
Cooperation Between Aflatoxin-Induced p53 Aberrations and Hepatitis B Virus in Hepatocellular Carcinoma



{kind=link}
{kind=link}
Abstract
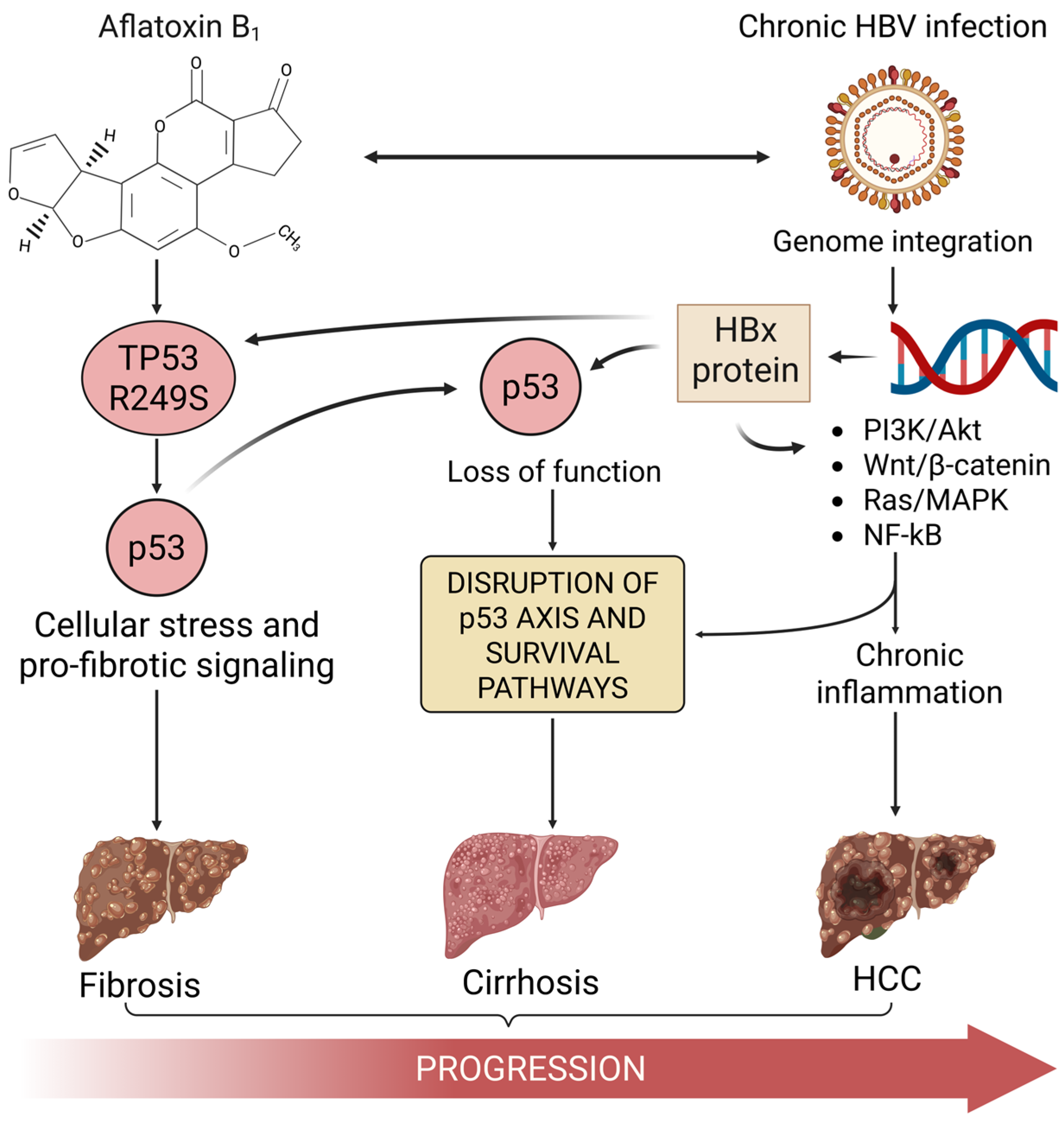
1. The Convergence of HBV, Aflatoxin, and p53 in HCC
2. The p53 Tumor Suppressor Pathway
3. Aflatoxin B1-Induced TP53 R249S Mutation: A Molecular Signature in HCC
4. Hepatitis B Virus and HBx-Mediated Disruption of p53 Function
5. Synergistic Disruption of p53 by Aflatoxin-Induced Mutation and HBV Activity
6. Conclusions: Aflatoxin-Induced p53 Mutation as a Key Driver in HBV-Associated HCC
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef]
- Li, Q.; Ding, C.; Cao, M.; Yang, F.; Yan, X.; He, S.; Zhang, S.; Teng, Y.; Tan, N.; Wang, J.; et al. Global epidemiology of liver cancer 2022: An emphasis on geographic disparities. Chin. Med. J. 2024, 137, 2334–2342. [Google Scholar] [CrossRef]
- Hudu, S.A.; Shinkafi, S.H.; Jimoh, A.O. A critical review of diagnostic and prognostic markers of chronic hepatitis B infection. Med. Rev. (2021) 2024, 4, 225–234. [Google Scholar] [CrossRef]
- Wu, H.C.; Santella, R. The role of aflatoxins in hepatocellular carcinoma. Hepat. Mon. 2012, 12, e7238. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, F. Global burden of aflatoxin-induced hepatocellular carcinoma: A risk assessment. Environ. Health Perspect. 2010, 118, 818–824. [Google Scholar] [CrossRef]
- Shabeer, S.; Asad, S.; Jamal, A.; Ali, A. Aflatoxin Contamination, Its Impact and Management Strategies: An Updated Review. Toxins 2022, 14, 307. [Google Scholar] [CrossRef]
- Ostry, V.; Malir, F.; Toman, J.; Grosse, Y. Mycotoxins as human carcinogens-the IARC Monographs classification. Mycotoxin Res. 2017, 33, 65–73. [Google Scholar] [CrossRef]
- Wu, F.; Stacy, S.L.; Kensler, T.W. Global risk assessment of aflatoxins in maize and peanuts: Are regulatory standards adequately protective? Toxicol. Sci. 2013, 135, 251–259. [Google Scholar] [CrossRef]
- Nugraha, A.; Khotimah, K.; Rietjens, I.M.C.M. Risk assessment of aflatoxin B1 exposure from maize and peanut consumption in Indonesia using the margin of exposure and liver cancer risk estimation approaches. Food Chem. Toxicol. 2018, 113, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Marchese, S.; Polo, A.; Ariano, A.; Velotto, S.; Costantini, S.; Severino, L. Aflatoxin B1 and M1: Biological Properties and Their Involvement in Cancer Development. Toxins 2018, 10, 214. [Google Scholar] [CrossRef] [PubMed]
- Seeger, C.; Mason, W.S. Molecular biology of hepatitis B virus infection. Virology 2015, 479–480, 672–686. [Google Scholar] [CrossRef] [PubMed]
- Ming, L.; Thorgeirsson, S.S.; Gail, M.H.; Lu, P.; Harris, C.C.; Wang, N.; Shao, Y.; Wu, Z.; Liu, G.; Wang, X.; et al. Dominant role of hepatitis B virus and cofactor role of aflatoxin in hepatocarcinogenesis in Qidong, China. Hepatology 2002, 36, 1214–1220. [Google Scholar] [CrossRef]
- Chen, C.J.; Hsu, W.L.; Yang, H.I.; Lee, M.H.; Chen, H.C.; Chien, Y.C.; You, S.L. Epidemiology of virus infection and human cancer. Recent Results Cancer Res. 2014, 193, 11–32. [Google Scholar] [CrossRef]
- Kew, M.C. Synergistic interaction between aflatoxin B1 and hepatitis B virus in hepatocarcinogenesis. Liver Int. 2003, 23, 405–409. [Google Scholar] [CrossRef]
- Hsu, I.C.; Metcalf, R.A.; Sun, T.; Welsh, J.A.; Wang, N.J.; Harris, C.C. Mutational hotspot in the p53 gene in human hepatocellular carcinomas. Nature 1991, 350, 427–428. [Google Scholar] [CrossRef]
- Ozturk, M. p53 mutation in hepatocellular carcinoma after aflatoxin exposure. Lancet 1991, 338, 1356–1359. [Google Scholar] [CrossRef]
- Iyer, S.; Groopman, J.D. Interaction of mutant hepatitis B X protein with p53 tumor suppressor protein affects both transcription and cell survival. Mol. Carcinog. 2011, 50, 972–980. [Google Scholar] [CrossRef] [PubMed]
- Che, L.; Du, Z.B.; Wang, W.H.; Wu, J.S.; Han, T.; Chen, Y.Y.; Han, P.Y.; Lei, Z.; Chen, X.X.; He, Y.; et al. Intracellular antibody targeting HBx suppresses invasion and metastasis in hepatitis B virus-related hepatocarcinogenesis via protein phosphatase 2A-B56γ-mediated dephosphorylation of protein kinase B. Cell Prolif. 2022, 55, e13304. [Google Scholar] [CrossRef]
- Kew, M.C. Hepatitis B virus x protein in the pathogenesis of hepatitis B virus-induced hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2011, 26 (Suppl. S1), 144–152. [Google Scholar] [CrossRef] [PubMed]
- Kong, G.; Zhang, J.; Zhang, S.; Shan, C.; Ye, L.; Zhang, X. Upregulated microRNA-29a by hepatitis B virus X protein enhances hepatoma cell migration by targeting PTEN in cell culture model. PLoS ONE 2011, 6, e19518. [Google Scholar] [CrossRef]
- Wu, H.C.; Wang, Q.; Yang, H.I.; Ahsan, H.; Tsai, W.Y.; Wang, L.Y.; Chen, S.Y.; Chen, C.J.; Santella, R.M. Aflatoxin B1 exposure, hepatitis B virus infection, and hepatocellular carcinoma in Taiwan. Cancer Epidemiol. Biomark. Prev. 2009, 18, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.Y.; Chen, C.J.; Chou, S.R.; Hsieh, L.L.; Wang, L.Y.; Tsai, W.Y.; Ahsan, H.; Santella, R.M. Association of aflatoxin B(1)-albumin adduct levels with hepatitis B surface antigen status among adolescents in Taiwan. Cancer Epidemiol. Biomark. Prev. 2001, 10, 1223–1226. [Google Scholar]
- Bannasch, P.; Khoshkhou, N.I.; Hacker, H.J.; Radaeva, S.; Mrozek, M.; Zillmann, U.; Kopp-Schneider, A.; Haberkorn, U.; Elgas, M.; Tolle, T. Synergistic hepatocarcinogenic effect of hepadnaviral infection and dietary aflatoxin B1 in woodchucks. Cancer Res. 1995, 55, 3318–3330. [Google Scholar] [PubMed]
- Kew, M.C. Aflatoxins as a cause of hepatocellular carcinoma. J. Gastrointest. Liver Dis. 2013, 22, 305–310. [Google Scholar]
- Schrenk, D.; Bignami, M.; Bodin, L.; Chipman, J.K.; Del Mazo, J.; Grasl-Kraupp, B.; Hogstrand, C.; Hoogenboom, L.R.; Leblanc, J.C.; Nebbia, C.S.; et al. Risk assessment of aflatoxins in food. EFSA J. 2020, 18, e06040. [Google Scholar] [CrossRef]
- Han, C.; Yu, T.; Qin, W.; Liao, X.; Huang, J.; Liu, Z.; Yu, L.; Liu, X.; Chen, Z.; Yang, C.; et al. Genome-wide association study of the TP53 R249S mutation in hepatocellular carcinoma with aflatoxin B1 exposure and infection with hepatitis B virus. J. Gastrointest. Oncol. 2020, 11, 1333–1349. [Google Scholar] [CrossRef]
- Alvarez, C.S.; Ortiz, J.; Bendfeldt-Avila, G.; Xie, Y.; Wang, M.; Wu, D.; Higson, H.; Lee, E.; Teshome, K.; Barnoya, J.; et al. Analysis of TP53 aflatoxin signature mutation in hepatocellular carcinomas from Guatemala: A cross-sectional study (2016–2017). Health Sci. Rep. 2020, 3, e155. [Google Scholar] [CrossRef] [PubMed]
- Gouas, D.A.; Villar, S.; Ortiz-Cuaran, S.; Legros, P.; Ferro, G.; Kirk, G.D.; Lesi, O.A.; Mendy, M.; Bah, E.; Friesen, M.D.; et al. TP53 R249S mutation, genetic variations in HBX and risk of hepatocellular carcinoma in The Gambia. Carcinogenesis 2012, 33, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Bressac, B.; Kew, M.; Wands, J.; Ozturk, M. Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature 1991, 350, 429–431. [Google Scholar] [CrossRef]
- Chu, Y.J.; Yang, H.I.; Wu, H.C.; Liu, J.; Wang, L.Y.; Lu, S.N.; Lee, M.H.; Jen, C.L.; You, S.L.; Santella, R.M.; et al. Aflatoxin B. Int. J. Cancer 2017, 141, 711–720. [Google Scholar] [CrossRef]
- Ortiz-Cuaran, S.; Villar, S.; Gouas, D.; Ferro, G.; Plymoth, A.; Khuhaprema, T.; Kalalak, A.; Sangrajrang, S.; Friesen, M.D.; Groopman, J.D.; et al. Association between HBX status, aflatoxin-induced R249S TP53 mutation and risk of hepatocellular carcinoma in a case-control study from Thailand. Cancer Lett. 2013, 331, 46–51. [Google Scholar] [CrossRef]
- Villar, S.; Le Roux-Goglin, E.; Gouas, D.A.; Plymoth, A.; Ferro, G.; Boniol, M.; Lereau, M.; Bah, E.; Hall, A.J.; Wild, C.P.; et al. Seasonal variation in TP53 R249S-mutated serum DNA with aflatoxin exposure and hepatitis B virus infection. Environ. Health Perspect. 2011, 119, 1635–1640. [Google Scholar] [CrossRef] [PubMed]
- Kuniholm, M.H.; Lesi, O.A.; Mendy, M.; Akano, A.O.; Sam, O.; Hall, A.J.; Whittle, H.; Bah, E.; Goedert, J.J.; Hainaut, P.; et al. Aflatoxin exposure and viral hepatitis in the etiology of liver cirrhosis in the Gambia, West Africa. Environ. Health Perspect. 2008, 116, 1553–1557. [Google Scholar] [CrossRef]
- Qin, W.; Han, C.; Mai, R.; Yu, T.; Shang, L.; Ye, X.; Zhu, G.; Su, H.; Liao, X.; Liu, Z.; et al. Establishment of a prognostic model for predicting short-term disease-free survival in cases of hepatitis B-related hepatocellular carcinoma with the TP53 249Ser mutation in southern China. Transl. Cancer Res. 2020, 9, 4517–4533. [Google Scholar] [CrossRef]
- Valdés-Peregrina, E.N.; Sánchez-Hernández, B.E.; Gamboa-Domínguez, A. Metabolic Syndrome Instead of Aflatoxin-Related TP53 R249S Mutation as a Hepatocellular Carcinoma Risk Factor. Rev. Investig. Clin. 2020, 72, 127–134. [Google Scholar] [CrossRef]
- Levine, A.J. p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer 2020, 20, 471–480. [Google Scholar] [CrossRef]
- Hernández Borrero, L.J.; El-Deiry, W.S. Tumor suppressor p53: Biology, signaling pathways, and therapeutic targeting. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188556. [Google Scholar] [CrossRef] [PubMed]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef]
- Rivlin, N.; Brosh, R.; Oren, M.; Rotter, V. Mutations in the p53 Tumor Suppressor Gene: Important Milestones at the Various Steps of Tumorigenesis. Genes Cancer 2011, 2, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Okechukwu, V.O.; Adelusi, O.A.; Kappo, A.P.; Njobeh, P.B.; Mamo, M.A. Aflatoxins: Occurrence, biosynthesis, mechanism of action and effects, conventional/emerging detection techniques. Food Chem. 2024, 436, 137775. [Google Scholar] [CrossRef] [PubMed]
- Hamid, A.S.; Tesfamariam, I.G.; Zhang, Y.; Zhang, Z.G. Aflatoxin B1-induced hepatocellular carcinoma in developing countries: Geographical distribution, mechanism of action and prevention. Oncol. Lett. 2013, 5, 1087–1092. [Google Scholar] [CrossRef]
- Hsia, C.C.; Kleiner, D.E.; Axiotis, C.A.; Di Bisceglie, A.; Nomura, A.M.; Stemmermann, G.N.; Tabor, E. Mutations of p53 gene in hepatocellular carcinoma: Roles of hepatitis B virus and aflatoxin contamination in the diet. J. Natl. Cancer Inst. 1992, 84, 1638–1641. [Google Scholar] [CrossRef]
- Gramantieri, L.; Gnudi, F.; Vasuri, F.; Mandrioli, D.; Fornari, F.; Tovoli, F.; Suzzi, F.; Vornoli, A.; D’Errico, A.; Piscaglia, F.; et al. Aflatoxin B1 DNA-Adducts in Hepatocellular Carcinoma from a Low Exposure Area. Nutrients 2022, 14, 1652. [Google Scholar] [CrossRef]
- Smela, M.E.; Hamm, M.L.; Henderson, P.T.; Harris, C.M.; Harris, T.M.; Essigmann, J.M. The aflatoxin B(1) formamidopyrimidine adduct plays a major role in causing the types of mutations observed in human hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2002, 99, 6655–6660. [Google Scholar] [CrossRef]
- de Oliveira, C.A.; Germano, P.M. Aflatoxins: Current concepts on mechanisms of toxicity and their involvement in the etiology of hepatocellular carcinoma. Rev. Saude Publica 1997, 31, 417–424. [Google Scholar] [CrossRef]
- Coursaget, P.; Depril, N.; Chabaud, M.; Nandi, R.; Mayelo, V.; LeCann, P.; Yvonnet, B. High prevalence of mutations at codon 249 of the p53 gene in hepatocellular carcinomas from Senegal. Br. J. Cancer 1993, 67, 1395–1397. [Google Scholar] [CrossRef]
- Pineau, P.; Marchio, A.; Battiston, C.; Cordina, E.; Russo, A.; Terris, B.; Qin, L.X.; Turlin, B.; Tang, Z.Y.; Mazzaferro, V.; et al. Chromosome instability in human hepatocellular carcinoma depends on p53 status and aflatoxin exposure. Mutat. Res. 2008, 653, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.P.; Schwank, J.; Staib, F.; Wang, X.W.; Harris, C.C. TP53 mutations and hepatocellular carcinoma: Insights into the etiology and pathogenesis of liver cancer. Oncogene 2007, 26, 2166–2176. [Google Scholar] [CrossRef]
- Qi, L.N.; Bai, T.; Chen, Z.S.; Wu, F.X.; Chen, Y.Y.; De Xiang, B.; Peng, T.; Han, Z.G.; Li, L.Q. The p53 mutation spectrum in hepatocellular carcinoma from Guangxi, China: Role of chronic hepatitis B virus infection and aflatoxin B1 exposure. Liver Int. 2015, 35, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Gouas, D.A.; Shi, H.; Hautefeuille, A.H.; Ortiz-Cuaran, S.L.; Legros, P.C.; Szymanska, K.J.; Galy, O.; Egevad, L.A.; Abedi-Ardekani, B.; Wiman, K.G.; et al. Effects of the TP53 p.R249S mutant on proliferation and clonogenic properties in human hepatocellular carcinoma cell lines: Interaction with hepatitis B virus X protein. Carcinogenesis 2010, 31, 1475–1482. [Google Scholar] [CrossRef]
- Lee, Y.I.; Lee, S.; Das, G.C.; Park, U.S.; Park, S.M. Activation of the insulin-like growth factor II transcription by aflatoxin B1 induced p53 mutant 249 is caused by activation of transcription complexes; implications for a gain-of-function during the formation of hepatocellular carcinoma. Oncogene 2000, 19, 3717–3726. [Google Scholar] [CrossRef]
- Su, F.; Schneider, R.J. Hepatitis B virus HBx protein sensitizes cells to apoptotic killing by tumor necrosis factor alpha. Proc. Natl. Acad. Sci. USA 1997, 94, 8744–8749. [Google Scholar] [CrossRef]
- Lam, Y.K.; Yu, J.; Huang, H.; Ding, X.; Wong, A.M.; Leung, H.H.; Chan, A.W.; Ng, K.K.; Xu, M.; Wang, X.; et al. TP53 R249S mutation in hepatic organoids captures the predisposing cancer risk. Hepatology 2023, 78, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.; Zeng, S.X.; Zhou, X.; Chen, T.; Zhou, F.; Cao, B.; Jung, J.H.; Del Sal, G.; Luo, S.; Lu, H. Mutant p53 Gains Its Function via c-Myc Activation upon CDK4 Phosphorylation at Serine 249 and Consequent PIN1 Binding. Mol. Cell 2017, 68, 1134–1146.e6. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, Y.; Jang, H.; Nussinov, R. Cell cycle progression mechanisms: Slower cyclin-D/CDK4 activation and faster cyclin-E/CDK2. bioRxiv 2023. [Google Scholar] [CrossRef]
- Fasullo, M. Cellular Responses to Aflatoxin-Associated DNA Adducts. In DNA Repair; Mognato, M., Ed.; IntechOpen: London, UK, 2018. [Google Scholar]
- Dias, J.D.; Sarica, N.; Cournac, A.; Koszul, R.; Neuveut, C. Crosstalk between Hepatitis B Virus and the 3D Genome Structure. Viruses 2022, 14, 445. [Google Scholar] [CrossRef]
- Yeh, S.H.; Li, C.L.; Lin, Y.Y.; Ho, M.C.; Wang, Y.C.; Tseng, S.T.; Chen, P.J. Hepatitis B Virus DNA Integration Drives Carcinogenesis and Provides a New Biomarker for HBV-related HCC. Cell Mol. Gastroenterol. Hepatol. 2023, 15, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Han, Q.; Zhao, H.; Zhang, J. The Mechanisms of HBV-Induced Hepatocellular Carcinoma. J. Hepatocell. Carcinoma 2021, 8, 435–450. [Google Scholar] [CrossRef]
- Yen, C.J.; Lin, Y.J.; Yen, C.S.; Tsai, H.W.; Tsai, T.F.; Chang, K.Y.; Huang, W.C.; Lin, P.W.; Chiang, C.W.; Chang, T.T. Hepatitis B virus X protein upregulates mTOR signaling through IKKβ to increase cell proliferation and VEGF production in hepatocellular carcinoma. PLoS ONE 2012, 7, e41931. [Google Scholar] [CrossRef]
- Li, D.; Hamadalnil, Y.; Tu, T. Hepatitis B Viral Protein HBx: Roles in Viral Replication and Hepatocarcinogenesis. Viruses 2024, 16, 1361. [Google Scholar] [CrossRef] [PubMed]
- Sivasudhan, E.; Blake, N.; Lu, Z.; Meng, J.; Rong, R. Hepatitis B Viral Protein HBx and the Molecular Mechanisms Modulating the Hallmarks of Hepatocellular Carcinoma: A Comprehensive Review. Cells 2022, 11, 741. [Google Scholar] [CrossRef]
- Truant, R.; Antunovic, J.; Greenblatt, J.; Prives, C.; Cromlish, J.A. Direct interaction of the hepatitis B virus HBx protein with p53 leads to inhibition by HBx of p53 response element-directed transactivation. J. Virol. 1995, 69, 1851–1859. [Google Scholar] [CrossRef]
- Xian, L.; Zhao, J.; Wang, J.; Fang, Z.; Peng, B.; Wang, W.; Ji, X.; Yu, L. p53 Promotes proteasome-dependent degradation of oncogenic protein HBx by transcription of MDM2. Mol. Biol. Rep. 2010, 37, 2935–2940. [Google Scholar] [CrossRef] [PubMed]
- Elmore, L.W.; Hancock, A.R.; Chang, S.F.; Wang, X.W.; Chang, S.; Callahan, C.P.; Geller, D.A.; Will, H.; Harris, C.C. Hepatitis B virus X protein and p53 tumor suppressor interactions in the modulation of apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 14707–14712. [Google Scholar] [CrossRef]
- Lim, K.H.; Choi, H.S.; Park, Y.K.; Park, E.S.; Shin, G.C.; Kim, D.H.; Ahn, S.H.; Kim, K.H. HBx-induced NF-κB signaling in liver cells is potentially mediated by the ternary complex of HBx with p22-FLIP and NEMO. PLoS ONE 2013, 8, e57331. [Google Scholar] [CrossRef]
- Chen, L.; Lin, X.; Lei, Y.; Xu, X.; Zhou, Q.; Chen, Y.; Liu, H.; Jiang, J.; Yang, Y.; Zheng, F.; et al. Aerobic glycolysis enhances HBx-initiated hepatocellular carcinogenesis via NF-κBp65/HK2 signalling. J. Exp. Clin. Cancer Res. 2022, 41, 329. [Google Scholar] [CrossRef]
- Tu, T.; Budzinska, M.A.; Shackel, N.A.; Urban, S. HBV DNA Integration: Molecular Mechanisms and Clinical Implications. Viruses 2017, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.J.; Huang, T.; Wang, L.; Sun, X.F.; Zhang, Y.M. HBX suppresses PTEN to promote the malignant progression of hepatocellular carcinoma through mi-R155 activation. Ann. Hepatol. 2022, 27, 100688. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, R.; Wang, Y.; Luo, J.; Wang, P.; Cheng, B. Notch1 is a potential therapeutic target for the treatment of human hepatitis B virus X protein-associated hepatocellular carcinoma. Oncol. Rep. 2014, 31, 933–939. [Google Scholar] [CrossRef]
- Gao, W.Y.; Li, D.; Cai, D.E.; Huang, X.Y.; Zheng, B.Y.; Huang, Y.H.; Chen, Z.X.; Wang, X.Z. Hepatitis B virus X protein sensitizes HL-7702 cells to oxidative stress-induced apoptosis through modulation of the mitochondrial permeability transition pore. Oncol. Rep. 2017, 37, 48–56. [Google Scholar] [CrossRef]
- Xie, B.; Hao, Q.; Zhou, X.; Chen, D. Inactivation of tumor suppressor TAp63 by hepatitis B virus X protein in hepatocellular carcinoma. Chin. Med. J. 2022, 135, 1728–1733. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Gu, S.; Liang, J.; Lin, Z.; Zheng, S.; Yan, J. The Biological Function of Hepatitis B Virus X Protein in Hepatocellular Carcinoma. Oncol. Res. 2019, 27, 509–514. [Google Scholar] [CrossRef]
- Madden, C.R.; Finegold, M.J.; Slagle, B.L. Altered DNA mutation spectrum in aflatoxin b1-treated transgenic mice that express the hepatitis B virus x protein. J. Virol. 2002, 76, 11770–11774. [Google Scholar] [CrossRef]
- Becker, S.A.; Lee, T.H.; Butel, J.S.; Slagle, B.L. Hepatitis B virus X protein interferes with cellular DNA repair. J. Virol. 1998, 72, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Sekiba, K.; Otsuka, M.; Funato, K.; Miyakawa, Y.; Tanaka, E.; Seimiya, T.; Yamagami, M.; Tsutsumi, T.; Okushin, K.; Miyakawa, K.; et al. HBx-induced degradation of Smc5/6 complex impairs homologous recombination-mediated repair of damaged DNA. J. Hepatol. 2022, 76, 53–62. [Google Scholar] [CrossRef]
- Choudhary, H.B.; Mandlik, S.K.; Mandlik, D.S. Role of p53 suppression in the pathogenesis of hepatocellular carcinoma. World J. Gastrointest. Pathophysiol. 2023, 14, 46–70. [Google Scholar] [CrossRef]
- Farazi, P.A.; DePinho, R.A. Hepatocellular carcinoma pathogenesis: From genes to environment. Nat. Rev. Cancer 2006, 6, 674–687. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, G.E.M.; Cabibbo, G.; Craxì, A. Hepatitis B Virus-Associated Hepatocellular Carcinoma. Viruses 2022, 14, 986. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno-León, C.; Aguayo, F. Cooperation Between Aflatoxin-Induced p53 Aberrations and Hepatitis B Virus in Hepatocellular Carcinoma. J. Xenobiot. 2025, 15, 96. https://doi.org/10.3390/jox15040096
Moreno-León C, Aguayo F. Cooperation Between Aflatoxin-Induced p53 Aberrations and Hepatitis B Virus in Hepatocellular Carcinoma. Journal of Xenobiotics. 2025; 15(4):96. https://doi.org/10.3390/jox15040096
Chicago/Turabian StyleMoreno-León, Carolina, and Francisco Aguayo. 2025. "Cooperation Between Aflatoxin-Induced p53 Aberrations and Hepatitis B Virus in Hepatocellular Carcinoma" Journal of Xenobiotics 15, no. 4: 96. https://doi.org/10.3390/jox15040096
APA StyleMoreno-León, C., & Aguayo, F. (2025). Cooperation Between Aflatoxin-Induced p53 Aberrations and Hepatitis B Virus in Hepatocellular Carcinoma. Journal of Xenobiotics, 15(4), 96. https://doi.org/10.3390/jox15040096