death. PLoS ONE 2012, 7, e47868. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef]
- Costa, D.C.S.; de Almeida, G.S.; Rabelo, V.W.-H.; Cabral, L.M.; Sathler, P.C.; Abreu, P.A.; Ferreira, V.F.; da Silva, L.C.R.P.; da Silva, F.C. Synthesis and evaluation of the cytotoxic activity of Furanaphthoquinones tethered to 1H-1,2,3-triazoles in Caco-2, Calu-3, MDA-MB231 cells. Eur. J. Med. Chem. 2018, 156, 524–533. [Google Scholar] [CrossRef]
- Zorzanelli, B.C.; Ouverney, G.; Pauli, F.P.; da Fonseca, A.C.C.; de Almeida, E.C.P.; de Carvalho, D.G.; Possik, P.A.; Rabelo, V.W.-H.; Abreu, P.A.; Pontes, B.; et al. Pro-Apoptotic Antitumoral Effect of Novel Acridine-Core Naphthoquinone Compounds against Oral Squamous Cell Carcinoma. Molecules 2022, 27, 5148. [Google Scholar] [CrossRef]
- Neves, A.P.; Barbosa, C.C.; Greco, S.J.; Vargas, M.D.; Visentin, L.C.; Pinheiro, C.B.; Mangrich, A.S.; Barbosa, J.P.; da Costa, G.L. Novel Aminonaphthoquinone Mannich Bases Derived from Lawsone and their Copper(II) Complexes: Synthesis, Characterization and Antibacterial Activity. J. Braz. Chem. Soc. 2009, 20, 712–727. [Google Scholar] [CrossRef]
- da Rocha, D.R.; de Souza, A.C.; Resende, J.A.; Santos, W.C.; dos Santos, E.A.; Pessoa, C.; de Moraes, M.O.; Costa-Lotufo, L.V.; Montenegro, R.C.; Ferreira, V.F. Synthesis of new 9-hydroxy-α- and 7-hydroxy-β-pyran naphthoquinones and cytotoxicity against cancer cell lines. Org. Biomol. Chem. 2011, 9, 4315–4322. [Google Scholar] [CrossRef]
- Ribeiro, R.C.B.; de Freitas, P.P.; Moreira, C.S.; de Moraes, L.G.C.; de Moraes, M.G.; da Silva, F.C.; Rocha, D.R.; Gimba, E.R.P.; Ferreira, V.F. A New Strategy for the Synthesis of Nonsymmetrical 3,3′-(Aryl/alkyl- methylene) bis-2-hydroxy-1,4-naphthoquinones and Their Cytotoxic Effects in PC3 Prostate Cancer Cells. J. Braz. Chem. Soc. 2020, 31, 288–297. [Google Scholar] [CrossRef]
- Hartner, L. Chemotherapy for Oral Cancer. Dent. Clin. N. Am. 2018, 62, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Ho, G.Y.; Woodward, N.; Coward, J.I.G. Cisplatin versus carboplatin: Comparative review of therapeutic management in solid malignancies. Crit. Rev. Oncol. Hematol. 2016, 102, 37–46. [Google Scholar] [CrossRef]
- Khasraw, M.; Bell, R.; Dang, C. Epirubicin: Is it like doxorubicin in breast cancer? A clinical review. Breast 2012, 21, 142–149. [Google Scholar] [CrossRef]
- Shafei, A.; El-Bakly, W.; Sobhy, A.; Wagdy, O.; Reda, A.; Aboelenin, O.; Marzouk, A.; El Habak, K.; Mostafa, R.; Ali, M.A.; et al. A review on the efficacy and toxicity of different doxorubicin nanoparticles for targeted therapy in metastatic breast cancer. Biomed. Pharmacother. 2017, 95, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Boulos, J.C.; Rahama, M.; Hegazy, M.-E.F.; Efferth, T. Shikonin derivatives for cancer prevention and therapy. Cancer Lett. 2019, 459, 248–267. [Google Scholar] [CrossRef] [PubMed]
- Epifano, F.; Genovese, S.; Fiorito, S.; Mathieu, V.; Kiss, R. Lapachol and its congeners as anticancer agents: A review. Phytochem. Rev. 2014, 13, 37–49. [Google Scholar] [CrossRef]
- Chipoline, I.C.; da Fonseca, A.C.C.; da Costa, G.R.M.; de Souza, M.P.; Rabelo, V.W.-H.; de Queiroz, L.N.; de Souza, T.L.F.; de Almeida, E.C.P.; Abreu, P.A.; Pontes, B.; et al. Molecular mechanism of action of new 1,4-naphthoquinones tethered to 1,2,3-1H-triazoles with cytotoxic and selective effect against oral squamous cell carcinoma. Bioorg. Chem. 2020, 101, 103984. [Google Scholar] [CrossRef]
- Zorzanelli, B.C.; de Queiroz, L.N.; Santos, R.M.; Menezes, L.M.; Gomes, F.C.; Ferreira, V.F.; da Silva, F.C.; Robbs, B.K. Potential cytotoxic and selective effect of new benzo [b] xanthenes against oral squamous cell carcinoma. Fut. Med. Chem. 2018, 10, 1141–1157. [Google Scholar] [CrossRef]
- Militello, C.; Rundo, L.; Minafra, L.; Cammarata, F.P.; Calvaruso, M.; Conti, V.; Russo, G. MF2C3: Multi-Feature Fuzzy Clustering to Enhance Cell Colony Detection in Automated Clonogenic Assay Evaluation. Symmetry 2020, 12, 773. [Google Scholar] [CrossRef]
- Pereira, V.S.S.; de Oliveira, C.B.S.; Fumagalli, F.; Emery, F.S.; da Silva, N.B.; de Andrade-Neto, V.F. Cytotoxicity, hemolysis and in vivo acute toxicity of 2-hydroxy-3-anilino-1,4-naphthoquinone derivatives. Toxicol. Rep. 2016, 3, 756–762. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M. Acute Toxicity (Lethal Dose 50 Calculation) of Herbal Drug Somina in Rats and Mice. Pharmacol. Pharm. 2015, 6, 185–189. [Google Scholar] [CrossRef]
- Liu, W.; Chen, X.; Ye, Q.; Hou, S.; Lou, L.; Xie, C. 3-Hydroxycarboplatin, a simple carboplatin derivative endowed with an improved toxicological profile. Platin. Met. Rev. 2012, 56, 248–256. [Google Scholar] [CrossRef]
- Boonyalai, N.; Sittikul, P.; Pradidphol, N.; Kongkathip, N. Biophysical and molecular docking studies of naphthoquinone derivatives on the ATPase domain of human Topoisomerase II. Biomed. Pharmacother. 2013, 67, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Delgado, J.L.; Hsieh, C.-M.; Chan, N.-L.; Hiasa, H. Topoisomerases as Anticancer Targets. Biochem. J. 2017, 176, 139–148. [Google Scholar] [CrossRef] [PubMed]
- da Silva, M.N.; Ferreira, V.F.; de Souza, M.C.B.V. Um panorama atual da química e da farmacologia de naftoquinonas, com ênfase na beta-lapachona e derivados. Quím. Nova 2003, 26, 407–416. [Google Scholar] [CrossRef]
- Steves-Souza, A.; Figueiredo, D.V.; Esteves, A.; Câmara, C.A.; Vargas, M.D.; Pinto, A.C.; Echevarria, A. Cytotoxic and DNA-topoisomerase effects of lapachol amine derivatives and interactions with DNA. Braz. J. Med. Biol. Res. 2007, 40, 1399–1402. [Google Scholar] [CrossRef]
- Ferreira, S.B.; Gonzaga, D.T.G.; Santos, W.C.; Araújo, K.G.L.; Ferreira, V.F. ß-Lapachone: Medicinal chemistry significance and structural modifications. Rev. Virtual Quím. 2010, 2, 140–160. [Google Scholar] [CrossRef]
- Gurbani, D.; Kukshal, V.; Laubenthal, J.; Kumar, A.; Pandey, A.; Tripathi, S.; Arora, A.; Jain, S.K.; Ramachandran, R.; Anderson, D.; et al. Mechanism of inhibition of the ATpase domain of human topoisomerase IIα by 1,4-benzoquinone, 1,2-naphthoquinone, 1,4-naphthoquinone, and 9,10-phenanthroquinone. Toxicol. Sci. 2012, 126, 372–390. [Google Scholar] [CrossRef]
- Wellington, K.W. Understanding cancer and the anticancer activities of naphthoquinones-a review. RSC Adv. 2015, 5, 20309–20338. [Google Scholar] [CrossRef]
- Chhipa, A.S.; Patel, S. Targeting pyruvate kinase muscle isoform 2 (PKM2) in cancer: What do we know so far? Life Sci. 2021, 280, 119694. [Google Scholar] [CrossRef] [PubMed]
- Babu, M.S.; Mahanta, S.; Lakhter, A.J.; Hato, T.; Paul, S.; Naidu, S.R. Lapachol inhibits glycolysis in cancer cells by targeting pyruvate kinase M2. PLoS ONE 2018, 13, e0191419. [Google Scholar]
- Chen, J.; Xie, J.; Jiang, Z.; Wang, B.; Wang, Y.; Hu, X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene 2011, 30, 4297–4306. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhu, Y.; Hu, J.; Jiang, L.; Li, L.; Jia, S.; Zen, K. Shikonin Inhibits Tumor Growth in Mice by Suppressing Pyruvate Kinase M2-mediated Aerobic Glycolysis. Sci. Rep. 2018, 8, 14517. [Google Scholar] [CrossRef] [PubMed]
- Larsen, T.M.; Benning, M.M.; Rayment, I.; Reed, G.H. Structure of the Bis(Mg2+)-ATP-oxalate complex of the rabbit muscle pyruvate kinase at 2.1 Å resolution: ATP binding over a barrel. Biochemistry 1998, 37, 6247–6255. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Cardoso, M.F.D.C.; Salomão, K.; Bombaça, A.C.; da Rocha, D.R.; da Silva, F.C.; Cavaleiro, J.A.S.; de Castro, S.L.; Ferreira, V.F. Synthesis and anti-Trypanosoma cruzi activity of new 3-phenylthio-nor-β-lapachone derivatives. Bioorg. Med. Chem. 2015, 23, 4763–4768. [Google Scholar] [CrossRef]
- Palm, K.; Stenberg, P.; Luthman, K.; Artursson, P. Polar Molecular Surface Properties Predict the Intestinal Absorption of Drugs in Humans. Pharm. Res. 1997, 14, 568–571. [Google Scholar] [CrossRef]
- Alrushaid, S.; Sayre, C.L.; Yáñez, J.A.; Forrest, M.L.; Senadheera, S.N.; Burczynski, F.J.; Löbenberg, R.; Davies, N.M. Pharmacokinetic and Toxicodynamic Characterization of a Novel Doxorubicin Derivative. Pharmaceutics 2017, 9, 35. [Google Scholar] [CrossRef]
- Oguri, S.; Sakakibara, T.; Mase, H.; Shimizu, T.; Ishikawa, K.; Kimura, K.; Smyth, R.D. Clinical Pharmacokinetics of Carboplatin. Clin. Pharmacol. 1988, 28, 2018–2215. [Google Scholar] [CrossRef]
- Huber, P.C.; Maruiama, C.H.; Almeida, W.P. Glicoproteína-P, resistência a múltiplas drogas (MDR) e relação estrutura-atividade de moduladores. Quím. Nova 2010, 33, 2148–2154. [Google Scholar] [CrossRef]
- Mansilla, S.; Llovera, L.; Portugal, J. Chemotherapeutic Targeting of Cell Death Pathways. Anticancer Agents Med. Chem. 2012, 12, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Lai, Y.; Hua, Z.C. Apoptosis and apoptotic body: Disease message and therapeutic target potentials. Biosci. Rep. 2019, 39, BSR20180992. [Google Scholar] [CrossRef]
- Jamier, V.; Ba, L.A.; Jacob, C. Selenium- and tellurium-containing multifunctional redox agents as biochemical redox modulators with selective cytotoxicity. Chem. Eur. J. 2010, 16, 10920–10928. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Wang, B.; Zheng, L.; Yang, K.; Li, Y.; Hu, M.; He, D. Target ROS to induce apoptosis and cell cycle arrest by 5,7-dimethoxy-1,4-naphthoquinone derivative. Bioorg. Med. Chem. Lett. 2018, 28, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Gong, K.; Zhang, Z.; Chen, Y.; Shu, H.-B.; Li, W. Extracellular signal-regulated kinase, receptor interacting protein, and reactive oxygen species regulate shikonin-induced autophagy in human hepatocellular carcinoma. Eur. J. Pharmacol. 2014, 738, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Cao, H. Shikonin promotes autophagy in BXPC-3 human pancreatic cancer cells through the PI3K/Akt signaling pathway. Oncol. Lett. 2014, 8, 1087–1089. [Google Scholar] [CrossRef]
- Lin, Y.; Chen, Y.; Wang, S.; Ma, J.; Peng, Y.; Yuan, X.; Lv, B.; Chen, W.; Wei, Y. Plumbagin induces autophagy and apoptosis of SMMC-7721 cells in vitro and in vivo. J. Cell. Biochem. 2019, 120, 9820–9830. [Google Scholar] [CrossRef]
- Ma, X.; Yin, X.; Liu, H.; Chen, Q.; Feng, Y.; Ma, X.; Liu, W. Antiproliferative activity of plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone) in human gastric carcinoma cells is facilitated via activation of autophagic pathway, mitochondrialmediated programmed cell death and inhibition of cell migration and invasion. J. BUON 2019, 24, 2000–2005. [Google Scholar]
- Zheng, Q.; Li, Q.; Zhao, G.; Zhang, J.; Yuan, H.; Gong, D.; Guo, Y.; Liu, X.; Li, K.; Lin, P. Alkannin induces cytotoxic autophagy and apoptosis by promoting ROS-mediated mitochondrial dysfunction and activation of JNK pathway. Biochem. Pharmacol. 2020, 180, 114167. [Google Scholar] [CrossRef]
- Li, J.; Pang, J.; Liu, Z.; Ge, X.; Zhen, Y.; Jiang, C.C.; Liu, Y.; Huo, Q.; Sun, Y.; Liu, H. Shikonin induces programmed death of fibroblast synovial cells in rheumatoid arthritis by inhibiting energy pathways. Sci. Rep. 2021, 11, 18263. [Google Scholar] [CrossRef] [PubMed]
- Azoitei, N.; Becher, A.; Steinestel, K.; Rouhi, A.; Diepold, K.; Genze, F.; Simmet, T.; Seufferlein, T. PKM2 promotes tumor angiogenesis by regulating HIF-1aα through NF-κB activation. Mol. Cancer 2016, 15, 3. [Google Scholar] [CrossRef] [PubMed]
- Kurihara-Shimomura, M.; Sasahira, T.; Nakashima, C.; Kuniyasu, H.; Shimomura, H.; Kirita, T. The multifarious functions of pyruvate kinase M2 in oral cancer cells. Int. J. Mol. Sci. 2018, 19, 2907. [Google Scholar] [CrossRef] [PubMed]
- Kurihara-Shimomura, M.; Sasahira, T.; Shimomura, H.; Kirita, T. Peroxidan plays a tumor-promoting role in oral squamous cell carcinoma. Int. J. Mol. Sci. 2020, 21, 5416. [Google Scholar] [CrossRef]
- Tanaka, F.; Yoshimoto, S.; Okamura, K.; Ikebe, T.; Hashimoto, S. Nuclear PKM2 promotes the progression of oral squamous cell carcinoma by inducing EMT and post-translationally repressing TGIF2. Oncotarget 2018, 9, 33745–33761. [Google Scholar] [CrossRef]
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef]
- Ryter, S.W.; Mizumura, K.; Choi, A.M.K. The impact of autophagy on cell death modalities. Int. J. Cell Biol. 2014, 2014, 502676. [Google Scholar] [CrossRef]
- Dey, P.; Kundu, A.; Sachan, R.; Park, J.H.; Ahn, M.Y.; Yoon, K.; Lee, J.; Kim, N.D.; Kim, I.S.; Lee, B.M.; et al. PKM2 knockdown induces autophagic cell death via AKT/mTOR pathway in human prostate cancer cells. Cell. Physiol. Biochem. 2019, 52, 1535–1552. [Google Scholar]
- Pa, S.T.; Qin, Y.; Zhou, Z.-W.; He, Z.-X.; Zhang, X.; Yang, T.; Yang, Y.-X.; Wang, D.; Qiu, J.-X.; Zhou, S.-F. Plumbagin induces G2/M arrest, apoptosis, and autophagy via p38 MAPK- and PI3K/Akt/mTOR-mediated pathways in human tongue squamous cell carcinoma cells. Drug Des. Devel. Ther. 2015, 9, 1601–1626. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | SCC9—Oral Cancer | Primary Gengival Fibroblast | Selective Index (S.I.) | ||
|---|---|---|---|---|---|
| IC50 (µM) | SD | IC50 (µM) | SD | ||
| 5a | 23.2 | 0.05 | 37.5 | 0.09 | 1.61 |
| 5b | 28.5 | 0.04 | 55.7 | 0.04 | 1.95 |
| 5c | 24.8 | 0.07 | 38.3 | 0.09 | 1.54 |
| 5d | 38.8 | 0.02 | 69.1 | 0.01 | 1.78 |
| 5e | 22.1 | 0.07 | 40.1 | 0.05 | 1.81 |
| 5f | 22.2 | 0.06 | 45.7 | 0.07 | 2.05 |
| 5g | 21.0 | 0.08 | 35.5 | 0.05 | 1.68 |
| 5h | 24.9 | 0.12 | 49.1 | 0.05 | 1.97 |
| 5i | 23.9 | 0.05 | 42.6 | 0.09 | 1.77 |
| 6a | 56.2 | 0.07 | 186.6 | 0.07 | 3.31 |
| 6b | 82.2 | 0.02 | 229.8 | 0.06 | 2.79 |
| 6c | 56.1 | 0.09 | 148.1 | 0.05 | 2.63 |
| 6d | 73.0 | 0.03 | 155.1 | 0.04 | 2.12 |
| 6e | 73.2 | 0.06 | 196.7 | 0.06 | 2.68 |
| 6h | 11.1 | 0.17 | 53.7 | 0.08 | 4.83 |
| 6i | 70.7 | 0.14 | 73.5 | 0.14 | 1.03 |
| Doxorrubicin | 7.2 | 0.04 | 18.68 | 0.40 | 2.57 |
| Carboplatin | 223.0 | 0.04 | 192.8 | 0.03 | 0.85 |
| Lapachol | 42.1 | 0.08 | 101.6 | 0.09 | 2.41 |
| Shikonin | 1.8 | 0.02 | 1.72 | 0.05 | 0.95 |
| Oral Tumor Cells | Primary Gingival Fibroblast | Average S.I. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | SCC9 | SCC25 | SCC4 | Mean | ||||||
| IC50 | S.D. | IC50 | S.D. | IC50 | S.D. | IC50 | S.D. | |||
| 6a | 56.24 | 0.07 | 87.28 | 0.04 | 92.81 | 0.06 | 78.70 | 186.60 | 0.07 | 2.36 |
| 6h | 11.10 | 0.17 | 56.60 | 0.05 | 98.39 | 0.03 | 44.58 | 53.73 | 0.08 | 1.29 |
| Carboplatin | 223.00 | 0.04 | 264.00 | 0.02 | 195.00 | 0.06 | 227.50 | 192.80 | 0.03 | 0.85 |
| Doxorubicin | 7.20 | 0.04 | 1.12 | 0.06 | 5.87 | 0.03 | 4.73 | 18.68 | 0.40 | 3.94 |
| Lapachol | 42.12 | 0.08 | 85.85 | 0.07 | 55.32 | 0.05 | 61.09 | 101.60 | 0.09 | 1.66 |
| Shikonin | 1.81 | 0.02 | 1.50 | 0.05 | 1.91 | 0.05 | 1.74 | 1.72 | 0.05 | 0.98 |
| Treatment | Dose mg/kg a | Change in Body Weight | Change in Food Consumption | Morbidity b | Mortality | Gross Necropsy c | Histology d |
|---|---|---|---|---|---|---|---|
| Control | 0 n = 3 | Absent | Absent | Absent | Absent | No alteration | Normal |
| 6a | 100 n = 3 | Absent | Absent | Absent | Absent | No alteration | Mild kidney and portal hyperemia and mild liver intracytoplasmic degeneration |
| 150 n = 4 | Absent | ND e | Low activity and back-arching | 50% of death | Precipitaded compound in loco | Mild arterial and venous hyperemia at the lung, kidney and liver portal and liver intracytoplasmic degeneration | |
| 200 n = 3 | ND e | ND e | Low activity and back-arching | 100% of death | Precipitaded compound in loco | Mild arterial and venous hyperemia at thelung, kidney and liver portal and liver intracytoplasmic degeneration at time of death |
| Compounds | cLogP | nON | nOH/NH | MW | Lipinski’s Violations a | TPSA (Å2) |
|---|---|---|---|---|---|---|
| 6a | 2.54 | 5 | 1 | 461 | 0 | 83.91 |
| Doxorubicin | −2.10 | 12 | 6 | 543 | 3 | 206.1 |
| Carboplatin | −1.79 | 6 | 4 | 371 | 0 | 126.6 |
| ADMET | 6a | Carboplatin | Doxorrubicin |
|---|---|---|---|
| Oral Bioavailability | +0.63 | −0.60 | −0.91 |
| P-glycoprotein inhibitor | +0.82 | −0.99 | −0.92 |
| P-glycoprotein substrate | −0.56 | −0.99 | +0.95 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borges, A.A.; de Souza, M.P.; da Fonseca, A.C.C.; Wermelinger, G.F.; Ribeiro, R.C.B.; Amaral, A.A.P.; de Carvalho, C.J.C.; Abreu, L.S.; de Queiroz, L.N.; de Almeida, E.C.P.; et al. Chemoselective Synthesis of Mannich Adducts from 1,4-Naphthoquinones and Profile as Autophagic Inducers in Oral Squamous Cell Carcinoma. Molecules 2023, 28, 309. https://doi.org/10.3390/molecules28010309
Borges AA, de Souza MP, da Fonseca ACC, Wermelinger GF, Ribeiro RCB, Amaral AAP, de Carvalho CJC, Abreu LS, de Queiroz LN, de Almeida ECP, et al. Chemoselective Synthesis of Mannich Adducts from 1,4-Naphthoquinones and Profile as Autophagic Inducers in Oral Squamous Cell Carcinoma. Molecules. 2023; 28(1):309. https://doi.org/10.3390/molecules28010309
Chicago/Turabian StyleBorges, Amanda A., Michele P. de Souza, Anna Carolina C. da Fonseca, Guilherme F. Wermelinger, Ruan C. B. Ribeiro, Adriane A. P. Amaral, Cláudio José C. de Carvalho, Lucas S. Abreu, Lucas Nicolau de Queiroz, Elan C. P. de Almeida, and et al. 2023. "Chemoselective Synthesis of Mannich Adducts from 1,4-Naphthoquinones and Profile as Autophagic Inducers in Oral Squamous Cell Carcinoma" Molecules 28, no. 1: 309. https://doi.org/10.3390/molecules28010309
APA StyleBorges, A. A., de Souza, M. P., da Fonseca, A. C. C., Wermelinger, G. F., Ribeiro, R. C. B., Amaral, A. A. P., de Carvalho, C. J. C., Abreu, L. S., de Queiroz, L. N., de Almeida, E. C. P., Rabelo, V. W., Abreu, P. A., Pontes, B., Ferreira, V. F., da Silva, F. d. C., Forezi, L. d. S. M., & Robbs, B. K. (2023). Chemoselective Synthesis of Mannich Adducts from 1,4-Naphthoquinones and Profile as Autophagic Inducers in Oral Squamous Cell Carcinoma. Molecules, 28(1), 309. https://doi.org/10.3390/molecules28010309