
Environmental Xenobiotics and Epigenetic Modifications: Implications for Human Health and Disease

 ,
,  ,
,  ,
,  , and
, and 
Abstract

1. Introduction
2. Classes of Environmental Xenobiotics and Human Exposures
2.1. Heavy Metals
2.2. Endocrine-Disrupting Chemicals
2.3. Pesticides
2.4. Air Pollutants
2.5. Nano- and Microplastics
2.6. Mycotoxins and Phycotoxins
3. Environmental Xenobiotics and Their Impact on Epigenetic Regulation
4. Xenobiotic-Induced Epigenetic Changes in Disease Pathogenesis
4.1. Cancer
4.2. Neurodegenerative Diseases
4.3. Cardiovascular Diseases
4.4. Immune Disorders
5. Critical Windows of Susceptibility
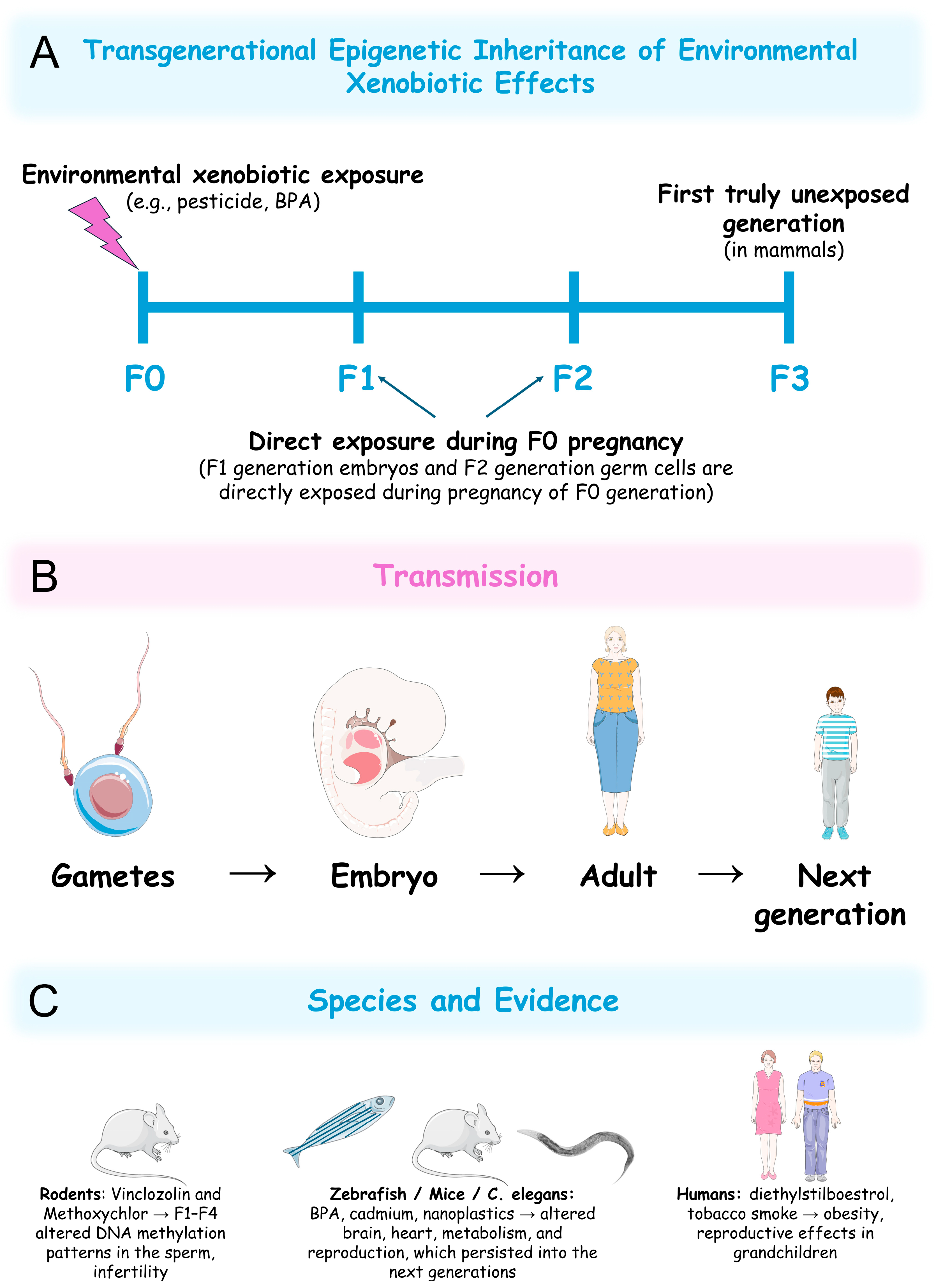
6. Transgenerational Epigenetic Effects of Xenobiotics
7. Reversibility of Xenobiotic-Induced Epigenetic Changes
8. Conclusions
9. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 2,4-D | 2,4-cichlorophenoxyacetic acid |
| 5mC | 5-Methylcytosine |
| Aβ | Amyloid beta |
| ACTA2 | Actin alpha 2 |
| AD | Alzheimer’s disease |
| ADP | Adenosine-5′-diphosphate |
| AKT | Protein kinase B (PKB) |
| ALSPAC | Avon longitudinal study of parents and children |
| Ano6 | Anoctamin 6 |
| Arap2 | Ankyrin repeat and PH domain 2 |
| ARTs | Adenosine-5′-diphosphate-ribosyltransferases |
| As | Arsenic |
| bdnf | Brain-derived neurotrophic factor |
| BPA | Bisphenol A |
| Cd | Cadmium |
| CDH13 | Cadherin 13 |
| CHRNA5 | Cholinergic receptor nicotinic alpha 5 subunit |
| CO | Carbon monoxide |
| CpG | Cytosine–phosphate–guanine |
| Cpne4 | Copine 4 |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| Cul2 | Cullin-2 |
| CVD | Cardiovascular diseases |
| DDT | Dichlorodiphenyltrichloroethane |
| DEHP | Di-(2-ethylhexyl) phthalate |
| DEPs | Diesel exhaust particles |
| dio3 | Deiodinase, iodothyronine type III |
| Dlg2 | discs large MAGUK scaffold protein 2 |
| DMPs | Differentially methylated positions |
| DMSO | Dimethyl sulfoxide |
| DNA | Deoxyribonucleic acid |
| DNMTs | Deoxyribonucleic acid methyltransferases |
| Dock3 | Dedicator of cytokinesis 3 |
| EDCs | Endocrine-disrupting chemicals |
| EPA | The U.S. Environmental Protection Agency |
| ESCs | Embryonic stem cells |
| ESCO1 | Establishment of cohesion 1 homologue 1 |
| EU | European Union |
| G9a | Euchromatic histone-lysine N-methyltransferase 2 |
| Gadd45 | Growth arrest and DNA damage-inducible 45 |
| GATA3 | GATA binding protein 3 |
| GBHs | glyphosate-based herbicides |
| GLAST | Astrocytic glutamate-aspartate transporter |
| GLT-1 | Glutamate transporter 1 |
| H2AK119Ub | Monoubiquitinated histone H2A at lysine 119 |
| H3K4me2 | Dimethylated histone H3 at lysine 4 |
| H3K4me3 | Trimethylated histone H3 at lysine 4 |
| H3K9 | Lysine 9 of histone H3 |
| H3K9ac | Acetylated histone H3 at lysine 9 |
| H3K9me2 | Dimethylated histone H3 at lysine 9 |
| H3K9me3 | Trimethylated histone H3 at lysine 9 |
| H3K14 | Lysine 14 of histone H3 |
| H3K27 | Lysine 27 of histone H3 |
| H3K27me3 | Trimethylated histone H3 at lysine 27 |
| H3K36me3 | Trimethylated histone H3 at lysine 36 |
| H3R2me2 | Dimethylated histone H3 at arginine 2 |
| H3S10ph | Phosphorylated histone H3 at serine 10 |
| H4K5ac | Acetylated histone H4 at lysine 5 |
| H4K20me2 | Dimethylated histone H4 at lysine 20 |
| H4K20me3 | Trimethylated histone H4 at lysine 20 |
| HATs | Histone acetyltransferases |
| HCB | Hexachlorobenzene |
| α-HCH | Alpha-hexachlorocyclohexane |
| β-HCH | Beta-hexachlorocyclohexane |
| HDAC2 | Histone deacetylase 2 |
| HDAC3 | Histone deacetylase 3 |
| HDACs | Histone deacetylases |
| Hells | Helicase, lymphoid specific |
| Hg | Mercury |
| IARC | International Agency for Research on Cancer |
| KAT2B | Lysine acetyltransferase 2B |
| KDM3A | Histone H3K9 demethylase lysine-specific demethylase 3A. |
| KDM5A | Histone H3K4 demethylase lysine-specific demethylase 5A |
| KDM5B | Histone H3K4 demethylase lysine-specific demethylase 5B |
| KLF6 | Kruppel-like factor 6 |
| LINE-1 | Long interspersed nuclear element-1 |
| lncRNA | Long non-coding ribonucleic acid |
| MBD2 | Methyl-CpG binding domain protein 2 |
| Mn | Manganese |
| mRNA | Messenger ribonucleic acid |
| miRNA | Micro ribonucleic acid |
| MPTP | 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MPP+ | 1-Methyl-4-phenylpyridinium |
| MSH5 | MutS homologue 5 |
| MPs | Microplastics |
| mTOR | Mammalian target of rapamycin |
| ncRNA | Non-coding ribonucleic acid |
| NFKB1 | Nuclear factor kappa B subunit 1 |
| NO | Nitric oxide |
| NO2 | Nitrogen dioxide |
| O3 | Ozone |
| Ogdh | Oxoglutarate dehydrogenase |
| OGG1 | 8-Oxoguanine deoxyribonucleic acid glycosylase 1 |
| NPs | Nanoplastics |
| p21 | Cyclin-dependent kinase inhibitor 1A |
| PADs | Peptidylarginine deiminases |
| PAHs | Polycyclic aromatic hydrocarbons |
| Pb | Lead |
| PBDEs | Polybrominated diphenyl ethers |
| PCBs | Polychlorinated biphenyls |
| PCDDs | Polychlorinated dibenzo-p-dioxins |
| PCDFs | Polychlorinated dibenzofurans |
| Pdpk1 | 3-Phosphoinositide-dependent protein kinase-1 |
| PFOS | Perfluorooctane sulfonic acid |
| PFOSF | Perfluorooctane sulfonyl fluoride |
| PD | Parkinson’s disease |
| PINK1 | PTEN-induced putative kinase 1 |
| PM | Particulate matter |
| POPs | persistent organic pollutants |
| PRMT6 | Protein arginine methyltransferase 6 |
| PTEN | Phosphatase and tensin homologue |
| PVC | polyvinyl chloride |
| R | Arginine |
| Rankl | Receptor activator of nuclear factor κB ligand |
| RC-LINE-1 | Retrotransposition-competent long interspersed nuclear element-1 |
| RNA | Ribonucleic acid |
| ROS | Reactive oxygen species |
| Rptor | Regulatory associated protein of mTOR complex 1 |
| SAM | S-adenosyl methionine |
| Sat2 | satellite 2 DNA |
| SETD8 | SET domain containing 8 or lysine methyltransferase 5A |
| Sirt1 | Sirtuin 1 |
| SLE | Systemic lupus erythematosus |
| SO2 | Sulphur dioxide |
| Spop | Speckle-type POZ protein |
| STK32A | Serine/threonine kinase 32A |
| SUMO | Small ubiquitin-like modifier |
| Tbc1d5 | TBC1 domain family member 5 |
| TCDD | 2,3,7,8-tetrachlorodibenzo-p-dioxin |
| TERT | Telomerase reverse transcriptase |
| TET | Ten-eleven translocation enzymes |
| TP53INP1 | Tumour protein p53 inducible nuclear protein 1 |
| Trap | Tartrate-resistant acid phosphatase |
| tRNA | Transfer RNA |
| TSA | Trichostatin A |
| USA | United States of America |
| VTI1A | Vesicle transport through interaction with T-SNAREs homologue 1A |
| WHO | World Health Organization |
| XIAP | X-linked inhibitor of apoptosis protein |
References
- Virolainen, S.J.; VonHandorf, A. Gene-environment interactions and their impact on human health. Genes. Immun. 2023, 24, 1–11. [Google Scholar] [CrossRef]
- Fu, W.; Guang, Y. Epidemiological perspectives on emerging contaminants and gout or hyperuricemia. Emerg. Contam. 2025, 11, 100485. [Google Scholar] [CrossRef]
- Briffa, J.; Sinagra, E. Heavy metal pollution in the environment and their toxicological effects on humans. Heliyon 2020, 6, e04691. [Google Scholar] [CrossRef]
- Metcalfe, C.D.; Bayen, S. An introduction to the sources, fate, occurrence and effects of endocrine disrupting chemicals released into the environment. Environ. Res. 2022, 207, 112658. [Google Scholar] [CrossRef] [PubMed]
- Marin, S.; Ramos, A.J. Mycotoxins: Occurrence, toxicology, and exposure assessment. Food Chem. Toxicol. 2013, 60, 218–237. [Google Scholar] [CrossRef] [PubMed]
- Pires, E.; Lana, P.D.C. Phycotoxins and marine annelids—A global review. Harmful Algae 2023, 122, 102373. [Google Scholar] [CrossRef]
- Pal, S.; Firdous, S.M. Unraveling the role of heavy metals xenobiotics in cancer: A critical review. Discov. Oncol. 2024, 15, 615. [Google Scholar] [CrossRef]
- Rock, K.D.; Patisaul, H.B. Environmental mechanisms of neurodevelopmental toxicity. Curr. Environ. Health Rep. 2018, 5, 145–157. [Google Scholar] [CrossRef]
- Zhang, J.; Jia, Q. The function of xenobiotic receptors in metabolic diseases. Drug Metab. Dispos. 2023, 51, 237–248. [Google Scholar] [CrossRef]
- Hussain, T.; Metwally, E. Redox mechanisms of environmental toxicants on male reproductive function. Front. Cell Dev. Biol. 2024, 12, 1333845. [Google Scholar] [CrossRef]
- Gibney, E.R.; Nolan, C.M. Epigenetics and gene expression. Heredity 2010, 105, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Bure, I.V.; Nemtsova, M.V. Histone modifications and non-coding RNAs: Mutual epigenetic regulation and role in pathogenesis. Int. J. Mol. Sci. 2022, 23, 5801. [Google Scholar] [CrossRef]
- Zhu, S.; Li, Z. VIRMA-mediated the m6A methylation of SCD facilitates wilms’ tumor progression via AMPK pathway. DNA Cell Biol. 2025, 44, 229–237. [Google Scholar] [CrossRef]
- Long, Z.Q.; Ding, R. Multi-omics characterization of genome-wide abnormal DNA methylation reveals FGF5 as a diagnosis of nasopharyngeal carcinoma recurrence after radiotherapy. Biomolecules 2025, 15, 283. [Google Scholar] [CrossRef] [PubMed]
- Rico-Méndez, M.A.; Trujillo-Rojas, M.A. MLH1 methylation status and microsatellite instability in patients with colorectal cancer. Genes 2025, 16, 182. [Google Scholar] [CrossRef]
- Zhang, R.; Nie, Y. A multicenter prospective clinical trial reveals cell free DNA methylation markers for early esophageal cancer. J. Clin. Investig. 2025, 135, e186816. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- McCabe, M.T.; Graves, A.P. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc. Natl. Acad. Sci. USA 2012, 109, 2989–2994. [Google Scholar] [CrossRef]
- Dai, Q.; Ye, Y. Development and validation of a novel histone acetylation-related gene signature for predicting the prognosis of ovarian cancer. Front. Cell Dev. Biol. 2022, 10, 793425. [Google Scholar] [CrossRef]
- Persico, G.; Casciaro, F. Histone H3 lysine 4 and 27 trimethylation landscape of human Alzheimer’s disease. Cells 2022, 11, 734. [Google Scholar] [CrossRef]
- Khazaei, S.; Chen, C.C.L. Single substitution in H3.3G34 alters DNMT3A recruitment to cause progressive neurodegeneration. Cell 2023, 186, 1162–1178.e20. [Google Scholar] [CrossRef]
- Shen, T.; Ji, F. Brain-specific deletion of histone variant H2A.z results in cortical neurogenesis defects and neurodevelopmental disorder. Nucleic Acids Res. 2018, 46, 2290–2307. [Google Scholar] [CrossRef]
- Lee, K.J.; Ahn, J.H. Non-coding RNA RMRP governs RAB31-dependent MMP secretion, enhancing ovarian cancer invasion. Biochim. Biophys. Acta Mol. Basis Dis. 2025, 1871, 167781. [Google Scholar] [CrossRef]
- Massone, S.; Ciarlo, E. NDM29, a RNA polymerase III-dependent non coding RNA, promotes amyloidogenic processing of APP and amyloid β secretion. Biochim. Biophys. Acta 2012, 1823, 1170–1177. [Google Scholar] [CrossRef]
- Senut, M.C.; Sen, A. Lead exposure disrupts global DNA methylation in human embryonic stem cells and alters their neuronal differentiation. Toxicol. Sci. 2014, 139, 142–161. [Google Scholar] [CrossRef] [PubMed]
- Demanelis, K.; Virani, S. Cadmium exposure and age-associated DNA methylation changes in non-smoking women from northern Thailand. Environ. Epigenet 2017, 3, dvx006. [Google Scholar] [CrossRef] [PubMed]
- Bailey, K.A.; Wu, M.C. Arsenic and the epigenome: Interindividual differences in arsenic metabolism related to distinct patterns of DNA methylation. J. Biochem. Mol. Toxicol. 2013, 27, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Deb, P.; Bhan, A. Endocrine disrupting chemical, bisphenol-A, induces breast cancer associated gene HOXB9 expression in vitro and in vivo. Gene 2016, 590, 234–243. [Google Scholar] [CrossRef]
- Koc, S.; Erdogmus, E. Prepubertal phthalate exposure can cause histopathological alterations, DNA methylation and histone acetylation changes in rat brain. Toxicol. Ind. Health 2025, 41, 163–175. [Google Scholar] [CrossRef]
- Schmitt, C.; Peterson, E. Transgenerational effects of developmental exposure to chlorpyrifos-oxon in zebrafish (DANIO RERIO). Toxicol. Appl. Pharmacol. 2020, 408, 115275. [Google Scholar] [CrossRef]
- Chiu, K.C.; Sisca, F. Prenatal chlorpyrifos exposure in association with PPARγ H3K4me3 and DNA methylation levels and child development. Environ. Pollut. 2021, 274, 116511. [Google Scholar] [CrossRef]
- Rossetti, M.F.; Canesini, G. Epigenetic changes associated with exposure to glyphosate-based herbicides in mammals. Front. Endocrinol. 2021, 12, 671991. [Google Scholar] [CrossRef] [PubMed]
- Kubsad, D.; Nilsson, E.E. Assessment of glyphosate induced epigenetic transgenerational inheritance of pathologies and sperm epimutations: Generational toxicology. Sci. Rep. 2019, 9, 6372. [Google Scholar] [CrossRef]
- Mukherjee, S.; Dasgupta, S. Air pollution-induced epigenetic changes: Disease development and a possible link with hypersensitivity pneumonitis. Environ. Sci. Pollut. Res. Int. 2021, 28, 55981–56002. [Google Scholar] [CrossRef] [PubMed]
- Krolevets, M.; Cate, V.T. DNA methylation and cardiovascular disease in humans: A systematic review and database of known CpG methylation sites. Clin. Epigenetics 2023, 15, 56. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Kang, H. A critical review on the bio-removal of hazardous heavy metals from contaminated soils: Issues, progress, eco-environmental concerns and opportunities. J. Hazard. Mater. 2010, 174, 1–8. [Google Scholar] [CrossRef]
- Pacyna, E.; Pacyna, J. Current and future emissions of selected heavy metals to the atmosphere from anthropogenic sources in Europe. Atmos. Environ. 2007, 41, 8557–8566. [Google Scholar] [CrossRef]
- Mitra, S.; Chakraborty, A.J. Impact of heavy metals on the environment and human health: Novel therapeutic insights to counter the toxicity. J. King Saud. Univ. Sci. 2022, 34, 101865. [Google Scholar] [CrossRef]
- Perrelli, M.; Wu, R. Heavy metals as risk factors for human diseases—A Bayesian network approach. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 9275–9310. [Google Scholar]
- Xu, J.; Bravo, A.G. Sources and remediation techniques for mercury contaminated soil. Environ. Int. 2015, 74, 42–53. [Google Scholar] [CrossRef]
- Gochfeld, M. Cases of mercury exposure, bioavailability, and absorption. Ecotoxicol. Environ. Saf. 2003, 56, 174–179. [Google Scholar] [CrossRef]
- Hightower, J.M.; O’Hare, A. Blood mercury reporting in NHANES: Identifying Asian, Pacific Islander, Native American, and multiracial groups. Environ. Health Perspect. 2006, 114, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Bellanger, M.; Pichery, C. Economic benefits of methylmercury exposure control in Europe: Monetary value of neurotoxicity prevention. Environ. Health 2013, 12, 3. [Google Scholar] [CrossRef] [PubMed]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Arsenic, metals, fibres, and dusts. IARC Monogr. Eval. Carcinog. Risks Hum. 2012, 100, 11–465. [Google Scholar]
- Frisbie, S.H.; Mitchell, E.J. Arsenic in drinking water: An analysis of global drinking water regulations and recommendations for updates to protect public health. PLoS ONE 2022, 17, e0263505. [Google Scholar] [CrossRef]
- Chakraborty, M.; Mukherjee, A. A Review of groundwater arsenic in the Bengal basin, Bangladesh and India: From source to sink. Curr. Pollut. Rep. 2015, 1, 220–247. [Google Scholar] [CrossRef]
- Genchi, G.; Sinicropi, M.S. The effects of cadmium toxicity. Int. J. Environ. Res. Public Health 2020, 17, 3782. [Google Scholar] [CrossRef]
- Rehman, K.; Fatima, F. Prevalence of exposure of heavy metals and their impact on health consequences. J. Cell Biochem. 2018, 119, 157–184. [Google Scholar] [CrossRef]
- Lone, I.; Saleem, S. Heavy metal contents of vegetables irrigated by sewage/tubewell water. Int. J. Agric. Biol. 2003, 5, 533–535. [Google Scholar]
- World Health Organization. Cadmium in Drinking-Water: Background Document for Development of WHO Guidelines for Drinking-Water Quality (WHO/SDE/WSH/03.04/80/Rev/1). Available online: https://cdn.who.int/media/docs/default-source/wash-documents/wash-chemicals/cadmium.pdf?sfvrsn=4dd545bd_4 (accessed on 15 March 2025).
- Casals-Casas, C.; Desvergne, B. Endocrine disruptors: From endocrine to metabolic disruption. Annu. Rev. Physiol. 2011, 73, 135–162. [Google Scholar] [CrossRef]
- Diamanti-Kandarakis, E.; Bourguignon, J.P. Endocrine-disrupting chemicals: An Endocrine Society scientific statement. Endocr. Rev. 2009, 30, 293–342. [Google Scholar] [CrossRef]
- Vandenberg, L.N.; Maffini, M.V. Bisphenol-A and the great divide: A review of controversies in the field of endocrine disruption. Endocr. Rev. 2009, 30, 75–95. [Google Scholar] [CrossRef] [PubMed]
- Geens, T.; Goeyens, L. Are potential sources for human exposure to bisphenol-A overlooked? Int. J. Hyg. Environ. Health 2011, 214, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, L.N.; Hauser, R. Human exposure to bisphenol A (BPA). Reprod. Toxicol. 2007, 24, 139–177. [Google Scholar] [CrossRef] [PubMed]
- Covaci, A.; Den Hond, E. Urinary BPA measurements in children and mothers from six European member states: Overall results and determinants of exposure. Environ. Res. 2015, 141, 77–85. [Google Scholar] [CrossRef]
- Adoamnei, E.; Mendiola, J. Urinary bisphenol A concentrations are associated with reproductive parameters in young men. Environ. Res. 2018, 161, 122–128. [Google Scholar] [CrossRef]
- He, Y.; Miao, M. Occupational exposure levels of bisphenol A among Chinese workers. J. Occup. Health 2009, 51, 432–436. [Google Scholar] [CrossRef]
- Mariana, M.; Castelo-Branco, M. Phthalates’ exposure leads to an increasing concern on cardiovascular health. J. Hazard. Mater. 2023, 457, 131680. [Google Scholar] [CrossRef]
- Koch, H.M.; Preuss, R. Di(2-ethylhexyl)phthalate (DEHP): Human metabolism and internal exposure—An update and latest results. Int. J. Androl. 2006, 29, 155–165. [Google Scholar] [CrossRef]
- Hoppin, J.A.; Brock, J.W. Reproducibility of urinary phthalate metabolites in first morning urine samples. Environ. Health Perspect. 2002, 110, 515–518. [Google Scholar] [CrossRef]
- Silva, M.J.; Barr, D.B. Urinary levels of seven phthalate metabolites in the U.S. population from the National Health and Nutrition Examination Survey (NHANES) 1999–2000. Environ. Health Perspect. 2004, 112, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Karlaganis, G.; Marioni, R. The elaboration of the ‘Stockholm convention’ on persistent organic pollutants (POPs): A negotiation process fraught with obstacles and opportunities. Environ. Sci. Pollut. Res. Int. 2001, 8, 216–221. [Google Scholar] [CrossRef]
- Wöhrnschimmel, H.; MacLeod, M. Emissions, fate and transport of persistent organic pollutants to the Arctic in a changing global climate. Environ. Sci. Technol. 2013, 47, 2323–2330. [Google Scholar] [CrossRef]
- Brigden, K.; Labunska, I.; Santillo, D.; Allsopp, M. Recycling of Electronic Wastes in China and India: Workplace and Environment Contamination; Greenpeace International: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Marinković, N.; Pašalić, D. Dioxins and human toxicity. Arh. Hig. Rada Toksikol. 2010, 61, 445–453. [Google Scholar] [CrossRef] [PubMed]
- McKay, G. Dioxin characterisation, formation and minimisation during municipal solid waste (MSW) incineration: Review. Chem. Eng. J. 2002, 86, 343–368. [Google Scholar] [CrossRef]
- Lorber, M.; Patterson, D. Evaluation of background exposures of Americans to dioxin-like compounds in the 1990s and the 2000s. Chemosphere 2009, 77, 640–651. [Google Scholar] [CrossRef] [PubMed]
- Sorg, O.; Zennegg, M. 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) poisoning in Victor Yushchenko: Identification and measurement of TCDD metabolites. Lancet 2009, 374, 1179–1185. [Google Scholar] [CrossRef]
- Geusau, A.; Abraham, K. Severe 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) intoxication: Clinical and laboratory effects. Environ. Health Perspect. 2001, 109, 865–869. [Google Scholar] [CrossRef]
- Holt, J.S. Herbicides. In Encyclopedia of Biodiversity, 2nd ed.; Levin, S.A., Ed.; Academic Press: Waltham, MA, USA, 2013; pp. 87–95. [Google Scholar]
- Chaufan, G.; Coalova, I. Glyphosate commercial formulation causes cytotoxicity, oxidative effects, and apoptosis on human cells: Differences with its active ingredient. Int. J. Toxicol. 2014, 33, 29–38. [Google Scholar] [CrossRef]
- Battaglin, W.A.; Meyer, M.T. Glyphosate and its degradation product AMPA occur frequently and widely in U.S. soils, surface water, groundwater, and precipitation. J. Am. Water Resour. Assoc. 2014, 50, 275–290. [Google Scholar] [CrossRef]
- Conrad, A.; Schröter-Kermani, C. Glyphosate in German adults—Time trend (2001 to 2015) of human exposure to a widely used herbicide. Int. J. Hyg. Environ. Health 2017, 220, 8–16. [Google Scholar] [CrossRef]
- Krueger, M.; Schrödl, W. Detection of glyphosate in malformed piglets. J. Environ. Anal. Toxicol. 2014, 04, 230. [Google Scholar] [CrossRef]
- Acquavella, J.F.; Alexander, B.H. Glyphosate biomonitoring for farmers and their families: Results from the Farm Family Exposure Study. Environ. Health Perspect. 2004, 112, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Van Bruggen, A.H.C.; He, M.M. Environmental and health effects of the herbicide glyphosate. Sci. Total Environ. 2018, 616–617, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, H. Signaling pathways involved in paraquat-induced pulmonary toxicity: Molecular mechanisms and potential therapeutic drugs. Int. Immunopharmacol. 2022, 113, 109301. [Google Scholar] [CrossRef]
- Buendía, J.A.; Chavarriaga, G.J.R. Burden of paraquat poisoning in the department of Antioquia, Colombia. BMC Pharmacol. Toxicol. 2019, 20, 11. [Google Scholar] [CrossRef]
- Dinis-Oliveira, R.J.; Duarte, J.A. Paraquat poisonings: Mechanisms of lung toxicity, clinical features, and treatment. Crit. Rev. Toxicol. 2008, 38, 13–71. [Google Scholar] [CrossRef]
- Greaves, A.K.; Letcher, R.J. Comparative body compartment composition and in ovo transfer of organophosphate flame retardants in North American Great Lakes herring gulls. Environ. Sci. Technol. 2014, 48, 7942–7950. [Google Scholar] [CrossRef]
- Agarwal, A.; Prajapati, R. Pesticide residue in water—A challenging task in India. Environ. Monit. Assess. 2015, 187, 54. [Google Scholar] [CrossRef]
- Masiá, A.; Campo, J. Pesticide monitoring in the basin of Llobregat River (Catalonia, Spain) and comparison with historical data. Sci. Total Environ. 2015, 503–504, 58–68. [Google Scholar] [CrossRef]
- Hou, R.; Xu, Y. Review of OPFRs in animals and humans: Absorption, bioaccumulation, metabolism, and internal exposure research. Chemosphere 2016, 153, 78–90. [Google Scholar] [CrossRef] [PubMed]
- U.S. EPA. Air Quality Criteria for Particulate Matter; US Environmental Protection Agency, Research Triangle Park: Gaithersburg, MD, USA, 2004.
- Ghio, A.J.; Smith, C.B. Diesel exhaust particles and airway inflammation. Curr. Opin. Pulm. Med. 2012, 18, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Benbrahim-Tallaa, L.; Baan, R.A. Carcinogenicity of diesel-engine and gasoline-engine exhausts and some nitroarenes. Lancet Oncol. 2012, 13, 663–664. [Google Scholar] [CrossRef] [PubMed]
- Kittelson, D.B. Engines and nanoparticles: A review. J. Aerosol Sci. 1998, 29, 575–588. [Google Scholar] [CrossRef]
- Hellier, P.; Ladommatos, N. The influence of biodiesel composition on compression ignition combustion and emissions. Proc. Inst. Mech. Eng. Part A J. Power Energy 2015, 229, 714–726. [Google Scholar] [CrossRef]
- Hellier, P.; Talibi, M. An overview of the effects of fuel molecular structure on the combustion and emissions characteristics of compression ignition engines. Proc. Inst. Mech. Eng. Part D 2018, 232, 90–105. [Google Scholar] [CrossRef]
- Swanson, K.J.; Madden, M.C. Biodiesel exhaust: The need for health effects research. Environ. Health Perspect. 2007, 115, 496–499. [Google Scholar] [CrossRef]
- Ogbunuzor, C.; Fransen, L.F.H. Biodiesel exhaust particle airway toxicity and the role of polycyclic aromatic hydrocarbons. Ecotoxicol. Environ. Saf. 2023, 259, 115013. [Google Scholar] [CrossRef]
- Hawrot-Paw, M.; Koniuszy, A. Ecotoxicity of soil contaminated with diesel fuel and biodiesel. Sci. Rep. 2020, 10, 16436. [Google Scholar] [CrossRef]
- Savadkoohi, M.; Pandolfi, M. Recommendations for reporting equivalent black carbon (eBC) mass concentrations based on long-term pan-European in-situ observations. Environ. Int. 2024, 185, 108553. [Google Scholar] [CrossRef]
- Bond, T.C.; Doherty, S.J. Bounding the role of black carbon in the climate system: A scientific assessment. J. Geophys. Res. 2013, 118, 5380–5552. [Google Scholar] [CrossRef]
- Zhang, Z.; Cheng, Y. The measurement of atmospheric black carbon: A review. Toxics 2023, 11, 975. [Google Scholar] [CrossRef]
- Zhu, X.; Liu, B. Short and long-term association of exposure to ambient black carbon with all-cause and cause-specific mortality: A systematic review and meta-analysis. Environ. Pollut. 2023, 324, 121086. [Google Scholar] [CrossRef]
- Patel, A.B.; Shaikh, S. Polycyclic aromatic hydrocarbons: Sources, toxicity, and remediation approaches. Front. Microbiol. 2020, 11, 562813. [Google Scholar] [CrossRef]
- Mojiri, A.; Zhou, J.L. Comprehensive review of polycyclic aromatic hydrocarbons in water sources, their effects and treatments. Sci. Total Environ. 2019, 696, 133971. [Google Scholar] [CrossRef] [PubMed]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Tobacco smoke and involuntary smoking. IARC Monogr. Eval. Carcinog. Risks Hum. 2004, 83, 1–1438.
- Yershova, K.; Yuan, J.M. Tobacco-specific N-nitrosamines and polycyclic aromatic hydrocarbons in cigarettes smoked by the participants of the Shanghai Cohort Study. Int. J. Cancer 2016, 139, 1261–1269. [Google Scholar] [CrossRef]
- Vaz-Ramos, J.; Le Calvé, S. Polycyclic aromatic hydrocarbons in water environments: Impact, legislation, depollution processes and challenges, and magnetic iron oxide/graphene-based nanocomposites as promising adsorbent solutions. J. Hazard. Mater. 2025, 490, 137726. [Google Scholar] [CrossRef]
- Manoli, E.; Samara, C. Polycyclic aromatic hydrocarbons in natural waters: Sources, occurrence and analysis. Trac-Trend Anal. Chem. 1999, 18, 417–428. [Google Scholar] [CrossRef]
- WHO. WHO Guidelines for Indoor Air Quality: Selected Pollutants; WHO Regional Office for Europe: Copenhagen, Denmark, 2010. [Google Scholar]
- Mallah, M.A.; Changxing, L. Polycyclic aromatic hydrocarbon and its effects on human health: An overeview. Chemosphere 2022, 296, 133948. [Google Scholar] [CrossRef]
- Ramsperger, A.; Bergamaschi, E. Nano- and microplastics: A comprehensive review on their exposure routes, translocation, and fate in humans. NanoImpact 2023, 29, 100441. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, N.B.; Hüffer, T. Are we speaking the same language? Recommendations for a definition and categorization framework for plastic debris. Environ. Sci. Technol. 2019, 53, 1039–1047. [Google Scholar] [CrossRef]
- Barnes, D.K.; Galgani, F. Accumulation and fragmentation of plastic debris in global environments. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 1985–1998. [Google Scholar] [CrossRef] [PubMed]
- Râpă, M.; Darie-Niță, R.N. Insights into anthropogenic micro- and nanoplastic accumulation in drinking water sources and their potential effects on human health. Polymers 2023, 15, 2425. [Google Scholar] [CrossRef]
- EFSA Panel on Contaminants in the Food Chain. Presence of microplastics and nanoplastics in food, with particular focus on seafood. EFSA J. 2016, 14, e04501. [Google Scholar]
- Wright, S.L.; Ulke, J. Atmospheric microplastic deposition in an urban environment and an evaluation of transport. Environ. Int. 2020, 136, 105411. [Google Scholar] [CrossRef]
- Amato-Lourenço, L.F.; Carvalho-Oliveira, R. Presence of airborne microplastics in human lung tissue. J. Hazard. Mater. 2021, 416, 126124. [Google Scholar] [CrossRef]
- Mou, L.; Wu, C. Rapid detection of microplastics/nanoplastics directly exposed to blood during intravenous injections via mie scattering spectra. J. Hazard. Mater. 2024, 480, 136193. [Google Scholar] [CrossRef]
- Rezaei Kahkha, M.R.; Piri, J. Investigation of heavy metals adsorbed on microplastics in drinking water and water resources of Zabol. Sci. Rep. 2025, 15, 14378. [Google Scholar] [CrossRef]
- Lan, T.; Wang, T. A comparative study on the adsorption behavior of pesticides by pristine and aged microplastics from agricultural polyethylene soil films. Ecotoxicol. Environ. Saf. 2021, 209, 111781. [Google Scholar] [CrossRef]
- Llorca, M.; Schirinzi, G. Adsorption of perfluoroalkyl substances on microplastics under environmental conditions. Environ. Pollut. 2018, 235, 680–691. [Google Scholar] [CrossRef]
- Berber, A.A.; Akinci Kenanoğlu, N. Genotoxic and cytotoxic effects of polystyrene nanoplastics on human lymphocytes: A comprehensive analysis. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2025, 902, 503850. [Google Scholar] [CrossRef]
- Pontecorvi, P.; Ceccarelli, S. Assessing the impact of polyethylene nano/microplastic exposure on human vaginal keratinocytes. Int. J. Mol. Sci. 2023, 24, 11379. [Google Scholar] [CrossRef]
- Niaz, W.; Iqbal, S.Z. Mycotoxins: A comprehensive review of its global trends in major cereals, advancements in chromatographic detections and future prospectives. Food Chem. X 2025, 27, 102350. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, R.; Crisci, A. Occurrence and co-occurrence of mycotoxins in cereal-based feed and food. Microorganisms 2020, 8, 74. [Google Scholar] [CrossRef]
- Ahmad, M.M.; Ahmad, M. Detection of Aspergillus flavus and Aspergillus parasiticus from aflatoxin-contaminated peanuts and their differentiation using PCR-RFLP. Ann. Microbiol. 2014, 64, 1597–1605. [Google Scholar] [CrossRef]
- Marchese, S.; Polo, A. Aflatoxin B1 and M1: Biological properties and their involvement in cancer development. Toxins 2018, 10, 214. [Google Scholar] [CrossRef]
- European Commission. Commission regulation (EU) 2023/915 of 25 April 2023 on maximum levels for certain contaminants in food and repealing regulation (EC) no 1881/2006. Off. J. Eur. Union L 2023, 119, 103–157. [Google Scholar]
- Kaale, L.D.; Kimanya, M.E. Aflatoxin contamination and recommendations to improve its control: A review. World Mycotoxin J. 2021, 14, 27–40. [Google Scholar] [CrossRef]
- Bredenkamp, M.W.; Dillen, J.L.M. Crystal structures and conformational analysis of ochratoxin A and B: Probing the chemical structure causing toxicity. J. Chem. Soc. Perkin Trans 1989, 2, 1835–1839. [Google Scholar] [CrossRef]
- Echodu, R.; Maxwell Malinga, G. Prevalence of aflatoxin, ochratoxin and deoxynivalenol in cereal grains in northern Uganda: Implication for food safety and health. Toxicol. Rep. 2019, 6, 1012–1017. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Dahal, S. Reduction of ochratoxin A in oat flakes by twin-screw extrusion processing. J. Food Prot. 2017, 80, 1628–1634. [Google Scholar] [CrossRef]
- Domijan, A.M. Fumonisin B(1): A neurotoxic mycotoxin. Arh. Hig. Rada Toksikol. 2012, 63, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Azam, M.S.; Ahmed, S. Critical assessment of mycotoxins in beverages and their control measures. Toxins 2021, 13, 323. [Google Scholar] [CrossRef]
- Rogowska, A.; Pomastowski, P. Zearalenone and its metabolites: Effect on human health, metabolism and neutralisation methods. Toxicon 2019, 162, 46–56. [Google Scholar] [CrossRef]
- Luo, Y.; Liu, X. Complicated interactions between bio-adsorbents and mycotoxins during mycotoxin adsorption: Current research and future prospects. Trends Food Sci. Technol. 2020, 96, 127–134. [Google Scholar] [CrossRef]
- López-Díaz, C.; Rahjoo, V. Fusaric acid contributes to virulence of Fusarium oxysporum on plant and mammalian hosts. Mol. Plant Pathol. 2018, 19, 440–453. [Google Scholar] [CrossRef]
- Stipanovic, R.D.; Puckhaber, L.S. Phytotoxicity of fusaric acid and analogs to cotton. Toxicon 2011, 57, 176–178. [Google Scholar] [CrossRef]
- Porter, J.K.; Bacon, C.W. Fusaric acid in Fusarium moniliforme cultures, corn, and feeds toxic to livestock and the neurochemical effects in the brain and pineal gland of rats. Nat. Toxins 1995, 3, 91–100. [Google Scholar] [CrossRef]
- Tang, J.; He, X. Occurrence and distribution of phycotoxins in the Antarctic Ocean. Mar. Pollut. Bull. 2024, 201, 116250. [Google Scholar] [CrossRef]
- Salter, C.; VanMensel, D. Investigating the microbial dynamics of microcystin-LR degradation in Lake Erie sand. Chemosphere 2021, 272, 129873. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.; Mou, X. A brief review of the structure, cytotoxicity, synthesis, and biodegradation of microcystins. Water 2021, 13, 2147. [Google Scholar] [CrossRef]
- Chen, L.; Giesy, J.P. The dose makes the poison. Sci. Total Environ. 2018, 621, 649–653. [Google Scholar] [CrossRef]
- Valdiglesias, V.; Prego-Faraldo, M.V. Okadaic acid: More than a diarrheic toxin. Mar. Drugs 2013, 11, 4328–4349. [Google Scholar] [CrossRef] [PubMed]
- Authority, E.F.S. Marine biotoxins in shellfish—Okadaic acid and analogues—Scientific Opinion of the Panel on Contaminants in the Food chain. EFSA J. 2008, 6, 589. [Google Scholar]
- Dietrich, J.; Sommersdorf, C. Okadaic acid activates Wnt/β-catenin-signaling in human HepaRG cells. Arch. Toxicol. 2019, 93, 1927–1939. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, W. Microcystin-leucine-arginine affects brain gene expression programs and behaviors of offspring through paternal epigenetic information. Sci. Total Environ. 2023, 857, 159032. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Huang, K. Aflatoxin B1-induced epigenetic alterations: An overview. Food Chem. Toxicol. 2017, 109, 683–689. [Google Scholar] [CrossRef]
- Ferreira, R.G.; Cardoso, M.V. Epigenetic alterations caused by aflatoxin b1: A public health risk in the induction of hepatocellular carcinoma. Transl. Res. 2019, 204, 51–71. [Google Scholar] [CrossRef]
- Karaman, E.F.; Zeybel, M. Evaluation of the epigenetic alterations and gene expression levels of HepG2 cells exposed to zearalenone and α-zearalenol. Toxicol. Lett. 2020, 326, 52–60. [Google Scholar] [CrossRef]
- Li, X.; Gao, J. Dynamic changes of global DNA methylation and hypermethylation of cell adhesion-related genes in rat kidneys in response to ochratoxin A. World Mycotoxin J. 2015, 8, 465–476. [Google Scholar] [CrossRef]
- Soni, P.; Ghufran, M.S. Epigenetic alterations induced by aflatoxin B(1): An in vitro and in vivo approach with emphasis on enhancer of zeste homologue-2/p21 axis. Sci. Total Environ. 2021, 762, 143175. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.C.; Wang, Q. Global DNA methylation in a population with aflatoxin B1 exposure. Epigenetics 2013, 8, 962–969. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Qin, W. Alterations in microRNA expression linked to microcystin-LR-induced tumorigenicity in human WRL-68 Cells. Mutat. Res. 2012, 743, 75–82. [Google Scholar] [CrossRef]
- Zamudio, N.; Barau, J. DNA methylation restrains transposons from adopting a chromatin signature permissive for meiotic recombination. Genes. Dev. 2015, 29, 1256–1270. [Google Scholar] [CrossRef]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef] [PubMed]
- Sharp, A.J.; Stathaki, E. DNA methylation profiles of human active and inactive X chromosomes. Genome Res. 2011, 21, 1592–1600. [Google Scholar] [CrossRef]
- Lee, A.V.; Nestler, K.A. Therapeutic targeting of DNA methylation alterations in cancer. Pharmacol. Ther. 2024, 258, 108640. [Google Scholar] [CrossRef]
- Luo, C.; Hajkova, P. Dynamic DNA methylation: In the right place at the right time. Science 2018, 361, 1336–1340. [Google Scholar] [CrossRef]
- Ai, T.; Zhang, J. DNA methylation profile is associated with the osteogenic potential of three distinct human odontogenic stem cells. Signal Transduct. Target. Ther. 2018, 3, 1. [Google Scholar] [CrossRef]
- Younesian, S.; Yousefi, A.M. The DNA methylation in neurological diseases. Cells 2022, 11, 3439. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.; Sun, D. Large conserved domains of low DNA methylation maintained by Dnmt3a. Nat. Genet. 2014, 46, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.; Marks, L. The genetic basis of disease. Essays Biochem. 2018, 62, 643–723. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Jeltsch, A.; Ehrenhofer-Murray, A. Mechanism and biological role of Dnmt2 in nucleic acid Methylation. RNA Biol. 2017, 14, 1108–1123. [Google Scholar] [CrossRef]
- Weber, M.; Davies, J.J. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat. Genet. 2005, 37, 853–862. [Google Scholar] [CrossRef]
- Sergeeva, A.; Davydova, K. Mechanisms of human DNA methylation, alteration of methylation patterns in physiological processes and oncology. Gene 2023, 875, 147487. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef]
- Lee, J.-H.; Park, S.-J. Differential landscape of non-CpG methylation in embryonic stem cells and neurons caused by DNMT3s. Sci. Rep. 2017, 7, 11295. [Google Scholar] [CrossRef]
- Messerschmidt, D.M.; Knowles, B.B. DNA methylation dynamics during epigenetic reprogramming in the germline and preimplantation embryos. Genes. Dev. 2014, 28, 812–828. [Google Scholar] [CrossRef]
- Takiguchi, M.; Achanzar, W.E. Effects of cadmium on DNA-(Cytosine-5) methyltransferase activity and DNA methylation status during cadmium-induced cellular transformation. Exp. Cell Res. 2003, 286, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Freydenzon, A.; Nabais, M.F. Association between DNA methylation variability and self-reported exposure to heavy metals. Sci. Rep. 2022, 12, 10582. [Google Scholar] [CrossRef] [PubMed]
- Bihaqi, S.W. Early life exposure to lead (Pb) and changes in DNA methylation: Relevance to Alzheimer’s disease. Rev. Environ. Health 2019, 34, 187–195. [Google Scholar] [CrossRef]
- Stepanyan, A.; Petrackova, A. Long-term environmental metal exposure is associated with hypomethylation of CpG sites in NFKB1 and other genes related to oncogenesis. Clin. Epigenetics 2023, 15, 126. [Google Scholar] [CrossRef]
- de Gannes, M.; Ko, C.I. Dioxin disrupts dynamic DNA methylation patterns in genes that govern cardiomyocyte maturation. Toxicol. Sci. 2020, 178, 325–337. [Google Scholar] [CrossRef]
- Kelsey, K.T.; Rytel, M. Serum dioxin and DNA methylation in the sperm of operation ranch hand veterans exposed to Agent Orange. Environ. Health 2019, 18, 91. [Google Scholar] [CrossRef]
- Khodasevich, D.; Holland, N. Prenatal exposure to environmental phenols and phthalates and altered patterns of DNA methylation in childhood. Environ. Int. 2024, 190, 108862. [Google Scholar] [CrossRef]
- Li, W.; Guo, L. Phthalates and phthalate metabolites in urine from Tianjin and implications for platelet mitochondrial DNA methylation. Front. Public Health 2023, 11, 1108555. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Sartor, M.A. Perinatal bisphenol A exposure promotes dose-dependent alterations of the mouse methylome. BMC Genom. 2014, 15, 30. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, Y. Tobacco smoking and methylation of genes related to lung cancer development. Oncotarget 2016, 7, 59017–59028. [Google Scholar] [CrossRef]
- Stading, R.; Gastelum, G. Molecular mechanisms of pulmonary carcinogenesis by polycyclic aromatic hydrocarbons (PAHs): Implications for human lung cancer. Semin. Cancer Biol. 2021, 76, 3–16. [Google Scholar] [CrossRef]
- Ye, F.; Xu, X.C. Benzo[a]pyrene diol epoxide suppresses retinoic acid receptor-beta2 expression by recruiting DNA (cytosine-5-)-methyltransferase 3A. Mol. Cancer 2010, 9, 93. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Kim, M.S. Nanoplastics induce epigenetic signatures of transgenerational impairments associated with reproduction in copepods under ocean acidification. J. Hazard. Mater. 2023, 449, 131037. [Google Scholar] [CrossRef] [PubMed]
- Braham, M.Y.; Franchi, A. Fatal 4-MEC intoxication: Case report and review of literature. Am. J. Forensic Med. Pathol. 2021, 42, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Rieswijk, L.; Claessen, S.M. Aflatoxin B1 induces persistent epigenomic effects in primary human hepatocytes associated with hepatocellular carcinoma. Toxicology 2016, 350–352, 31–39. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Q.C. Aflatoxin B1 is toxic to porcine oocyte maturation. Mutagenesis 2015, 30, 527–535. [Google Scholar] [CrossRef]
- Kharboush, T.G.; Ahmed, I.A. Epigenetic alterations of miR-155 and global DNA methylation as potential mediators of ochratoxin A cytotoxicity and carcinogenicity in human lung fibroblasts. Environ. Sci. Pollut. Res. Int. 2024, 31, 5473–5483. [Google Scholar] [CrossRef]
- Ozden, S.; Turgut Kara, N. Assessment of global and gene-specific DNA methylation in rat liver and kidney in response to non-genotoxic carcinogen exposure. Toxicol. Appl. Pharmacol. 2015, 289, 203–212. [Google Scholar] [CrossRef]
- Zheng, J.; Zhang, Y. Zinc protects HepG2 cells against the oxidative damage and DNA damage induced by ochratoxin A. Toxicol. Appl. Pharmacol. 2013, 268, 123–131. [Google Scholar] [CrossRef]
- Zhu, C.C.; Hou, Y.J. Effect of mycotoxin-containing diets on epigenetic modifications of mouse oocytes by fluorescence microscopy analysis. Microsc. Microanal. 2014, 20, 1158–1166. [Google Scholar] [CrossRef]
- Karaman, E.F.; Ozden, S. Alterations in global DNA methylation and metabolism-related genes caused by zearalenone in MCF7 and MCF10F cells. Mycotoxin Res. 2019, 35, 309–320. [Google Scholar] [CrossRef]
- So, M.Y.; Tian, Z. Gene expression profile and toxic effects in human bronchial epithelial cells exposed to zearalenone. PLoS ONE 2014, 9, e96404. [Google Scholar] [CrossRef] [PubMed]
- Kouadio, J.H.; Dano, S.D. Effects of combinations of Fusarium mycotoxins on the inhibition of macromolecular synthesis, malondialdehyde levels, DNA methylation and fragmentation, and viability in Caco-2 cells. Toxicon 2007, 49, 306–317. [Google Scholar] [CrossRef]
- Liu, A.; Hu, S. Dietary exposure of deoxynivalenol affected cytochrome P450 and growth related-gene expression via DNA methylation in piglet liver. Res. Sq. 2020. [Google Scholar]
- Ghazi, T.; Nagiah, S. Fusaric acid-induced promoter methylation of DNA methyltransferases triggers DNA hypomethylation in human hepatocellular carcinoma (HepG2) cells. Epigenetics 2019, 14, 804–817. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Q.; Zhao, J. Gene expression network regulated by DNA methylation and microRNA during microcystin-leucine arginine induced malignant transformation in human hepatocyte L02 cells. Toxicol. Lett. 2018, 289, 42–53. [Google Scholar] [CrossRef]
- Matias, W.G.; Creppy, E.E. 5-Methyldeoxycytosine as a biological marker of DNA damage induced by okadaic acid in vero cells. Environ. Toxicol. Water Qual. 1998, 13, 83–88. [Google Scholar] [CrossRef]
- Rhie, S.K.; Hazelett, D.J. Nucleosome positioning and histone modifications define relationships between regulatory elements and nearby gene expression in breast epithelial cells. BMC Genom. 2014, 15, 331. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Furukawa, A.; Wakamori, M. Acetylated histone H4 tail enhances histone H3 tail acetylation by altering their mutual dynamics in the nucleosome. Proc. Natl. Acad. Sci. USA 2020, 117, 19661–19663. [Google Scholar] [CrossRef]
- Neganova, M.E.; Klochkov, S.G. Histone modifications in epigenetic regulation of cancer: Perspectives and achieved progress. Semin. Cancer Biol. 2022, 83, 452–471. [Google Scholar] [CrossRef] [PubMed]
- Brancolini, C.; Gagliano, T. HDACs and the epigenetic plasticity of cancer cells: Target the complexity. Pharmacol. Ther. 2022, 238, 108190. [Google Scholar] [CrossRef]
- Huang, H.; Weng, H. Histone H3 trimethylation at lysine 36 guides m(6)A RNA modification co-transcriptionally. Nature 2019, 567, 414–419. [Google Scholar] [CrossRef]
- Jambhekar, A.; Dhall, A. Roles and regulation of histone methylation in animal development. Nat. Rev. Mol. Cell Biol. 2019, 20, 625–641. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, T.; Chakravarti, D. A peek into the complex realm of histone phosphorylation. Mol. Cell Biol. 2011, 31, 4858–4873. [Google Scholar] [CrossRef]
- Cao, J.; Yan, Q. Histone ubiquitination and deubiquitination in transcription, DNA damage response, and cancer. Front. Oncol. 2012, 2, 26. [Google Scholar] [CrossRef]
- Rosonina, E.; Akhter, A. Regulation of transcription factors by sumoylation. Transcription 2017, 8, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, C.; Purkey, L.R. PARticular MARks: Histone ADP-ribosylation and the DNA damage response. DNA Repair 2024, 140, 103711. [Google Scholar] [CrossRef]
- Zhai, Q.; Wang, L. Role of citrullination modification catalyzed by peptidylarginine deiminase 4 in gene transcriptional regulation. Acta Biochim. Biophys. Sin. 2017, 49, 567–572. [Google Scholar] [CrossRef]
- Xiao, C.; Liu, Y. Cadmium induces histone H3 lysine methylation by inhibiting histone demethylase activity. Toxicol. Sci. 2015, 145, 80–89. [Google Scholar] [CrossRef]
- Sun, H.; Zhou, X. Modulation of histone methylation and MLH1 gene silencing by hexavalent chromium. Toxicol. Appl. Pharmacol. 2009, 237, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, H. Mechanism by which HDAC3 regulates manganese induced H3K27ac in SH-SY5Y cells and intervention by curcumin. Arch. Biochem. Biophys. 2024, 752, 109878. [Google Scholar] [CrossRef]
- Song, C.; Kanthasamy, A. Paraquat induces epigenetic changes by promoting histone acetylation in cell culture models of dopaminergic degeneration. Neurotoxicology 2011, 32, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Kanthasamy, A. Environmental neurotoxic pesticide increases histone acetylation to promote apoptosis in dopaminergic neuronal cells: Relevance to epigenetic mechanisms of neurodegeneration. Mol. Pharmacol. 2010, 77, 621–632. [Google Scholar] [CrossRef]
- Sundar, I.K.; Nevid, M.Z. Cigarette smoke induces distinct histone modifications in lung cells: Implications for the pathogenesis of COPD and lung cancer. J. Proteome Res. 2014, 13, 982–996. [Google Scholar] [CrossRef] [PubMed]
- Marwick, J.A.; Stevenson, C.S. Cigarette smoke exposure alters mSin3a and Mi-2alpha/beta expression; implications in the control of pro-inflammatory gene transcription and glucocorticoid function. J. Inflamm. 2010, 7, 33. [Google Scholar] [CrossRef]
- Limbeck, E.; Vanselow, J.T. Linking site-specific loss of histone acetylation to repression of gene expression by the mycotoxin ochratoxin A. Arch. Toxicol. 2018, 92, 995–1014. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, C. Vitamin C protects porcine oocytes from microcystin-LR toxicity during maturation. Front. Cell Dev. Biol. 2020, 8, 582715. [Google Scholar] [CrossRef] [PubMed]
- Ajiro, K.; Yoda, K. Alteration of cell cycle-dependent histone phosphorylations by okadaic acid. Induction of mitosis-specific H3 phosphorylation and chromatin condensation in mammalian interphase cells. J. Biol. Chem. 1996, 271, 13197–13201. [Google Scholar] [CrossRef]
- Mahadevan, L.C.; Willis, A.C. Rapid histone H3 phosphorylation in response to growth factors, phorbol esters, okadaic acid, and protein synthesis inhibitors. Cell 1991, 65, 775–783. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, P. Histone modification is involved in okadaic acid (OA) induced DNA damage response and G2-M transition arrest in maize. PLoS ONE 2016, 11, e0155852. [Google Scholar] [CrossRef] [PubMed]
- Sharma, H.; Valentine, M.N.Z. Decryption of sequence, structure, and functional features of SINE repeat elements in SINEUP non-coding RNA-mediated post-transcriptional gene regulation. Nat. Commun. 2024, 15, 1400. [Google Scholar] [CrossRef] [PubMed]
- Soni, D.K.; Biswas, R. Role of non-coding RNAs in post-transcriptional regulation of lung diseases. Front. Genet. 2021, 12, 767348. [Google Scholar] [CrossRef]
- Tan, J.; Sun, M. Arsenic exposure increased expression of HOTAIR and LincRNA-p21 in vivo and vitro. Environ. Sci. Pollut. Res. Int. 2021, 28, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Huang, Z. LncRNA-ENST00000446135 is a novel biomarker of cadmium toxicity in 16HBE cells, rats, and Cd-exposed workers and regulates DNA damage and repair. Toxicol. Res. 2020, 9, 823–834. [Google Scholar] [CrossRef] [PubMed]
- Sevim, C.; Tsatsakis, A. Investigation of the miRNA levels changes to acceptable daily intake dose pesticide mixture exposure on rat mesentery and pancreas. Chemosphere 2024, 349, 140712. [Google Scholar] [CrossRef]
- Kalinina, T.; Kononchuk, V. Effects of endocrine disruptors o,p’-dichlorodiphenyltrichloroethane, p,p’-dichlorodiphenyltrichloroethane, and endosulfan on the expression of estradiol-, progesterone-, and testosterone-responsive microRNAs and their target genes in MCF-7 cells. Toxics 2022, 10, 25. [Google Scholar] [CrossRef]
- Souki, R.; Amosse, J. Small RNA-sequencing reveals the involvement of microRNA-132 in benzo[a]pyrene-induced toxicity in primary human blood cells. Environ. Pollut. 2023, 328, 121653. [Google Scholar] [CrossRef]
- Ge, Y.; Gu, P. Benzo[a]pyrene stimulates miR-650 expression to promote the pathogenesis of fatty liver disease and hepatocellular carcinoma via SOCS3/JAK/STAT3 cascades. J. Mol. Cell Biol. 2021, 13, 556–564. [Google Scholar] [CrossRef]
- Zhu, L.; Gao, J. miR-34a screened by miRNA profiling negatively regulates Wnt/β-catenin signaling pathway in Aflatoxin B1 induced hepatotoxicity. Sci. Rep. 2015, 5, 16732. [Google Scholar] [CrossRef]
- Lv, J.; Yu, Y.Q. Aflatoxin B1 promotes cell growth and invasion in hepatocellular carcinoma HepG2 cells through H19 and E2F1. Asian Pac. J. Cancer Prev. 2014, 15, 2565–2570. [Google Scholar] [CrossRef]
- Yang, S.; Chen, L. MicroRNA expression profiling involved in MC-LR-induced hepatotoxicity using high-throughput sequencing analysis. J. Toxicol. Environ. Health A 2018, 81, 89–97. [Google Scholar] [CrossRef]
- Ma, J.; Li, Y. Analysis of microRNA expression profiling involved in MC-LR-induced cytotoxicity by high-throughput sequencing. Toxins 2017, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Lin, P. Exosomal miRNA analysis provides new insights into exposure to nanoplastics and okadaic acid. Sci. Total Environ. 2023, 905, 167010. [Google Scholar] [CrossRef] [PubMed]
- Farsetti, A.; Illi, B. How epigenetics impacts on human diseases. Eur. J. Intern. Med. 2023, 114, 15–22. [Google Scholar] [CrossRef]
- Costa, C.; Teodoro, M. MicroRNAs alteration as early biomarkers for cancer and neurodegenerative diseases: New challenges in pesticides exposure. Toxicol. Rep. 2020, 7, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Mezynska, M.; Brzoska, M.M. Environmental exposure to cadmium-a risk for health of the general population in industrialized countries and preventive strategies. Environ. Sci. Pollut. Res. Int. 2018, 25, 3211–3232. [Google Scholar] [CrossRef]
- Damiani, L.A.; Yingling, C.M. Carcinogen-induced gene promoter hypermethylation is mediated by DNMT1 and causal for transformation of immortalized bronchial epithelial cells. Cancer Res. 2008, 68, 9005–9014. [Google Scholar] [CrossRef]
- Ye, M.; Huang, T. Role of CDH13 promoter methylation in the carcinogenesis, progression, and prognosis of colorectal cancer: A systematic meta-analysis under PRISMA guidelines. Medicine 2017, 96, e5956. [Google Scholar] [CrossRef]
- Sakai, M.; Hibi, K. Frequent promoter methylation and gene silencing of CDH13 in pancreatic cancer. Cancer Sci. 2004, 95, 588–591. [Google Scholar] [CrossRef]
- Tseng, C.H.; Tsuang, B.J. The relationship between air pollution and lung cancer in nonsmokers in Taiwan. J. Thorac. Oncol. 2019, 14, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Mengersen, K. Short-term association between ambient air pollution and lung cancer mortality. Environ. Res. 2019, 179, 108748. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Kato, A. Aberrant methylation patterns of the Rassf1a gene in rat lung adenocarcinomas induced by N-nitrosobis(2-hydroxypropyl)amine. Mol. Carcinog. 2006, 45, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Rajput, S.; Jain, S. Role of cyanotoxins in the development and promotion of cancer. Toxicol. Rep. 2024, 13, 101798. [Google Scholar] [CrossRef]
- Mungamuri, S.K.; Mavuduru, V.A. Role of epigenetic alterations in aflatoxin-induced hepatocellular carcinoma. Liver Cancer Int. 2020, 1, 41–50. [Google Scholar] [CrossRef]
- Zhao, Y.; Xie, P. Genomic profiling of microRNAs and proteomics reveals an early molecular alteration associated with tumorigenesis induced by MC-LR in mice. Environ. Sci. Technol. 2012, 46, 34–41. [Google Scholar] [CrossRef]
- Qiu, J.; Gu, R. Long noncoding RNA ZFAS1: A novel anti-apoptotic target in Fuchs endothelial corneal dystrophy. Exp. Eye Res. 2024, 241, 109832. [Google Scholar] [CrossRef]
- Shi, J.; He, J. Distinct response of the hepatic transcriptome to Aflatoxin B1 induced hepatocellular carcinogenesis and resistance in rats. Sci. Rep. 2016, 6, 31898. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, L. Systemic analyses of cuproptosis-related lncRNAs in pancreatic adenocarcinoma, with a focus on the molecular mechanism of LINC00853. Int. J. Mol. Sci. 2023, 24, 7923. [Google Scholar] [CrossRef]
- Kovalchuk, O. Epigenetic research sheds new light on the nature of interactions between organisms and their environment. Environ. Mol. Mutagen. 2008, 49, 1–3. [Google Scholar] [CrossRef]
- Marques, S.C.; Oliveira, C.R. Epigenetics in neurodegeneration: A new layer of complexity. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Altuna, M.; Urdanoz-Casado, A. DNA methylation signature of human hippocampus in Alzheimer’s disease is linked to neurogenesis. Clin. Epigenet. 2019, 11, 91. [Google Scholar] [CrossRef] [PubMed]
- Calderón-Garcidueñas, L.; Herrera-Soto, A. Reduced repressive epigenetic marks, increased DNA damage and Alzheimer’s disease hallmarks in the brain of humans and mice exposed to particulate urban air pollution. Environ. Res. 2020, 183, 109226. [Google Scholar] [CrossRef]
- Lee, E.; Sidoryk-Wegrzynowicz, M. Transforming growth factor-α mediates estrogen-induced upregulation of glutamate transporter GLT-1 in rat primary astrocytes. Glia 2012, 60, 1024–1036. [Google Scholar] [CrossRef]
- Erikson, K.; Aschner, M. Manganese causes differential regulation of glutamate transporter (GLAST) taurine transporter and metallothionein in cultured rat astrocytes. Neurotoxicology 2002, 23, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Tarale, P.; Sivanesan, S. Global DNA methylation profiling of manganese-exposed human neuroblastoma SH-SY5Y cells reveals epigenetic alterations in Parkinson’s disease-associated genes. Arch. Toxicol. 2017, 91, 2629–2641. [Google Scholar] [CrossRef]
- Issah, I.; Arko-Mensah, J. Global DNA (LINE-1) methylation is associated with lead exposure and certain job tasks performed by electronic waste workers. Int. Arch. Occup. Environ. Health 2021, 94, 1931–1944. [Google Scholar] [CrossRef]
- Pfaff, A.L.; Bubb, V.J. An increased burden of highly active retrotransposition competent L1s is associated with Parkinson’s disease risk and progression in the PPMI cohort. Int. J. Mol. Sci. 2020, 21, 6562. [Google Scholar] [CrossRef]
- Sharma, P.; Mittal, P. Paraquat (herbicide) as a cause of Parkinson’s Disease. Parkinsonism Relat. Disord. 2024, 119, 105932. [Google Scholar] [CrossRef]
- Wang, Q.; Ren, N. Paraquat and MPTP induce neurodegeneration and alteration in the expression profile of microRNAs: The role of transcription factor Nrf2. NPJ Parkinsons Dis. 2017, 3, 31. [Google Scholar] [CrossRef]
- Rostamian Delavar, M.; Baghi, M. Differential expression of miR-34a, miR-141, and miR-9 in MPP+-treated differentiated PC12 cells as a model of Parkinson’s disease. Gene 2018, 662, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Jiang, E.X.; Domingo-Relloso, A. Arsenic Exposure and Epigenetic Aging: The Association with Cardiovascular Disease and All-Cause Mortality in the Strong Heart Study. Environ. Health Perspect. 2023, 131, 127016. [Google Scholar] [CrossRef]
- Domingo-Relloso, A.; Makhani, K. Arsenic exposure, blood DNA methylation, and cardiovascular disease. Circ. Res. 2022, 131, e51–e69. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Braun, D. Evaluating the impact of long-term exposure to fine particulate matter on mortality among the elderly. Sci. Adv. 2020, 6, eaba5692. [Google Scholar] [CrossRef]
- Roth, G.A.; Mensah, G.A. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Montiel-Davalos, A.; Ibarra-Sanchez Mde, J. Oxidative stress and apoptosis are induced in human endothelial cells exposed to urban particulate matter. Toxicol. In Vitro 2010, 24, 135–141. [Google Scholar] [CrossRef]
- Traboulsi, H.; Guerrina, N. Inhaled pollutants: The molecular scene behind respiratory and systemic diseases associated with ultrafine particulate matter. Int. J. Mol. Sci. 2017, 18, 243. [Google Scholar] [CrossRef]
- Tobaldini, E.; Bollati, V. Acute particulate matter affects cardiovascular autonomic modulation and IFN-gamma methylation in healthy volunteers. Environ. Res. 2018, 161, 97–103. [Google Scholar] [CrossRef]
- Wu, X.; Pan, B. In utero exposure to PM2.5 during gestation caused adult cardiac hypertrophy through histone acetylation modification. J. Cell Biochem. 2019, 120, 4375–4384. [Google Scholar] [CrossRef]
- Feil, R.; Fraga, M.F. Epigenetics and the environment: Emerging patterns and implications. Nat. Rev. Genet. 2012, 13, 97–109. [Google Scholar] [CrossRef]
- Zhang, L.; Lu, Q. Epigenetics in Health and Disease. Adv. Exp. Med. Biol. 2020, 1253, 3–55. [Google Scholar] [PubMed]
- Strickland, F.M.; Hewagama, A. Environmental exposure, estrogen and two X chromosomes are required for disease development in an epigenetic model of lupus. J. Autoimmun. 2012, 38, J135–J143. [Google Scholar] [CrossRef] [PubMed]
- Gorelik, G.; Richardson, B. Key role of ERK pathway signaling in lupus. Autoimmunity 2010, 43, 17–22. [Google Scholar] [CrossRef]
- Perricone, C.; Versini, M. Smoke and autoimmunity: The fire behind the disease. Autoimmun. Rev. 2016, 15, 354–374. [Google Scholar] [CrossRef]
- Somma, D.; Kok, F.O. Defining the role of nuclear factor (NF)-κB p105 subunit in human macrophage by transcriptomic analysis of NFKB1 knockout THP1 cells. Front. Immunol. 2021, 12, 669906. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, A.L.; Zorzan, I. Epigenetic regulation of early human embryo development. Cell Stem Cell 2023, 30, 1569–1584. [Google Scholar] [CrossRef]
- Gicquel, C.; El-Osta, A. Epigenetic regulation and fetal programming. Best. Pract. Res. Clin. Endocrinol. Metab. 2008, 22, 1–16. [Google Scholar] [CrossRef]
- Barker, D.J. The fetal and infant origins of adult disease. BMJ 1990, 301, 1111. [Google Scholar] [CrossRef]
- Barker, D.J.; Osmond, C. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ 1989, 298, 564–567. [Google Scholar] [CrossRef]
- Barker, D.J. Developmental origins of adult health and disease. J. Epidemiol. Community Health 2004, 58, 114–115. [Google Scholar] [CrossRef]
- Law, C.M.; Barker, D.J. Early growth and abdominal fatness in adult life. J. Epidemiol. Community Health 1992, 46, 184–186. [Google Scholar] [CrossRef]
- McMillen, I.C.; Robinson, J.S. Developmental origins of the metabolic syndrome: Prediction, plasticity, and programming. Physiol. Rev. 2005, 85, 571–633. [Google Scholar] [CrossRef] [PubMed]
- Dolinoy, D.C.; Huang, D. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc. Natl. Acad. Sci. USA 2007, 104, 13056–13061. [Google Scholar] [CrossRef]
- Dolinoy, D.C.; Weidman, J.R. Maternal genistein alters coat color and protects Avy mouse offspring from obesity by modifying the fetal epigenome. Environ. Health Perspect. 2006, 114, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Schrott, R.; Song, A. Epigenetics as a biomarker for early-life environmental exposure. Curr. Environ. Health Rep. 2022, 9, 604–624. [Google Scholar] [CrossRef]
- Perera, F.; Herbstman, J. Prenatal environmental exposures, epigenetics, and disease. Reprod. Toxicol. 2011, 31, 363–373. [Google Scholar] [CrossRef]
- Gómez-Roig, M.D.; Pascal, R. Environmental exposure during pregnancy: Influence on prenatal development and early life: A comprehensive review. Fetal Diagn. Ther. 2021, 48, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Breton, C.V.; Byun, H.M. Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. Am. J. Respir. Crit. Care Med. 2009, 180, 462–467. [Google Scholar] [CrossRef]
- Neophytou, A.M.; Oh, S.S. In utero tobacco smoke exposure, DNA methylation, and asthma in Latino children. Environ. Epidemiol. 2019, 3, e048. [Google Scholar] [CrossRef]
- Gao, L.; Liu, X. Self-reported prenatal tobacco smoke exposure, AXL gene-body methylation, and childhood asthma phenotypes. Clin. Epigenetics 2018, 10, 98. [Google Scholar] [CrossRef]
- Lee, K.W.; Richmond, R. Prenatal exposure to maternal cigarette smoking and DNA methylation: Epigenome-wide association in a discovery sample of adolescents and replication in an independent cohort at birth through 17 years of age. Environ. Health Perspect. 2015, 123, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Perera, F.; Tang, W.Y. Relation of DNA methylation of 5’-CpG island of ACSL3 to transplacental exposure to airborne polycyclic aromatic hydrocarbons and childhood asthma. PLoS ONE 2009, 4, e4488. [Google Scholar] [CrossRef]
- Grova, N.; Schroeder, H. Epigenetic and neurological impairments associated with early life exposure to persistent organic pollutants. Int. J. Genom. 2019, 2019, 2085496. [Google Scholar] [CrossRef] [PubMed]
- Neven, K.Y.; Saenen, N.D. Placental promoter methylation of DNA repair genes and prenatal exposure to particulate air pollution: An ENVIRONAGE cohort study. Lancet Planet. Health 2018, 2, e174–e183. [Google Scholar] [CrossRef]
- Wang, X.; Zhong, C. The influence of DNA repair genes and prenatal tobacco exposure on risk of childhood acute lymphoblastic leukemia: A gene-environment interaction study. Cancer Epidemiol. Biomark. Prev. 2025, 34, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Khokhlova, E.V.; Fesenko, Z.S. Features of DNA repair in the early stages of mammalian embryonic development. Genes 2020, 11, 1138. [Google Scholar] [CrossRef]
- Wu, G.; Bazer, F.W. Maternal nutrition and fetal development. J. Nutr. 2004, 134, 2169–2172. [Google Scholar]
- Olsen, A.K.; Li, D. Explore the dosimetric relationship between the intake of chemical contaminants and their occurrence in blood and urine. Environ. Sci. Technol. 2023, 57, 9526–9537. [Google Scholar] [CrossRef]
- Sly, P.D.; Flack, F. Susceptibility of children to environmental pollutants. Ann. N. Y Acad. Sci. 2008, 1140, 163–183. [Google Scholar] [CrossRef]
- Hines, R.N. The ontogeny of drug metabolism enzymes and implications for adverse drug events. Pharmacol. Ther. 2008, 118, 250–267. [Google Scholar] [CrossRef]
- Makri, A.; Goveia, M. Children’s susceptibility to chemicals: A review by developmental stage. J. Toxicol. Environ. Health B Crit. Rev. 2004, 7, 417–435. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, M.A.; Harrad, S. Hexabromocyclododecanes and tetrabromobisphenol-A in indoor air and dust in Birmingham, U.K: Implications for human exposure. Environ. Sci. Technol. 2008, 42, 6855–6861. [Google Scholar] [CrossRef]
- Abdallah, M.A.; Harrad, S. Personal exposure to HBCDs and its degradation products via ingestion of indoor dust. Environ. Int. 2009, 35, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Kundakovic, M.; Jaric, I. The epigenetic link between prenatal adverse environments and neurodevelopmental disorders. Genes 2017, 8, 104. [Google Scholar] [CrossRef]
- Xin, F.; Susiarjo, M. Multigenerational and transgenerational effects of endocrine disrupting chemicals: A role for altered epigenetic regulation? Semin. Cell Dev. Biol. 2015, 43, 66–75. [Google Scholar] [CrossRef]
- Jiménez-Chillarón, J.C.; Nijland, M.J. Back to the future: Transgenerational transmission of xenobiotic-induced epigenetic remodeling. Epigenetics 2015, 10, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Terrazas-Salgado, L.; García-Gasca, A. Epigenetic transgenerational modifications induced by xenobiotic exposure in Zebrafish. Front. Cell Dev. Biol. 2022, 10, 832982. [Google Scholar] [CrossRef]
- Anway, M.D.; Cupp, A.S. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 2005, 308, 1466–1469. [Google Scholar] [CrossRef]
- González-Rojo, S.; Lombó, M. Male exposure to bisphenol a impairs spermatogenesis and triggers histone hyperacetylation in zebrafish testes. Environ. Pollut. 2019, 248, 368–379. [Google Scholar] [CrossRef]
- Hao, L.; Ru, S. Transgenerational effects of parental bisphenol S exposure on zebrafish (Danio rerio) reproduction. Food Chem. Toxicol. 2022, 165, 113142. [Google Scholar] [CrossRef]
- Lombó, M.; Herráez, M.P. Paternal inheritance of bisphenol A cardiotoxic effects: The implications of sperm epigenome. Int. J. Mol. Sci. 2021, 22, 2125. [Google Scholar] [CrossRef]
- Lombó, M.; Fernández-Díez, C. Transgenerational inheritance of heart disorders caused by paternal bisphenol A exposure. Environ. Pollut. 2015, 206, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Drobná, Z.; Henriksen, A.D. Transgenerational effects of bisphenol A on gene expression and DNA methylation of imprinted genes in brain. Endocrinology 2018, 159, 132–144. [Google Scholar] [CrossRef]
- Yu, C.W.; Luk, T.C. Long-term nanoplastics exposure results in multi and trans-generational reproduction decline associated with germline toxicity and epigenetic regulation in Caenorhabditis elegans. J. Hazard. Mater. 2021, 412, 125173. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Shen, L. The phenotypic and behavioral defects can be transferred from zinc-exposed nematodes to their progeny. Environ. Toxicol. Pharmacol. 2007, 24, 223–230. [Google Scholar] [CrossRef]
- Wang, D.; Wang, Y. Nickel sulfate induces numerous defects in Caenorhabditis elegans that can also be transferred to progeny. Environ. Pollut. 2008, 151, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Y.; Yang, P. Multi-biological defects caused by lead exposure exhibit transferable properties from exposed parents to their progeny in Caenorhabditis elegans. J. Environ. Sci. 2007, 19, 1367–1372. [Google Scholar] [CrossRef]
- Wang, M.; Nie, Y. Transgenerational effects of diesel particulate matter on Caenorhabditis elegans through maternal and multigenerational exposure. Ecotoxicol. Environ. Saf. 2019, 170, 635–643. [Google Scholar] [CrossRef]
- Wang, Y.; Xie, W. Transferable properties of multi-biological toxicity caused by cobalt exposure in Caenorhabditis elegans. Environ. Toxicol. Chem. 2007, 26, 2405–2412. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, J. A review of transgenerational and multigenerational toxicology in the in vivo model animal Caenorhabditis elegans. J. Appl. Toxicol. 2023, 43, 122–145. [Google Scholar] [CrossRef]
- Titus, L.; Hatch, E.E. Reproductive and hormone-related outcomes in women whose mothers were exposed in utero to diethylstilbestrol (DES): A report from the US National Cancer Institute DES Third Generation Study. Reprod. Toxicol. 2019, 84, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Golding, J.; Gregory, S. Human transgenerational observations of regular smoking before puberty on fat mass in grandchildren and great-grandchildren. Sci. Rep. 2022, 12, 1139. [Google Scholar] [CrossRef] [PubMed]
- Svanes, C.; Bertelsen, R.J. Exposures during the prepuberty period and future offspring’s health: Evidence from human cohort studies. Biol. Reprod. 2021, 105, 667–680. [Google Scholar] [CrossRef] [PubMed]
- Burton, N.O.; Greer, E.L. Multigenerational epigenetic inheritance: Transmitting information across generations. Semin. Cell Dev. Biol. 2022, 127, 121–132. [Google Scholar] [CrossRef]
- Toth, M. Mechanisms of non-genetic inheritance and psychiatric disorders. Neuropsychopharmacology 2015, 40, 129–140. [Google Scholar] [CrossRef]
- Ouni, M.; Schürmann, A. Epigenetic contribution to obesity. Mamm. Genome 2020, 31, 134–145. [Google Scholar] [CrossRef]
- Guerrero-Bosagna, C.; Settles, M. Epigenetic transgenerational actions of vinclozolin on promoter regions of the sperm epigenome. PLoS ONE 2010, 5, e13100. [Google Scholar] [CrossRef]
- Skinner, M.K.; Ben Maamar, M. Alterations in sperm DNA methylation, non-coding RNA and histone retention associate with DDT-induced epigenetic transgenerational inheritance of disease. Epigenetics Chromatin 2018, 11, 8. [Google Scholar] [CrossRef]
- Handy, D.E.; Castro, R. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef]
- Bhatia, S.; Yan, Y. Sex- and OGG1-dependent reversal of in utero ethanol-initiated changes in postnatal behaviour by neonatal treatment with the histone deacetylase inhibitor trichostatin A (TSA) in oxoguanine glycosylase 1 (Ogg1) knockout mice. Toxicol. Lett. 2022, 356, 121–131. [Google Scholar] [CrossRef]
- Hosseini, H.; Teimouri, M. Resveratrol alleviates non-alcoholic fatty liver disease through epigenetic modification of the Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 2020, 119, 105667. [Google Scholar] [CrossRef]
- Chatterjee, B.; Ghosh, K. Resveratrol modulates epigenetic regulators of promoter histone methylation and acetylation that restores BRCA1, p53, p21(CIP1) in human breast cancer cell lines. Biofactors 2019, 45, 818–829. [Google Scholar] [CrossRef]
- Lameira, A.G., Jr.; Françoso, B.G.; Absy, S.; Pecorari, V.G.; Casati, M.Z.; Ribeiro, F.V.; Andia, D.C. Resveratrol reverts epigenetic and transcription changes caused by smoke inhalation on bone-related genes in rats. DNA Cell Biol. 2018, 37, 670–679. [Google Scholar] [CrossRef]
- Richmond, R.C.; Simpkin, A.J. Prenatal exposure to maternal smoking and offspring DNA methylation across the lifecourse: Findings from the Avon Longitudinal Study of Parents and Children (ALSPAC). Hum. Mol. Genet. 2015, 24, 2201–2217. [Google Scholar] [CrossRef] [PubMed]
- Thaler, R.; Spitzer, S. DMSO is a strong inducer of DNA hydroxymethylation in pre-osteoblastic MC3T3-E1 cells. Epigenetics 2012, 7, 635–651. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Y. TET (Ten-eleven translocation) family proteins: Structure, biological functions and applications. Signal Transduct. Target. Ther. 2023, 8, 297. [Google Scholar] [CrossRef]
- Santiago, M.; Antunes, C. TET enzymes and DNA hydroxymethylation in neural development and function—How critical are they? Genomics 2014, 104, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Labonté, B.; Jeong, Y.H. Gadd45b mediates depressive-like role through DNA demethylation. Sci. Rep. 2019, 9, 4615. [Google Scholar] [CrossRef]
- Ng, J.W.; Barrett, L.M. The role of longitudinal cohort studies in epigenetic epidemiology: Challenges and opportunities. Genome Biol. 2012, 13, 246. [Google Scholar] [CrossRef]
- Montrose, L.; Padmanabhan, V. Maternal levels of endocrine disrupting chemicals in the first trimester of pregnancy are associated with infant cord blood DNA methylation. Epigenetics 2018, 13, 301–309. [Google Scholar] [CrossRef]
- Curtis, S.W.; Cobb, D.O. Exposure to polybrominated biphenyl (PBB) associates with genome-wide DNA methylation differences in peripheral blood. Epigenetics 2019, 14, 52–66. [Google Scholar] [CrossRef] [PubMed]
- Winterbottom, E.F.; Moroishi, Y. Prenatal arsenic exposure alters the placental expression of multiple epigenetic regulators in a sex-dependent manner. Environ. Health 2019, 18, 18. [Google Scholar] [CrossRef]
- Klibaner-Schiff, E.; Simonin, E.M. Environmental exposures influence multigenerational epigenetic transmission. Clin. Epigenetics 2024, 16, 145. [Google Scholar] [CrossRef]
- Song, J.; Kim, C. Transgenerational effects of polyethylene microplastic fragments containing benzophenone-3 additive in Daphnia magna. J. Hazard. Mater. 2022, 436, 129225. [Google Scholar] [CrossRef] [PubMed]
- Harney, E.; Paterson, S. Pollution induces epigenetic effects that are stably transmitted across multiple generations. Evol. Lett. 2022, 6, 118–135. [Google Scholar] [CrossRef]
- Bruner-Tran, K.L.; Osteen, K.G. Developmental exposure to TCDD reduces fertility and negatively affects pregnancy outcomes across multiple generations. Reprod. Toxicol. 2011, 31, 344–350. [Google Scholar] [CrossRef]
- Wolstenholme, J.T.; Edwards, M. Gestational exposure to bisphenol a produces transgenerational changes in behaviors and gene expression. Endocrinology 2012, 153, 3828–3838. [Google Scholar] [CrossRef] [PubMed]
- Lowe, R.; Shirley, N. Transcriptomics technologies. PLoS Comput. Biol. 2017, 13, e1005457. [Google Scholar] [CrossRef]
- Zheng, F.; Gonçalves, F.M. Redox toxicology of environmental chemicals causing oxidative stress. Redox Biol. 2020, 34, 101475. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Xenobiotic | Effect on DNA Methylation | Mechanism | References |
|---|---|---|---|
| Cadmium (acute exposure—24 h to 1 week) | Decreased DNA methylation | Inhibition of DNMTs | [166] |
| Cadmium (prolonged exposure—10 weeks) | Increased DNA methylation | Increased activity of DNMTs | [166] |
| Heavy metals (e.g., arsenic, lead, aluminium) | DNA methylation alterations, especially in promoter regions | Interference with DNMTs | [167,168] |
| Heavy metals | Hypomethylation of NFKB1 gene | ----- | [169] |
| Dioxins | Hypomethylation and alterations in gene expression | Interaction with aryl hydrocarbon receptor, affecting DNMT expression | [170,171] |
| Phthalates | Alterations in DNA methylation, particularly in reproductive tissues | Disruption of epigenetic signalling and hormonal pathways | [172,173] |
| Bisphenol A | Alterations in methylation of genes involved in hormonal function | Interaction with hormonal receptors and DNMT activity | [172,174] |
| Tobacco | DNA hypomethylation | ----- | [175] |
| Benzo(a)pyrene | DNA hypomethylation | DNMTs inhibition | [176] |
| Benzo(a)pyrene diol epoxide | DNA hypermethylation | Recruitment of DNMT3A | [177] |
| Polystyrene nanoplastics | DNA hypomethylation/hypermethylation | ----- | [178] |
| Aflatoxin B1 | Hypermethylation of p21 promotor | ----- | [179] |
| DNA hypermethylation | ----- | [180,181] | |
| DNA hypomethylation | ----- | [180] | |
| Decreased LINE-1 and Sat2 promotor methylation | ----- | [148] | |
| Ochratoxin A | Global DNA hypermethylation | ---- | [182] |
| Global DNA hypermethylation | Increased DNMT1, and DNMT3B expression, but decreased DNMT3A expression | [146] | |
| DNA hypermethylation (Tbc1d5, Arap2, Ano6, Cul2, and Dlg2 gene promoters) | ----- | [183] | |
| DNA hypomethylation (Cpne4, Pdpk1, Spop, Ogdh, Dock3, and Rptor gene promoters) | ----- | ||
| DNA hypomethylation | ----- | [184] | |
| Mixture of aflatoxin, zearalenone, and deoxynivalenol | DNA hypermethylation | ----- | [185] |
| Zearalenone | Global DNA hypermethylation | Increased DNMT1 expression | [186] |
| ----- | [145] | ||
| Global DNA hypomethylation | ----- | [187] | |
| Zearalenone, fumonisin B1, and deoxynivalenol, individually or in a mixture | Global DNA hypermethylation | ----- | [188] |
| Deoxynivalenol | Global DNA hypermethylation | Increased DNMT1 and DNMT3B expression | [189] |
| Fusaric acid | DNA hypomethylation | Decrease DNMT1, DNMT3A, and DNMT3B expression, and increase MBD2 expression | [190] |
| Microcystin-LR | DNA hypermethylation | Increased DNMT3A and DNMT3B expression | [191] |
| Global DNA hypermethylation and increased DNA methylation of bdnf gene promoter | Increased expression of DNMTs | [142] | |
| DNA hypomethylation (dio3 and gad1 gene promoters) in F1 generation | ----- | [142] | |
| Okadaic acid | DNA hypermethylation | ----- | [192] |
| Xenobiotic(s) | Effects on Histones | Mechanism | References |
|---|---|---|---|
| Cadmium | Increased H3K4me3 and H3K9me2 levels | Reduced activities of H3K4 and H3K9 demethylases; no changes on KDM5A and KDM3A | [205] |
| Hexavalent chromium [Cr(VI)] | Increased levels of H3K9me2, H3K9me3, H3K4me2, and H3K4me3 | Increased expression of G9a histone methyltransferase | [206] |
| Decreased levels of H3K27me3, and H3R2me2 | ----- | ||
| Manganese | H3K27 hypoacetylation | Increased expression of HDAC3 | [207] |
| Paraquat | Acetylation of histone H3 | Decreased HDAC expression | [208] |
| Dieldrin | Histones H3 and H4 hyperacetylation | Reduced proteasomal degradation of HATs | [209] |
| Cigarette smoke | Increased acetylation and methylation of histone H3 and histone H4 | Alteration of HATs, HDACs, and DNMT activity | [210] |
| Decreased histone acetylation | Reduced HDAC2 activity | [211] | |
| Aflatoxin B1 | Increased levels of H3K9me3 | ----- | [181] |
| Decreased levels of H3K27me3 and H3K4me2 | ----- | ||
| Increased levels of H3K27me3 and H2AK119Ub | ----- | [147] | |
| Ochratoxin A | Decreased acetylation at H3K9 and H3K14 | Inhibition of HATs | [212] |
| Mixture of aflatoxin, zearalenone, and deoxynivalenol | Increased levels of H3K9me3 and H4K20me3, and decreased levels of H3K27me3 and H4K20me2 | ----- | [185] |
| Zearalenone | Increased levels of H3K9me3, H3K9ac, and H3K27me3 | Increased expression of HAT1, KAT2B, ESCO1, PRMT6, and SETD8 | [145] |
| Microcystin-LR | Decreased levels of H3K4me2, H3K4me3, and H3K36me3 | Increased expression of KDM5B | [213] |
| Okadaic acid | Increased phosphorylation of histones H1 and H3 | Likely inhibition of phosphatases 1 and 2A | [214] |
| Hyperphosphorylation of histone H3 | Likely inhibition of phosphatases 1 and 2A | [215] | |
| Increased levels of H4K5ac and altered spatial distribution of H3S10ph | ----- | [216] |
| Xenobiotic | Effects on ncRNAs | References |
|---|---|---|
| Arsenic | Increase expression of lncRNA-p21 | [219] |
| Cadmium | Increased expression of lncRNA-ENST00000446135 | [220] |
| Mixture of pesticides (chlormequat chloride, pirimiphos-methyl, glyphosate, tebuconazole, chlorpyrifos-methyl, and deltamethrin) | Downregulation of miRNA-146a | [221] |
| p,p’-DDT | Upregulation of miRNA-19b, miRNA-27a, miRNA-126, miRNA-190a, miRNA-193b, and miRNA-378 | [222] |
| o,p’-DDT | Upregulation of miRNA-126, miRNA-190b, miRNA-193b, miRNA-324, miRNA-342, miRNA-378, and miRNA-423 | [222] |
| Downregulation of miRNA-190a | ||
| Benzo(a)pyrene | Upregulation of miRNA-132 | [223] |
| Upregulation of miRNA-650 | [224] | |
| Aflatoxin B1 | Upregulation of miRNA-19b, miRNA-19a, miRNA-34a, miRNA-99a, miRNA-190a, and miRNA-16 | [225] |
| Downregulation of miRNA-1307, miRNA-99b, and miRNA-100-5p | ||
| Aflatoxin B1 | Upregulation of lncRNA-H19 | [226] |
| Ochratoxin A | Upregulation of miRNA-155 | [182] |
| Microcystin-LR | Upregulation of miRNA-15b-3p | [227] |
| Upregulation of miRNA-21, and miRNA-221 | [149] | |
| Upregulation of miRNA-149-3p, miRNA-449c-5p, and miRNA-454-3p | [228] | |
| Downregulation of miRNA-122 | [149] | |
| Downregulation of miRNA-500a-3p, miRNA-500a-5p, miRNA-500b-5p, and miRNA-4286 | [228] | |
| Downregulation of miRNA-4521 | [227] | |
| Okadaic acid | Downregulation of miRNA-4492 and miRNA-4497 | [229] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sobral, A.F.; Cunha, A.; Costa, I.; Silva-Carvalho, M.; Silva, R.; Barbosa, D.J. Environmental Xenobiotics and Epigenetic Modifications: Implications for Human Health and Disease. J. Xenobiot. 2025, 15, 118. https://doi.org/10.3390/jox15040118
Sobral AF, Cunha A, Costa I, Silva-Carvalho M, Silva R, Barbosa DJ. Environmental Xenobiotics and Epigenetic Modifications: Implications for Human Health and Disease. Journal of Xenobiotics. 2025; 15(4):118. https://doi.org/10.3390/jox15040118
Chicago/Turabian StyleSobral, Ana Filipa, Andrea Cunha, Inês Costa, Mariana Silva-Carvalho, Renata Silva, and Daniel José Barbosa. 2025. "Environmental Xenobiotics and Epigenetic Modifications: Implications for Human Health and Disease" Journal of Xenobiotics 15, no. 4: 118. https://doi.org/10.3390/jox15040118
APA StyleSobral, A. F., Cunha, A., Costa, I., Silva-Carvalho, M., Silva, R., & Barbosa, D. J. (2025). Environmental Xenobiotics and Epigenetic Modifications: Implications for Human Health and Disease. Journal of Xenobiotics, 15(4), 118. https://doi.org/10.3390/jox15040118