
The Role of Immune Checkpoint Inhibitors in Cancer Therapy


Abstract
1. Introduction
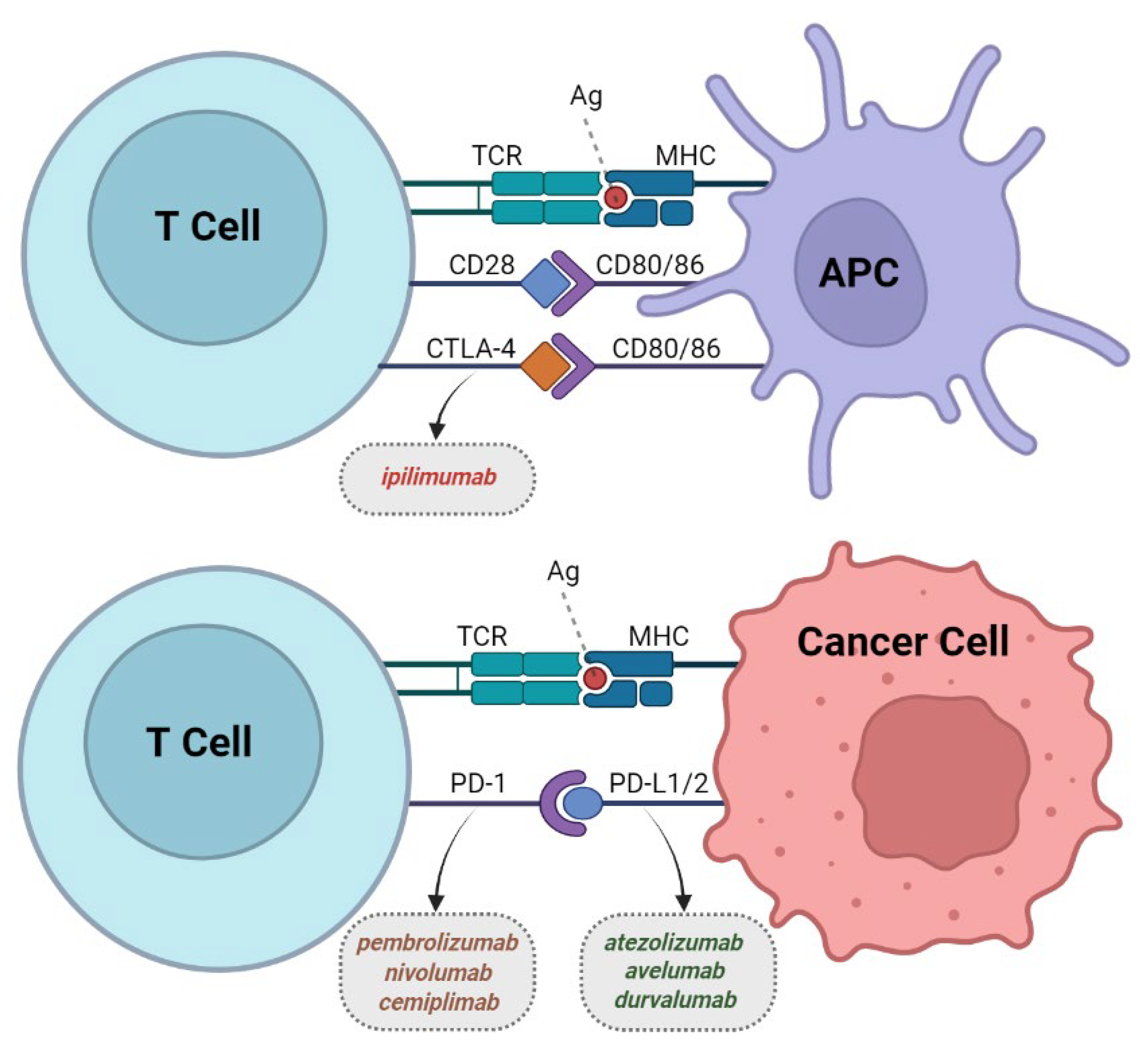
2. Immune Checkpoint Inhibitors (CPIs)
2.1. Development of CPIs
2.2. CTLA-4 Inhibitors
2.3. PD-1 Inhibitors
2.4. PD-L1 Inhibitors
3. Predictive Biomarkers for Clinical Response to CPIs
3.1. PD-L1 Expression
3.2. Tumor Mutational Burden (TMB)
3.3. Defective DNA Mismatch Repair (dMMR) and Microsatellite Instability (MSI)
3.4. Tumor Microenvironment (TME)
3.5. Microbiota
4. Toxicity and Management of Adverse Effects
5. Novel Delivery Approaches
6. Conclusions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Burnet, M. Cancer—A biological approach. I. The processes of control. Br. Med. J. 1957, 1, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Burnet, F.M. The Concept of Immunological Surveillance. Prog. Exp. Tumor Res. 1970, 13, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L. On immunosurveillance in human cancer. Yale J. Biol. Med. 1982, 55, 329. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2596448/?report=abstract (accessed on 9 October 2022). [PubMed]
- Bretscher, P.; Cohn, M. A theory of self-nonself discrimination. Science 1970, 169, 1042–1049. [Google Scholar] [CrossRef]
- Jenkins, M.K.; Schwartz, R.H. Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo. J. Exp. Med. 1987, 165, 302–319. [Google Scholar] [CrossRef]
- Altmann, D.M. A Nobel Prize-worthy pursuit: Cancer immunology and harnessing immunity to tumour neoantigens. Immunology 2018, 155, 283–284. [Google Scholar] [CrossRef]
- Lee, J.B.; Ha, S.J.; Kim, H.R. Clinical Insights into Novel Immune Checkpoint Inhibitors. Front. Pharmacol. 2021, 12, 2074. [Google Scholar] [CrossRef]
- Giannini, E.G.; Aglitti, A.; Borzio, M.; Gambato, M.; Guarino, M.; Iavarone, M.; Lai, Q.; Sandri, G.B.L.; Melandro, F.; Morisco, F.; et al. Overview of Immune Checkpoint Inhibitors Therapy for Hepatocellular Carcinoma, and The ITA.LI.CA Cohort Derived Estimate of Amenability Rate to Immune Checkpoint Inhibitors in Clinical Practice. Cancers 2019, 11, 1689. [Google Scholar] [CrossRef]
- Pala, L.; Sala, I.; Oriecuia, C.; De Pas, T.; Queirolo, P.; Specchia, C.; Cocorocchio, E.; Ferrucci, P.; Patanè, D.; Saponara, M.; et al. Association of Anticancer Immune Checkpoint Inhibitors with Patient-Reported Outcomes Assessed in Randomized Clinical Trials: A Systematic Review and Meta-analysis. JAMA Netw. Open 2022, 5, e2226252. [Google Scholar] [CrossRef]
- Marin-Acevedo, J.A.; Kimbrough, E.M.O.; Lou, Y. Next generation of immune checkpoint inhibitors and beyond. J. Hematol. Oncol. 2021, 14, 45. [Google Scholar] [CrossRef]
- Esfahani, K.; Roudaia, L.; Buhlaiga, N.; Del Rincon, S.V.; Papneja, N.; Miller, W.H. A review of cancer immunotherapy: From the past, to the present, to the future. Curr. Oncol. 2020, 27, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Shiravand, Y.; Khodadadi, F.; Kashani, S.M.A.; Hosseini-Fard, S.R.; Hosseini, S.; Sadeghirad, H.; Ladwa, R.; O’byrne, K.; Kulasinghe, A. Immune Checkpoint Inhibitors in Cancer Therapy. Curr. Oncol. 2022, 29, 3044–3060. [Google Scholar] [CrossRef]
- Riley, R.S.; June, C.H.; Langer, R.; Mitchell, M.J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 2019, 18, 175–196. [Google Scholar] [CrossRef] [PubMed]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Webb, E.S.; Liu, P.; Baleeiro, R.; Lemoine, N.R.; Yuan, M.; Wang, Y. Immune checkpoint inhibitors in cancer therapy. J. Biomed. Res. 2018, 32, 317. [Google Scholar] [CrossRef]
- Granier, C.; De Guillebon, E.; Blanc, C.; Roussel, H.; Badoual, C.; Colin, E.; Saldmann, A.; Gey, A.; Oudard, S.; Tartour, E. Mechanisms of action and rationale for the use of checkpoint inhibitors in cancer. ESMO Open 2017, 2, e000213. [Google Scholar] [CrossRef] [PubMed]
- Saleh, R.; Toor, S.M.; Khalaf, S.; Elkord, E. Breast Cancer Cells and PD-1/PD-L1 Blockade Upregulate the Expression of PD-1, CTLA-4, TIM-3 and LAG-3 Immune Checkpoints in CD4+ T Cells. Vaccines 2019, 7, 149. [Google Scholar] [CrossRef]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef]
- Mohsenzadegan, M.; Bavandpour, P.; Nowroozi, M.R.; Amini, E.; Kourosh-Arami, M.; Momeni, S.A.; Bokaie, S.; Sharifi, L. The Potential of T Cell Immunoglobulin and Mucin-Domain Containing-3 (Tim-3) in Designing Novel Immunotherapy for Bladder Cancer. Endocr. Metab. Immune Disord. Drug Targets 2021, 21, 2131–2146. [Google Scholar] [CrossRef]
- Hoos, A.; Ibrahim, R.; Korman, A.; Abdallah, K.; Berman, D.; Shahabi, V.; Chin, K.; Canetta, R.; Humphrey, R. Development of Ipilimumab: Contribution to a New Paradigm for Cancer Immunotherapy. Semin. Oncol. 2010, 37, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [PubMed]
- Iwai, Y.; Terawaki, S.; Honjo, T. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int. Immunol. 2005, 17, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Littman, D.R. Releasing the Brakes on Cancer Immunotherapy. Cell 2015, 162, 1186–1190. [Google Scholar] [CrossRef] [PubMed]
- Naluai, Å.T.; Nilsson, S.; Samuelsson, L.; Gudjónsdóttir, A.H.; Ascher, H.; Ek, J.; Hallberg, B.; Kristiansson, B.; Martinsson, T.; Nerman, O.; et al. The CTLA4/CD28 gene region on chromosome 2q33 confers susceptibility to celiac disease in a way possibly distinct from that of type 1 diabetes and other chronic inflammatory disorders. Tissue Antigens. 2000, 56, 350–355. [Google Scholar] [CrossRef]
- Rudd, C.E.; Taylor, A.; Schneider, H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol. Rev. 2009, 229, 12. [Google Scholar] [CrossRef]
- Alegre, M.L.; Frauwirth, K.A.; Thompson, C.B. T-cell regulation by CD28 and CTLA-4. Nat. Rev. Immunol. 2001, 1, 220–228. [Google Scholar] [CrossRef]
- Fife, B.T.; Bluestone, J.A. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol. Rev. 2008, 224, 166–182. [Google Scholar] [CrossRef]
- Van Elsas, A.; Hurwitz, A.A.; Allison, J.P. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J. Exp. Med. 1999, 190, 355–366. [Google Scholar] [CrossRef]
- Kwon, E.D.; Hurwitz, A.A.; Foster, B.A.; Madias, C.; Feldhaus, A.L.; Greenberg, N.M.; Burg, M.B.; Allison, J.P. Manipulation of T cell costimulatory and inhibitory signals for immunotherapy of prostate cancer. Proc. Natl. Acad. Sci. USA 1997, 94, 8099. [Google Scholar] [CrossRef]
- Van Ginderachter, J.A.; Liu, Y.; Geldhof, A.B.; Brijs, L.; Thielemans, K.; De Baetselier, P.; Raes, G. B7-1, IFNγ and anti-CTLA-4 co-operate to prevent T-cell tolerization during immunotherapy against a murine T-lymphoma. J. Cancer 2000, 87, 539–547. [Google Scholar] [CrossRef]
- Saha, A.; Chatterjee, S.K. Combination of CTL-associated antigen-4 blockade and depletion of CD25 regulatory T cells enhance tumour immunity of dendritic cell-based vaccine in a mouse model of colon cancer. Scand. J. Immunol. 2010, 71, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Sutmuller, R.P.M.; Van Duivenvoorde, L.M.; Van Elsas, A.; Schumacher, T.N.M.; Wildenberg, M.E.; Allison, J.P.; Toes, R.E.M.; Offringa, R.; Melief, C.J.M. Synergism of cytotoxic T lymphocyte-associated antigen 4 blockade and depletion of CD25(+) regulatory T cells in antitumor therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte responses. J. Exp. Med. 2001, 194, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Boasberg, P.; Hamid, O.; O’Day, S. Ipilimumab: Unleashing the power of the immune system through CTLA-4 blockade. Semin. Oncol. 2010, 37, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.-F.; Testori, A.; Grob, J.-J.; et al. Wolchok, Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Riella, L.V.; Paterson, A.M.; Sharpe, A.H.; Chandraker, A. Role of the PD-1 pathway in the immune response. Am. J. Transplant. 2012, 12, 2575–2587. [Google Scholar] [CrossRef]
- Davis, A.A.; Patel, V.G. The role of PD-L1 expression as a predictive biomarker: An analysis of all US Food and Drug Administration (FDA) approvals of immune checkpoint inhibitors. J. Immunother. Cancer 2019, 7, 278. [Google Scholar] [CrossRef]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef]
- Amarnath, S.; Mangus, C.W.; Wang, J.C.M.; Wei, F.; He, A.; Kapoor, V.; Foley, J.E.; Massey, P.R.; Felizardo, T.C.; Riley, J.L.; et al. The PDL1-PD1 Axis Converts Human Th1 Cells into Regulatory T Cells. Sci. Transl. Med. 2011, 3, 111ra120. [Google Scholar] [CrossRef]
- Dermani, F.K.; Samadi, P.; Rahmani, G.; Kohlan, A.K.; Najafi, R. PD-1/PD-L1 immune checkpoint: Potential target for cancer therapy. J. Cell. Physiol. 2019, 234, 1313–1325. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Vanroey, M.; Wang, C.; Chen, T.H.T.; Korman, A.; Jooss, K. Anti-programmed death-1 synergizes with granulocyte macrophage colony-stimulating factor--secreting tumor cell immunotherapy providing therapeutic benefit to mice with established tumors. Clin. Cancer Res. 2009, 15, 1623–1634. [Google Scholar] [CrossRef] [PubMed]
- Soares, K.C.; Rucki, A.A.; Wu, A.A.; Olino, K.; Xiao, Q.; Chai, Y.; Wamwea, A.; Bigelow, E.; Lutz, E.; Liu, L.; et al. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors. J. Immunother. 2015, 38, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Raedler, L.A. Keytruda (Pembrolizumab): First PD-1 Inhibitor Approved for Previously Treated Unresectable or Metastatic Melanoma. Am. Health Drug Benefits 2015, 8, 96. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4665064/ (accessed on 18 October 2022).
- Kang, S.P.; Gergich, K.; Lubiniecki, G.M.; de Alwis, D.P.; Chen, C.; Tice, M.A.B.; Rubin, E.H. Pembrolizumab KEYNOTE-001: An adaptive study leading to accelerated approval for two indications and a companion diagnostic. Ann. Oncol. 2017, 28, 1388. [Google Scholar] [CrossRef]
- Burtness, B.; Harrington, K.J.; Greil, R.; Soulières, D.; Tahara, M.; de Castro, G.; Psyrri, A.; Basté, N.; Neupane, P.; Bratland, Å.; et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): A randomised, open-label, phase 3 study. Lancet 2019, 394, 1915–1928. [Google Scholar] [CrossRef]
- Marcus, L.; Lemery, S.J.; Keegan, P.; Pazdur, R. FDA Approval Summary: Pembrolizumab for the Treatment of Microsatellite Instability-High Solid Tumors. Clin. Cancer Res. 2019, 25, 3753–3758. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Doi, T.; Jang, R.W.; Muro, K.; Satoh, T.; Machado, M.; Sun, W.; Jalal, S.I.; Shah, M.A.; Metges, J.P.; et al. Safety and Efficacy of Pembrolizumab Monotherapy in Patients with Previously Treated Advanced Gastric and Gastroesophageal Junction Cancer: Phase 2 Clinical KEYNOTE-059 Trial. JAMA Oncol. 2018, 4, e180013. [Google Scholar] [CrossRef]
- Bang, Y.J.; Kang, Y.K.; Catenacci, D.V.; Muro, K.; Fuchs, C.S.; Geva, R.; Hara, H.; Golan, T.; Garrido, M.; Jalal, S.I.; et al. Pembrolizumab alone or in combination with chemotherapy as first-line therapy for patients with advanced gastric or gas-troesophageal junction adenocarcinoma: Results from the phase II nonrandomized KEYNOTE-059 study. Gastric Cancer 2019, 22, 828–837. [Google Scholar] [CrossRef]
- Chung, H.C.; Ros, W.; Delord, J.P.; Perets, R.; Italiano, A.; Shapira-Frommer, R.; Manzuk, L.; Piha-Paul, S.A.; Xu, L.; Zeigenfuss, S.; et al. Efficacy and Safety of Pembrolizumab in Previously Treated Advanced Cervical Cancer: Results from the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2019, 37, 1470–1478. [Google Scholar] [CrossRef]
- Borcoman, E.; Le Tourneau, C. Keynote-158 study, FDA granted accelerated approval of pembrolizumab for the treatment of patients with advanced PD-L1-positive cervical cancer. Ann. Transl. Med. 2020, 8, 1611. [Google Scholar] [CrossRef] [PubMed]
- Vuky, J.; Balar, A.V.; Castellano, D.; O’Donnell, P.H.; Grivas, P.; Bellmunt, J.; Powles, T.; Bajorin, D.; Hahn, N.M.; Savage, M.J.; et al. Long-Term Outcomes in KEYNOTE-052: Phase II Study Investigating First-Line Pembrolizumab in Cisplatin-Ineligible Patients with Locally Advanced or Metastatic Urothelial Cancer. J. Clin. Oncol. 2020, 38, 2658–2666. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Salgado, R.; Park, Y.H.; Muñoz-Couselo, E.; Kim, S.B.; Sohn, J.; Im, S.A.; Foukakis, T.; Kuemmel, S.; Dent, R.; et al. Pembrolizumab plus chemotherapy as neoadjuvant treatment of high-risk, early-stage triple-negative breast cancer: Results from the phase 1b open-label, multicohort KEYNOTE-173 study. Ann. Oncol. 2020, 31, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Nanda, R.; Chow, L.Q.M.; Dees, E.C.; Berger, R.; Gupta, S.; Geva, R.; Pusztai, L.; Pathiraja, K.; Aktan, G.; Cheng, J.D.; et al. Pembrolizumab in Patients with Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 Study. J. Clin. Oncol. 2016, 34, 2460–2467. [Google Scholar] [CrossRef]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H.; et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: Prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef]
- O’Malley, D.M.; Bariani, G.M.; Cassier, P.A.; Marabelle, A.; Hansen, A.R.; De Jesus Acosta, A.; Miller, W.H.; Safra, T.; Italiano, A.; Mileshkin, L.; et al. Pembrolizumab in Patients with Microsatellite Instability-High Advanced Endometrial Cancer: Results From the KEYNOTE-158 Study. J. Clin. Oncol. 2022, 40, 752–761. [Google Scholar] [CrossRef]
- Raedler, L.A. Opdivo (Nivolumab): Second PD-1 Inhibitor Receives FDA Approval for Unresectable or Metastatic Melanoma. Am. Health Drug Benefits 2015, 8, 180. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4665056/ (accessed on 19 October 2022).
- Rizvi, N.A.; Mazières, J.; Planchard, D.; Stinchcombe, T.E.; Dy, G.K.; Antonia, S.J.; Horn, L.; Lena, H.; Minenza, E.; Mennecier, B.; et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): A phase 2, single-arm trial. Lancet Oncol. 2015, 16, 257–265. [Google Scholar] [CrossRef]
- Albiges, L.; Tannir, N.M.; Burotto, M.; McDermott, D.; Plimack, E.R.; Barthélémy, P.; Porta, C.; Powles, T.; Donskov, F.; George, S.; et al. Nivolumab plus ipilimumab versus sunitinib for first-line treatment of advanced renal cell carcinoma: Extended 4-year follow-up of the phase III CheckMate 214 trial. ESMO Open 2020, 5, e001079. [Google Scholar] [CrossRef]
- Ramchandren, R.; Domingo-Domènech, E.; Rueda, A.; Trněný, M.; Feldman, T.A.; Lee, H.J.; Provencio, M.; Sillaber, C.; Cohen, J.B.; Savage, K.J.; et al. Nivolumab for Newly Diagnosed Advanced-Stage Classic Hodgkin Lymphoma: Safety and Efficacy in the Phase II CheckMate 205 Study. J. Clin. Oncol. 2019, 37, 1997–2007. [Google Scholar] [CrossRef]
- Borel, C.; Jung, A.C.; Burgy, M. Immunotherapy Breakthroughs in the Treatment of Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma. Cancers 2020, 12, 2691. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.; Kang, Y.K.; Kim, T.Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.M.; Matilla, A.; et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients with Advanced Hepatocellular Carcinoma Previously Treated with Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Cho, B.C.; Takahashi, M.; Okada, M.; Lin, C.Y.; Chin, K.; Kadowaki, S.; Ahn, M.J.; Hamamoto, Y.; Doki, Y.; et al. Nivolumab versus chemotherapy in patients with advanced oesophageal squamous cell carcinoma refractory or intolerant to previous chemotherapy (ATTRACTION-3): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019, 20, 1506–1517. [Google Scholar] [CrossRef] [PubMed]
- Wright, K. FDA Approves Nivolumab Plus Ipilimumab for Previously Untreated Unresectable Malignant Pleural Mesothelioma. Oncology 2020, 34, 502–503. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Castillo Gutiérrez, E.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N. Engl. J. Med. 2022, 386, 24–34. [Google Scholar] [CrossRef]
- Forde, P.M.; Spicer, J.; Lu, S.; Provencio, M.; Mitsudomi, T.; Awad, M.M.; Felip, E.; Broderick, S.R.; Brahmer, J.R.; Swanson, S.J.; et al. Neoadjuvant nivolumab plus chemotherapy in resectable lung cancer. N. Engl. J. Med. 2022, 386, 1973–1985. [Google Scholar] [CrossRef]
- Doki, Y.; Ajani, J.A.; Kato, K.; Xu, J.; Wyrwicz, L.; Motoyama, S.; Ogata, T.; Kawakami, H.; Hsu, C.H.; Adenis, A.; et al. Nivolumab combination therapy in advanced esophageal squamous-cell carcinoma. N. Engl. J. Med. 2022, 386, 449–462. [Google Scholar] [CrossRef]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 Blockade with Cemi-plimab in Advanced Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef]
- Keeping, S.; Xu, Y.; Chen, C.I.; Cope, S.; Mojebi, A.; Kuznik, A.; Konidaris, G.; Ayers, D.; Sasane, M.; Allen, R.; et al. Comparative Efficacy of Cemi-Plimab versus Other Systemic Treatments for Advanced Cutaneous Squamous Cell Carcinoma. Future Oncol. 2021, 17, 611–627. [Google Scholar] [CrossRef]
- Dong, Y.; Sun, Q.; Zhang, X. PD-1 and its ligands are important immune checkpoints in cancer. Oncotarget 2017, 8, 2171. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Atezolizumab: First Global Approval. Drugs 2016, 76, 1227–1232. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Giaccone, G.; de Marinis, F.; Reinmuth, N.; Vergnenegre, A.; Barrios, C.H.; Morise, M.; Felip, E.; Andric, Z.; Geater, S.; et al. Atezolizumab for First-Line Treatment of PD-L1-Selected Patients with NSCLC. N. Engl. J. Med. 2020, 383, 1328–1339. [Google Scholar] [CrossRef] [PubMed]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef]
- Mathieu, L.; Shah, S.; Pai-Scherf, L.; Larkins, E.; Vallejo, J.; Li, X.; Rodriguez, L.; Mishra-Kalyani, P.; Goldberg, K.B.; Kluetz, P.G.; et al. FDA Approval Summary: Atezolizumab and Durvalumab in Combination with Platinum-Based Chemotherapy in Extensive Stage Small Cell Lung Cancer. Oncologist 2021, 26, 433. [Google Scholar] [CrossRef]
- Gutzmer, R.; Stroyakovskiy, D.; Gogas, H.; Robert, C.; Lewis, K.; Protsenko, S.; Pereira, R.P.; Eigentler, T.; Rutkowski, P.; Demidov, L.; et al. Atezolizumab, vemu-rafenib, and cobimetinib as first-line treatment for unresectable advanced BRAFV600 mutation-positive melanoma (IMspire150): Primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 395, 1835–1844. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Syed, Y.Y. Durvalumab: First Global Approval. Drugs 2017, 77, 1369. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Kurata, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Overall Survival with Durvalumab after Chemoradiotherapy in Stage III NSCLC. N. Engl. J. Med. 2018, 379, 2342–2350. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Cho, B.C.; Reinmuth, N.; Lee, K.H.; Luft, A.; Ahn, M.J.; Van Den Heuvel, M.M.; Cobo, M.; Vicente, D.; Smolin, A.; et al. Durvalumab with or without Tremelimumab vs Standard Chemotherapy in First-line Treatment of Metastatic Non-Small Cell Lung Cancer: The MYSTIC Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 661–674. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Dvorkin, M.; Chen, Y.; Reinmuth, N.; Hotta, K.; Trukhin, D.; Statsenko, G.; Hochmair, M.J.; Özgüroğlu, M.; Ji, J.H.; et al. Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): A randomised, controlled, open-label, phase 3 trial. Lancet 2019, 394, 1929–1939. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.-Y.; He, A.R.; Qin, S.; Chen, L.-T.; Okusaka, T.; Vogel, A.; Kim, J.W.; Suksombooncharoen, T.; Lee, M.A.; Kitano, M.; et al. Durvalumab plus Gemcitabine and Cisplatin in Advanced Biliary Tract Cancer. NEJM Evid. 2022, 1, EVIDoa2200015. [Google Scholar] [CrossRef]
- Walker, J.W.; Lebbé, C.; Grignani, G.; Nathan, P.; Dirix, L.; Fenig, E.; Ascierto, P.A.; Sandhu, S.; Munhoz, R.; Benincasa, E.; et al. Efficacy and safety of avelumab treatment in patients with metastatic Merkel cell carcinoma: Experience from a global expanded access program. J. Immunother. Cancer 2020, 8, e000313. [Google Scholar] [CrossRef] [PubMed]
- Apolo, A.B.; Ellerton, J.A.; Infante, J.R.; Agrawal, M.; Gordon, M.S.; Aljumaily, R.; Gourdin, T.; Dirix, L.; Lee, K.W.; Taylor, M.H.; et al. Avelumab as second-line therapy for metastatic, platinum-treated urothelial carcinoma in the phase Ib JAVELIN Solid Tumor study: 2-year updated efficacy and safety analysis. J. Immunother. Cancer 2020, 8, e001246. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Larkin, J.; Oya, M.; Thistlethwaite, F.; Martignoni, M.; Nathan, P.; Powles, T.; McDermott, D.; Robbins, P.B.; Chism, D.D.; et al. Preliminary results for avelumab plus axitinib as first-line therapy in patients with advanced clear-cell renal-cell carcinoma (JAVELIN Renal 100): An open-label, dose-finding and dose-expansion, phase 1b trial. Lancet Oncol. 2018, 19, 451–460. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Nigro, O.; Pinotti, G.; De Galitiis, F.; Di Pietro, F.R.; Giusti, R.; Filetti, M.; Bersanelli, M.; Lazzarin, A.; Bordi, P.; Catino, A. Late immune-related adverse events in long-term responders to PD-1/PD-L1 checkpoint inhibitors: A multicentre study. Eur. J. Cancer 2020, 134, 19–28. [Google Scholar] [CrossRef]
- Paucek, R.D.; Baltimore, D.; Li, G. The Cellular Immunotherapy Revolution: Arming the Immune System for Precision Therapy. Trends Immunol. 2019, 40, 292–309. [Google Scholar] [CrossRef]
- Patel, S.P.; Kurzrock, R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef]
- Bai, R.; Lv, Z.; Xu, D.; Cui, J. Predictive biomarkers for cancer immunotherapy with immune checkpoint inhibitors. Biomark. Res. 2020, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csoszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Updated Analysis of KEYNOTE-024: Pembrolizumab Versus Platinum-Based Chemotherapy for Advanced Non-Small-Cell Lung Cancer with PD-L1 Tumor Proportion Score of 50% or Greater. J. Clin. Oncol. 2019, 37, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Yi, M.; Qin, S.; Chu, Q.; Zheng, X.; Wu, K. The efficacy and safety of combination of PD-1 and CTLA-4 inhibitors: A meta-analysis. Exp. Hematol. Oncol. 2019, 8, 26. [Google Scholar] [CrossRef] [PubMed]
- Nishino, M.; Ramaiya, N.H.; Hatabu, H.; Hodi, F.S. Monitoring immune-checkpoint blockade: Response evaluation and biomarker development. Nat. Rev. Clin. Oncol. 2017, 14, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Luchini, C.; Bibeau, F.; Ligtenberg, M.J.L.; Singh, N.; Nottegar, A.; Bosse, T.; Miller, R.; Riaz, N.; Douillard, J.Y.; Andre, F.; et al. ESMO recommendations on microsatellite instability testing for immunotherapy in cancer, and its relationship with PD-1/PD-L1 expression and tumour mutational burden: A systematic review-based approach. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 1232–1243. [Google Scholar] [CrossRef] [PubMed]
- Schrock, A.B.; Ouyang, C.; Sandhu, J.; Sokol, E.; Jin, D.; Ross, J.S.; Miller, V.A.; Lim, D.; Amanam, I.; Chao, J.; et al. Tumor mutational burden is predictive of response to immune checkpoint inhibitors in MSI-high metastatic colorectal cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 1096–1103. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Callahan, M.K.; Awad, M.M.; Calvo, E.; Ascierto, P.A.; Atmaca, A.; Rizvi, N.A.; Hirsch, F.R.; Selvaggi, G.; Szustakowski, J.D.; et al. Tumor Mutational Burden and Efficacy of Nivolumab Monotherapy and in Combination with Ipilimumab in Small-Cell Lung Cancer. Cancer Cell 2018, 33, 853–861. [Google Scholar] [CrossRef]
- Marabelle, A.; Fakih, M.G.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.; et al. Association of Tumor Mutational Burden with Outcomes in Patients with Select Advanced Solid Tumors Treated with Pembrolizumab in KEYNOTE-158. Ann. Oncol. 2019, 30, v477–v478. [Google Scholar] [CrossRef]
- Yarchoan, M.; Albacker, L.A.; Hopkins, A.C.; Montesion, M.; Murugesan, K.; Vithayathil, T.T.; Zaidi, N.; Azad, N.S.; Laheru, D.A.; Frampton, G.M.; et al. PD-L1 Expression and Tumor Mutational Burden are Independent Biomarkers in Most Cancers. JCI Insight 2019, 4, e126908. [Google Scholar] [CrossRef] [PubMed]
- Strickler, J.H.; Hanks, B.A.; Khasraw, M. Tumor Mutational Burden as a Predictor of Immunotherapy Response: Is More Always Better? Clin. Cancer Res. 2021, 27, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- Lyer, R.R.; Pluciennik, A.; Burdett, V.; Modrich, P.L. DNA Mismatch Repair: Functions and Mechanisms. Chem. Rev. 2006, 106, 302–323. [Google Scholar] [CrossRef]
- Fabrizio, D.A.; George, T.J.; Dunne, R.F.; Frampton, G.; Sun, J.; Gowen, K.; Kennedy, M.; Greenbowe, J.; Schrock, A.B.; Hezel, A.F.; et al. Beyond Microsatellite Testing: Assessment of Tumor Mutational Burden Identifies Subsets of Colorectal Cancer Who May Respond to Immune Checkpoint Inhibition. J. Gastrointest. Oncol. 2018, 9, 610. [Google Scholar] [CrossRef]
- Kruger, S.; Ilmer, M.; Kobold, S.; Cadilha, B.L.; Endres, S.; Ormanns, S.; Schuebbe, G.; Renz, B.W.; D’Haese, J.G.; Schloesser, H.; et al. Advances in Cancer Immunotherapy 2019—Latest Trends. J. Exp. Clin. Cancer Res. 2019, 38, 268. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the Tumor Immune Microenvironment (TIME) for Effective Therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Pagès, F.; Mlecnik, B.; Marliot, F.; Bindea, G.; Ou, F.S.; Bifulco, C.; Lugli, A.; Zlobec, I.; Rau, T.T.; Berger, M.D.; et al. International Validation of the Consensus Immunoscore for the Classification of Colon Cancer: A Prognostic and Accuracy Study. Lancet 2018, 391, 2128–2139. [Google Scholar] [CrossRef]
- Galon, J.; Mlecnik, B.; Bindea, G.; Angell, H.K.; Berger, A.; Lagorce, C.; Lugli, A.; Zlobec, I.; Hartmann, A.; Bifulco, C.; et al. Towards the Introduction of the “Immunoscore” in the Classification of Malignant Tumours. J. Pathol. 2014, 232, 199–209. [Google Scholar] [CrossRef]
- Mahmoud, S.M.A.; Paish, E.C.; Powe, D.G.; Macmillan, R.D.; Grainge, M.J.; Lee, A.H.S.; Ellis, I.O.; Green, A.R. Tumor-Infiltrating CD8+ Lymphocytes Predict Clinical Outcome in Breast Cancer. J. Clin. Oncol. 2011, 29, 1949–1955. [Google Scholar] [CrossRef]
- Gao, Q.; Qiu, S.J.; Fan, J.; Zhou, J.; Wang, X.Y.; Xiao, Y.S.; Xu, Y.; Li, Y.W.; Tang, Z.Y. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J. Clin. Oncol. 2007, 25, 2586–2593. [Google Scholar] [CrossRef] [PubMed]
- Petitprez, F.; Meylan, M.; de Reyniès, A.; Sautès-Fridman, C.; Fridman, W.H. The Tumor Microenvironment in the Response to Immune Checkpoint Blockade Therapies. Front. Immunol. 2020, 11, 784. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, P.; Curtis, N. The influence of the intestinal microbiome on vaccine responses. Vaccine 2018, 36, 4433–4439. [Google Scholar] [CrossRef]
- Yi, M.; Qin, S.; Chu, Q.; Wu, K. The role of gut microbiota in immune checkpoint inhibitor therapy. Hepatobiliary Surg. Nutr. 2018, 7, 48183–48483. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Lv, J.; Guo, F.; Li, J.; Jia, Y.; Jiang, D.; Wang, N.; Zhang, C.; Kong, L.; Liu, Y.; et al. Gut Microbiome Influences the Efficacy of PD-1 Antibody Immunotherapy on MSS-Type Colorectal Cancer via Metabolic Pathway. Front. Microbiol. 2020, 11, 814. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Naqash, A.R.; Kihn-Alarcón, A.J.; Stavraka, C.; Kerrigan, K.; Vareki, S.M.; Pinato, D.J.; Puri, S. The role of gut microbiome in modulating response to immune checkpoint inhibitor therapy in cancer. Ann. Transl. Med. 2021, 9, 1034. [Google Scholar] [CrossRef]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Marin-Acevedo, J.A.; Chirila, R.M.; Dronca, R.S. Immune Checkpoint Inhibitor Toxicities. Mayo Clin. Proc. 2019, 94, 1321–1329. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Naidoo, J.; Page, D.B.; Li, B.T.; Connell, L.C.; Schindler, K.; Lacouture, M.E.; Postow, M.A.; Wolchok, J.D. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann. Oncol. 2015, 26, 2375. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Yao, Z.; Bai, H.; Duan, J.; Wang, Z.; Wang, X.; Zhang, X.; Xu, J.; Fei, K.; Zhang, Z.; et al. Treatment-related adverse events of PD-1 and PD-L1 inhibitor-based combination therapies in clinical trials: A systematic review and meta-analysis. Lancet Oncol. 2021, 22, 1265–1274. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Lacchetti, C.; Schneider, B.J.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; Chau, I.; Ernstoff, M.S.; Gardner, J.M.; Ginex, P.; et al. Thompson, Management of Immune-Related Adverse Events in Patients Treated with Immune Checkpoint Inhibitor Therapy: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2018, 36, 1714–1768. [Google Scholar] [CrossRef] [PubMed]
- Haanen, J.B.A.G.; Carbonnel, F.; Robert, C.; Kerr, K.M.; Peters, S.; Larkin, J.; Jordan, K. Management of toxicities from immunotherapy: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, iv119–iv142. [Google Scholar] [CrossRef]
- Arbour, K.C.; Mezquita, L.; Long, N.; Rizvi, H.; Auclin, E.; Ni, A.; Martínez-Bernal, G.; Ferrara, R.; Victoria Lai, W.; Hendriks, L.E.L.; et al. Impact of Baseline Steroids on Efficacy of Programmed Cell Death-1 and Programmed Death-Ligand 1 Blockade in Patients with Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 2872–2878. [Google Scholar] [CrossRef]
- Faje, A.T.; Lawrence, D.; Flaherty, K.; Freedman, C.; Fadden, R.; Rubin, K.; Cohen, J.; Sullivan, R.J. High-dose glucocorticoids for the treatment of ipilimumab-induced hypophysitis is associated with reduced survival in patients with melanoma. Cancer 2018, 124, 3706–3714. [Google Scholar] [CrossRef]
- Von Pawel, J.; Syrigos, K.; Mazieres, J.; Cortinovis, D.; Dziadziuszko, R.; Gandara, D.R.; Conkling, P.; Goldschmidt, J.; Thomas, C.A.; Bordoni, R.; et al. Association between immune-related adverse events (irAEs) and atezolizumab efficacy in advanced NSCLC: Analyses from the phase III study OAK. Ann. Oncol. 2017, 28, v469. [Google Scholar] [CrossRef]
- Weber, J.S.; Hodi, F.S.; Wolchok, J.D.; Topalian, S.L.; Schadendorf, D.; Larkin, J.; Sznol, M.; Long, G.V.; Li, H.; Waxman, I.M.; et al. Safety profile of nivolumab monotherapy: A pooled analysis of patients with advanced melanoma. J. Clin. Oncol. 2017, 35, 785–792. [Google Scholar] [CrossRef]
- Schneider, B.J.; Naidoo, J.; Santomasso, B.D.; Lacchetti, C.; Adkins, S.; Anadkat, M.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; Chau, I.; et al. Management of Immune-Related Adverse Events in Patients Treated with Immune Checkpoint Inhibitor Therapy: ASCO Guideline Update. J. Clin. Oncol. 2021, 39, 4073–4126. [Google Scholar] [CrossRef]
- Song, W.; Shen, L.; Wang, Y.; Liu, Q.; Goodwin, T.J.; Li, J.; Dorosheva, O.; Liu, T.; Liu, R.; Huang, L. Synergistic and low adverse effect cancer immunotherapy by immunogenic chemotherapy and locally expressed PD-L1 trap. Nat. Commun. 2018, 9, 2237. [Google Scholar] [CrossRef]
- Schmid, D.; Park, C.G.; Hartl, C.A.; Subedi, N.; Cartwright, A.N.; Puerto, R.B.; Zheng, Y.; Maiarana, J.; Freeman, G.J.; Wucherpfennig, K.W.; et al. T cell-targeting nanoparticles focus delivery of immunotherapy to improve antitumor immunity. Nat. Commun. 2017, 8, 1747. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.; Rao, P.; Natarajan, S.; Goldman, A.; Sabbisetti, V.S.; Khater, Y.; Korimerla, N.; Chandrasekar, V.; Mashelkar, R.A.; Sengupta, S. Reporter nanoparticle that monitors its anticancer efficacy in real time. Proc. Natl. Acad. Sci. USA 2016, 113, E2104–E2113. [Google Scholar] [CrossRef]
- Ishihara, J.; Fukunaga, K.; Ishihara, A.; Larsson, H.M.; Potin, L.; Hosseinchi, P.; Galliverti, G.; Swartz, M.A.; Hubbell, J.A. Matrix-binding checkpoint immunotherapies enhance antitumor efficacy and reduce adverse events. Sci. Transl. Med. 2017, 9, eaan0401. [Google Scholar] [CrossRef] [PubMed]
- Lamichhane, P.; Deshmukh, R.; Brown, J.A.; Jakubski, S.; Parajuli, P.; Nolan, T.; Raja, D.; Badawy, M.; Yoon, T.; Zmiyiwsky, M.; et al. Novel Delivery Systems for Checkpoint Inhibitors. Medicines 2019, 6, 74. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, J.; Hu, L.; Han, X.; Zou, X.; Chen, Q.; Chen, Y.; Liu, Z.; Wang, C. In Situ Formed Fibrin Scaffold with Cyclophosphamide to Synergize with Immune Checkpoint Blockade for Inhibition of Cancer Recurrence after Surgery. Adv. Funct. Mater. 2020, 30, 1906922. [Google Scholar] [CrossRef]
- Wang, C.; Wang, J.; Zhang, X.; Yu, S.; Wen, D.; Hu, Q.; Ye, Y.; Bomba, H.; Hu, X.; Liu, Z.; et al. In situ formed reactive oxygen species-responsive scaffold with gemcitabine and checkpoint inhibitor for combination therapy. Sci. Transl. Med. 2018, 10, eaan3682. [Google Scholar] [CrossRef]
- Wang, C.; Ye, Y.; Hochu, G.M.; Sadeghifar, H.; Gu, Z. Enhanced Cancer Immunotherapy by Microneedle Patch-Assisted Delivery of Anti-PD1 Antibody. Nano Lett. 2016, 16, 2334–2340. [Google Scholar] [CrossRef]
- Lin, A.H.; Twitty, C.G.; Burnett, R.; Hofacre, A.; Mitchell, L.A.; Espinoza, F.L.; Gruber, H.E.; Jolly, D. Retroviral Replicating Vector Delivery of miR-PDL1 Inhibits Immune Checkpoint PDL1 and Enhances Immune Responses In Vitro. Mol. Ther. Nucleic Acids 2016, 6, 221–232. [Google Scholar] [CrossRef]
- Reul, J.; Frisch, J.; Engeland, C.E.; Thalheimer, F.B.; Hartmann, J.; Ungerechts, G.; Buchholz, C.J. Tumor-specific delivery of immune checkpoint inhibitors by engineered AAV vectors. Front. Oncol. 2019, 9, 52. [Google Scholar] [CrossRef]
- Shi, T.; Song, X.; Wang, Y.; Liu, F.; Wei, J. Combining Oncolytic Viruses with Cancer Immunotherapy: Establishing a New Generation of Cancer Treatment. Front. Immunol. 2020, 11, 683. [Google Scholar] [CrossRef]
- Gurbatri, C.R.; Lia, I.; Vincent, R.; Coker, C.; Castro, S.; Treuting, P.M.; Hinchliffe, T.E.; Arpaia, N.; Danino, T. Engineered probiotics for local tumor delivery of checkpoint blockade nanobodies. Sci. Transl. Med. 2020, 12, eaax0876. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| CPI Target | Drug | Class | 1st Approval Year | Indication(s) |
|---|---|---|---|---|
| CTLA-4 | Ipilimumab | IgG1 mAb | 2010 | Melanoma. |
| PD-1 | Pembrolizumab | IgG4 mAb | 2016 | Melanoma, NSCLC, HNSCC, MSI-H solid tumors, GC, cervical cancer, urothelial carcinoma, TNBC, TMB-H/dMMR endometrial carcinoma. |
| Nivolumab | IgG4 mAb | 2014 | Melanoma, NSCLC, RCC, Hodgkin’s lymphoma, HNSCC, HCC, ESCC, pleural mesothelioma, MSI-H/dMMR CRC. | |
| Cemiplimab | IgG4 mAb | 2018 | Cutaneous squamous cell carcinoma. | |
| PD-L1 | Atezolizumab | IgG1 mAb | 2016 | Urothelial carcinoma, NSCLC, SCLC, melanoma, HCC. |
| Durvalumab | IgG1 mAb | 2017 | Urothelial bladder cancer, NSCLC, SCLC, BTC. | |
| Avelumab | IgG1 mAb | 2017 | Merkel cell carcinoma, urothelial carcinoma, RCC. |
| Rank | NCT Number | Title | Acronym | Status | Conditions | Interventions | Phase | Location(s) |
|---|---|---|---|---|---|---|---|---|
| 1 | 5335928 | Abatacept in Immune Checkpoint Inhibitor Myocarditis | ATRIUM | Recruiting | Myocarditis Acute, cancer | Drug: abatacept plus, drug: placebo | 3 | United States |
| 2 | 4338269 | A Study of Atezolizumab in Combination with Cabozantinib Compared to Cabozantinib Alone in Participants With Advanced Renal Cell Carcinoma After Immune Checkpoint Inhibitor Treatment | CONTACT-03 | Active, not recruiting | Carcinoma, renal cell | Drug: atezolizumab, drug: cabozantinib | 3 | United States, Argentina, Australia, Canada, Denmark, France, Germany, Greece, Italy, Japan, Korea, Poland, Russian Federation, Russian Federation, Spain, United Kingdom |
| 3 | 2654587 | Study of OSE2101 Versus Standard Treatment as 2nd or 3rd Line in HLA-A2 Positive Patients with Advanced NSCLC After Failure of Immune Checkpoint Inhibitor | ATALANTE 1 | Active, not recruiting | Non-small-cell lung cancer | Drug: OSE2101, drug: docetaxel, drug: pemetrexed | 3 | United States, Czechia, France, Germany, Hungary, Israel, Italy, Poland, Spain, United Kingdom |
| 4 | 4987203 | Study to Compare Tivozanib in Combination with Nivolumab to Tivozanib Monotherapy in Subjects With Renal Cell Carcinoma | NA | Recruiting | Renal cell carcinoma | Drug: tivozanib, drug: nivolumab | 3 | United States, Argentina, Australia, Belgium, Brazil, Canada, Chile, Czechia, France, Germany, Italy, Mexico, Poland, Portugal, Spain, United Kingdom |
| 5 | 3469960 | Double Immune Checkpoint Inhibitors in PD-L1-positive Stage IV Non-Small Lung Cancer | DICIPLE | Active, not recruiting | Non-small-cell lung cancer metastatic | Drug: ipilimumab, drug: nivolumab | 3 | France |
| 6 | 4590963 | Assessment of Efficacy and Safety of Monalizumab Plus Cetuximab Compared to Placebo Plus Cetuximab in Recurrent or Metastatic Head and Neck Cancer | INTERLINK-1 | Active, not recruiting | Squamous cell carcinoma of the head and neck | Drug: monalizumab, drug: cetuximab | Phase 3 | United States, Argentina, Australia, Belgium, Brazil, Bulgaria, Canada, France, Germany, Greece, Italy, Japan, Korea, Netherlands, Philippines, Portugal, Russian Federation, Spain, Switzerland, Taiwan, United Kingdom |
| 7 | 5106335 | A Study to Evaluate Camrelizumab Combined with Famitinib as Subsequent Therapy in Patients With Advanced NSCLC | NA | Recruiting | Advanced NSCLC | Drug: camrelizumab + famitinib, drug: famitinib, drug: docetaxel | 3 | China |
| 8 | 4059887 | Evaluation of Blood TMB for the Efficacy of Atezolizumab [BUDDY] | BUDDY | Recruiting | Lung neoplasm Malignant | Drug: atezolizumab injection (Tecentriq) | 4 | Korea |
| 9 | 3427827 | PD-1 Antibody Versus Best Supportive Care After Chemoradiation in Locoregionally Advanced Nasopharyngeal Carcinoma | PACIFIC-NPC | Recruiting | Nasopharyngeal neoplasms | Drug: camrelizumab | 3 | China |
| 10 | 4907370 | PD-1 Blockade Combined with De-intensification Radical Chemoradiotherapy in Nasopharyngeal Carcinoma | TIRA | Recruiting | Nasopharyngeal carcinoma | Drug: PD-1 blocking antibody, drug: gemcitabine, drug: cisplatin (80 mg/m2), drug: cisplatin (100 mg/m2), radiation: intensity-modulated radiotherapy | 3 | China |
| 11 | 4334759 | Durvalumab With Chemotherapy as First Line Treatment in Advanced Pleural Mesothelioma | DREAM3R | Recruiting | Mesothelioma, pleural mesothelioma, malignant pleural mesothelioma | Drug: durvalumab, drug: standard chemotherapy, drug: ipilimumab and nivolumab | 3 | United States, Australia, New Zealand |
| 12 | 4799249 | Trilaciclib, a CDK 4/6 Inhibitor, in Patients Receiving Gemcitabine and Carboplatin for Metastatic Triple-Negative Breast Cancer (TNBC) | PRESERVE 2 | Active, not recruiting | Triple-negative breast cancer (TNBC), breast cancer | Drug: trilaciclib, drug: placebo, drug: gemcitabine, drug: carboplatin | 3 | United States, Australia, Bulgaria, China, France, Georgia, Moldova, Poland, Russian Federation, Serbia, Spain, Ukraine |
| 13 | 2973789 | Effect of Tumor Treating Fields (TTFields) (150 kHz) Concurrent with Standard of Care Therapies for Treatment of Stage 4 Non-Small Cell Lung Cancer (NSCLC) Following Platinum Failure (LUNAR) | NA | Active, not recruiting | Non-small-cell lung cancer (NSCLC) | Device: NovoTTF-200T, drug: immune checkpoint inhibitors or docetaxel | 3 | United States, Austria, Belgium, Bulgaria, Canada, China, Czechia, France, Germany, Hong Kong, Italy, Netherlands, Poland, Serbia, Spain, Switzerland |
| Adverse Event Grade | Adverse Event | Frequency %, [95% Confidence Interval] |
|---|---|---|
| All-Grade | Anemia | 45 [32.4–59.1] |
| Fatigue | 34.3 [27.5–41.9] | |
| Dysphagia | 30 [18.7–44.5] | |
| Grade 3 or Higher | Neutropenia | 19.6 [13.5–27.7] |
| Hypertension | 9.3 [5.7–14.9] | |
| Lipase increase | 7.2 [5.2–9.9] | |
| Lymphopenia | 10.3 [4.5–21.8] |
| Grade | Management |
|---|---|
| 1 | CPI therapy should be continued with close monitoring for grade 1 toxicities, except for some neurologic, hematologic, and cardiac toxicities. |
| 2 | CPIs therapy may be suspended for most grade 2 toxicities, with consideration of resuming when symptoms revert ≤ grade 1. |
| 3 | Generally, warrant suspension of ICPs and the initiation of high-dose corticosteroids (should be tapered over the course of at least 4–6 weeks). |
| 4 | In general, permanent discontinuation of ICPis is recommended with grade 4 toxicities, except for endocrinopathies that have been controlled by hormone replacement. |
| Approach | Advantages | Limitations |
|---|---|---|
| Nanoparticles | Stimulate tumor immunogenicity, localized delivery, cargo protection. | High manufacturing cost, design complexity, possible toxicity, efficacy needs further characterization. |
| Matrix-binding molecular conjugates | Prolonged retention, localized delivery, efficient targeting of ECM. | Targeting restricted ECM can be challenging, ECM expression in healthy tissue may cause off-targeting and toxicity. |
| Engineered biomaterials | Adjustable design, localized delivery, drug controlled-release, | Acute/chronic inflammatory reactions, biocompatibility and biodegradability are not fully characterized, difficulties with hydrophobic drugs. |
| Viral vector systems | Multiple clinical trials using OVs, easily genetically modifiable, high level of expression. | Antiviral response can be a limiting factor, need complex design for improved efficacy, high regulatory standards. |
| Bacteria | Natural colonization of tumor sites, easily genetically engineered, potential for oral delivery (noninvasive). | The approach still in infancy, issues with microtumors, can be difficult to apply in immunocompromised individuals. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basudan, A.M. The Role of Immune Checkpoint Inhibitors in Cancer Therapy. Clin. Pract. 2023, 13, 22-40. https://doi.org/10.3390/clinpract13010003
Basudan AM. The Role of Immune Checkpoint Inhibitors in Cancer Therapy. Clinics and Practice. 2023; 13(1):22-40. https://doi.org/10.3390/clinpract13010003
Chicago/Turabian StyleBasudan, Ahmed M. 2023. "The Role of Immune Checkpoint Inhibitors in Cancer Therapy" Clinics and Practice 13, no. 1: 22-40. https://doi.org/10.3390/clinpract13010003
APA StyleBasudan, A. M. (2023). The Role of Immune Checkpoint Inhibitors in Cancer Therapy. Clinics and Practice, 13(1), 22-40. https://doi.org/10.3390/clinpract13010003