
Associations of Environmental Exposure to Arsenic, Manganese, Lead, and Cadmium with Alzheimer’s Disease: A Review of Recent Evidence from Mechanistic Studies



Abstract

1. Introduction
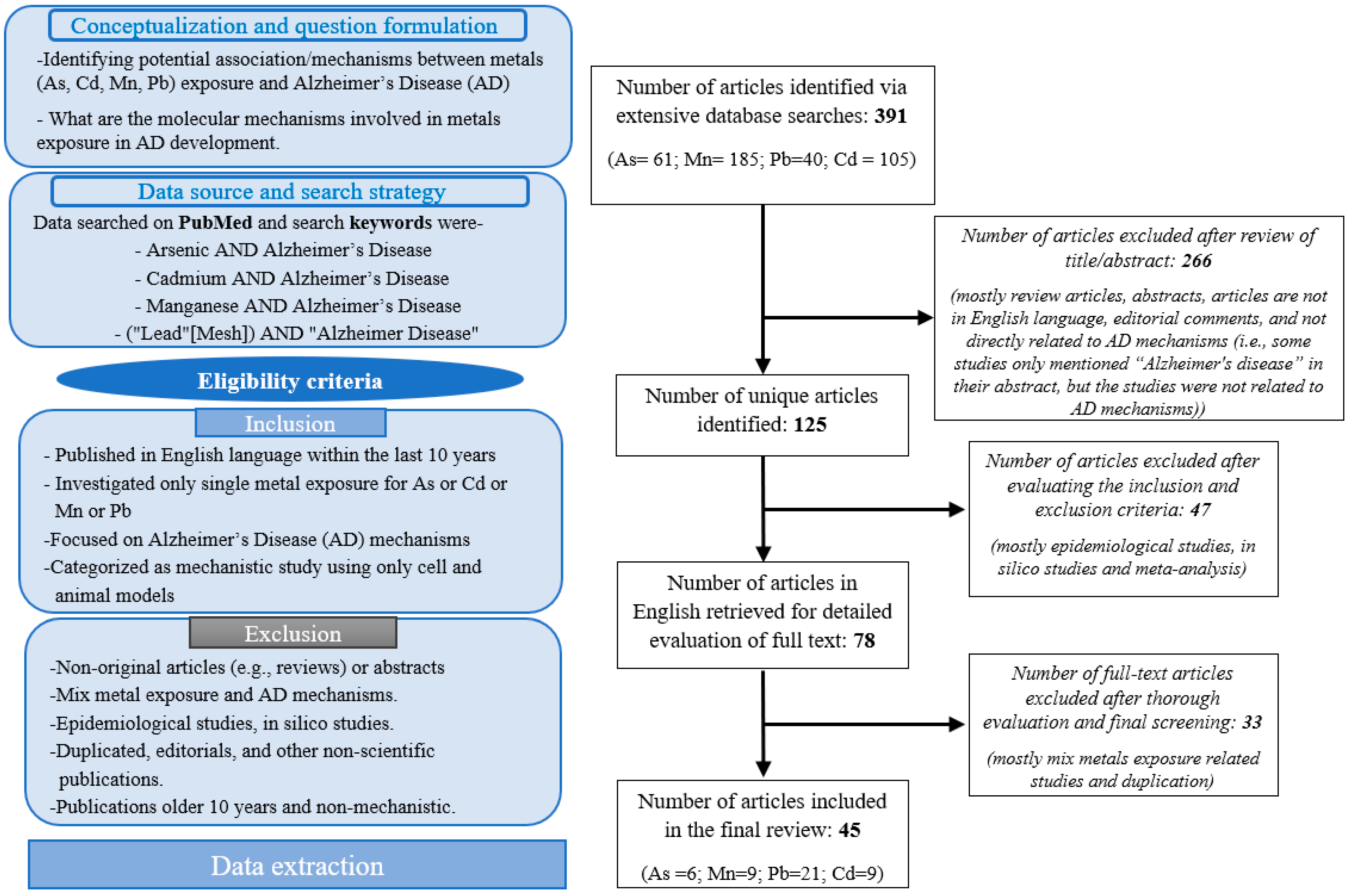
2. Methods
2.1. Search Strategy
2.2. Inclusion Criteria
2.3. Exclusion Criterion
2.4. Data Extraction
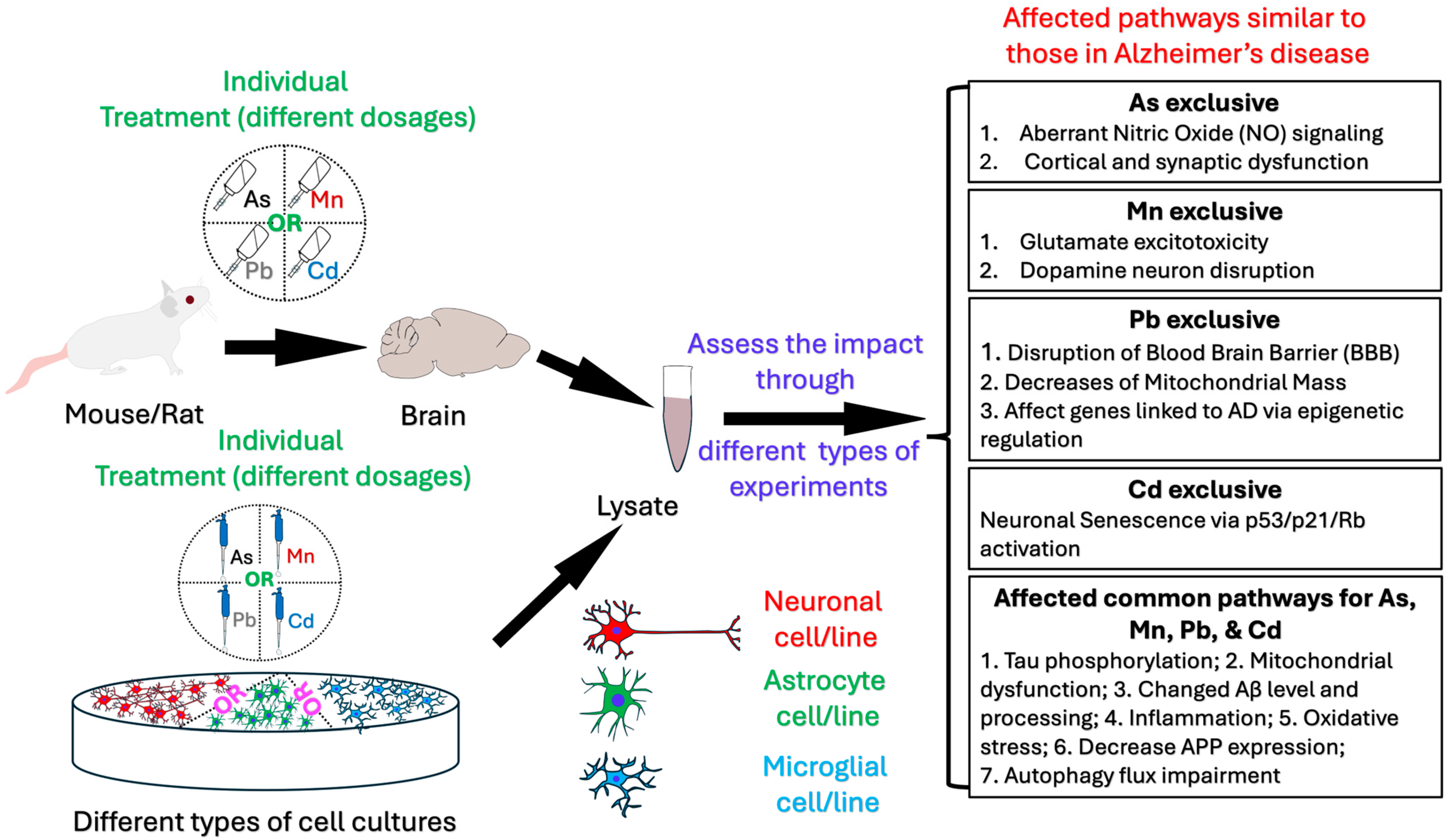
3. Results
3.1. Mechanisms and Pathobiology of Alzheimer’s Disease Development Related to Arsenic Exposure

{kind=link}
{kind=link}
{kind=link}
| SL | References | Study Objective | Study Type | Exposure | Key Findings |
|---|---|---|---|---|---|
| 1 | Tripathi et al., 2022 [41] | Investigated nitric oxide signaling in As neurotoxicity using mice model. | Mice model | Mice—drinking water (0.1 ppm and 1 ppm for one month) | As disrupts nitric oxide (NO) regulation, antioxidant defense, and S-nitrosylation (SNO) signaling in the striatum and hippocampus, resembling features of ASD and AD pathobiology. |
| 2 | Niño et al., 2021 [72] | Investigated synaptic structure (cortical microstructure and synapses) in chronic As exposure using both a triple-transgenic AD model and Wistar rats. | Triple-transgenic AD model and Wistar rats | Rats—drinking water (3 ppm; until 2 and 4 months of age) | As exposure changes cortical microstructure, decreases synaptic connectivity, increases spine density as well as causes structural changes in the cortex and synaptic regions. |
| 3 | Wisessaowapak et al., 2021 [74] | Investigated if prolonged exposure to As affected the phosphorylation of wild-type tau in the neuronal cell model (SH-SY5Y Cells). | SH-SY5Y Cells | Cells (1, 5, or 10 μM for 1, 3, or 5 days) | As induces tau hyperphosphorylation, mediated by GSK3β and ERK1/2 activation, which may contribute to the development of sporadic AD. |
| 4 | Pakzad et al., 2021 [70] | Investigated the correlation between arsenic trioxide exposure and its impact on the tau protein Ser262 phosphorylation in male mice. | Mice model | Mice—drinking water (1 and 10 ppm for 3 months) | As exposure increases tau phosphorylation at Ser262. |
| 5 | Niño et al., 2018a [71] | Investigated As exposure and the pathophysiological progress of AD using the 3xTgAD mouse model. | 3xTgAD mouse model | Mice—drinking water (3 ppm; until 6 months of age) | As exposure accelerates neurodegenerative processes via mitochondrial dysfunction (bioenergetic dysfunction) and increases oxidative stress, key factors to AD pathology. |
| 6 | Niño et al., 2018b [73] | Investigated the effects of chronic As exposure on the production and elimination of Amyloid-β (Aβ) in Wistar rats. | Male Wistar rats | Rats—drinking water (3 ppm; until 4 months of age) | As exposure causes behavioral deficits and increases in cleaved Aβ (1–42) levels in brain lysates, potentially disrupting Aβ processing and promoting amyloid accumulation in the brain, hallmark of AD. |
3.1.1. Arsenic and Bioenergetic Dysfunction in Alzheimer’s Disease
3.1.2. Arsenic and Tau Phosphorylation: A Key Marker of Alzheimer’s Disease
3.1.3. Arsenic-Induced Nitric Oxide Dysregulation and Neurotoxicity
3.1.4. Neurotoxicity and Amyloid-β Production, Synaptic, and Cortical Changes
3.1.5. Tau Hyperphosphorylation and Aggregation
3.2. Mechanisms and Pathobiology of Alzheimer’s Disease Development Related to Manganese Exposure
3.2.1. Drp1 Role in Neuroprotection and Manganese-Induced Toxicity
3.2.2. Impact of Manganese Exposure on Autophagy
3.2.3. Manganese Exposure Impairs Astrocytic Glutamate Transporter, EAAT2
3.2.4. REST Protects Dopaminergic Neurons Against Manganese-Induced Neurotoxicity
3.2.5. Glutamate Excitotoxicity in Manganese-Induced Neurotoxicity
3.2.6. Role of Manganese Exposure and NF-κB in Neuroinflammation
3.2.7. Manganese-Induced Toxicity Impairs Glutamatergic Signaling
3.2.8. Astrocytic REST (Repressor Element-1 Silencing Transcription Factor) Role in Mn-Induced Neurotoxicity
3.2.9. Manganese-Induced Dysregulation Amyloid Precursor Protein (APP) Processing and Cognitive Impairment
| SL | References | Study Objective | Study Type | Exposure | Key Findings |
|---|---|---|---|---|---|
| 1 | Fan et al. 2024 [82] | Investigated the role of Drp1 in Mn-exposure-induced autophagy and mitochondrial function. Determined if Drp1 inhibition improves autophagy independent of mitochondria. | HeLa cells, N27 neuronal cells and mice | Cells and mice—oral gavage (for cell: 62.5μM to 2mM for 24 or 48 h; for mice: ~4.2 mg absolute Mn/kg/day for 30 days) | A partial Drp1 loss of function appears to be safe and sufficient to confer neuroprotection against Mn-induced autophagy flux. |
| 2 | Spitznagel et al., 2023 [40] | Examined acute Mn exposure effect on glutamatergic neurotransmission, astrocytic glutamate clearance, behavior in AD models, and pre-symptomatic AD vulnerabilities. | Mice and primary astrocytes | Mice and primary astrocytes—injections (13.8 mg/kg for 1 week (injected on days 1, 4, and 7)) | Mn exposure disrupts glutamate clearance, elevates GLAST (Glutamate Aspartate Transporter), and increases seizure susceptibility in APP/PSEN1 mice, potentially aiding early AD. |
| 3 | Pajarillo et al., 2022 [83] | Investigated astrocytic REST’s role in Mn-induced neurotoxicity, assessed locomotor and cognitive function impairment, and evaluated the impact of astrocytic REST deletion on proinflammatory factors. | Mice and primary astrocytes | Mice and primary astrocytes—nostril (30 mg/kg, 1 μL per nostril in both nostrils, daily for 21 days) | Astrocytic REST (Repressor Element-1 Silencing Transcription Factor) deletion worsens Mn toxicity, causing dopamine dysfunction, motor deficits, cognitive decline, and inflammation. |
| 4 | Xu et al., 2021 [78] | Investigated Mn-induced autophagy dysfunction in N2a cells, PP2Ac methylation in autophagy regulation, the effects of ABL-127 on PP2Ac methylation, and the impact of LCMT1 overexpression on autophagy. | N2a cells | N2a cells (250, 500, 1000, 2000 μM for 6, 12, and 24 h) | Mn cytotoxicity disrupts autophagy in neuronal N2a cells, a hall mark of AD. Regulating PP2Ac methylation may help prevent Mn neurotoxicity and neurodegenerative diseases like AD. |
| 5 | Rizor et al., 2021 [75] | Investigated Mn-induced YY1 activation via the NF-kB pathway and mechanisms impairing EAAT2 function in astrocytes. | H4 human astrocyte cells | Cells (250 μM, 3 h) | Mn exposure triggers oxidative stress, TNF-α production, IKK-β activation, YY1 upregulation, and EAAT2 repression. |
| 6 | Yang et al., 2021 [84] | Investigated Mn-induced cognitive impairment mechanisms by assessing the role of APP in cognitive deficits, APP’s secretase processing in neurotoxicity, and synaptic dysfunction by using both in vivo mouse model and in vitro cell culture (N2a cells). | Astrocyte cell | Mice and N2a cells—gastric gavage (25, 50, or 100 mg/kg for 90 days) | Mn exposure impairs cognition in mouse models and inhibits APP expression and α-secretase activity. |
| 7 | Pajarillo et al., 2020 [79] | Investigated the role of RE1-silencing transcription factor (REST) in dopaminergic neurons against Mn-induced toxicity and the enhancement of the expression of the dopamine-synthesizing enzyme tyrosine hydroxylase (TH). | Neuronal cell lines (Mouse CAD cell line and LUHMES (CRL-2927) cell line) | Cells (50, 100, 250 μM for 3, 6, 12 h) | REST enhances TH expression, protects dopaminergic neurons from Mn toxicity, reduces oxidative stress, regulates apoptosis, promotes antioxidants, and its dysfunction links to Parkinson’s and Alzheimer’s disease. |
| 8 | Lu et al., 2018 [80] | Investigated the impact of Mn-induced toxicity on the function of the gap junctional intercellular communication (GJIC) by examining the Cx43 expression, excitotoxicity cell death mechanisms, and glutamate homeostasis disruption. | Primary astrocytes | Primary astrocytes (125, 250, 500, 1000 μM for 4 and 16 h) | Mn exposure impairs astrocyte viability, increased apoptosis, disrupts glutamate homeostasis, increases intracellular glutamate levels, and downregulates glutamate transporter expression. |
| 9 | Kirkley et al., 2017 [81] | Investigated the role of microglia and glial crosstalk in Mn-induced neurodegeneration. | Mixed glial cultures from whole brain (astrocytes and microglia) | Cells (10, 30, 100 μM for 2, 4, 6, 8, 12, 24 h) | NF-κB signaling in microglia plays an essential role in inflammatory responses to Mn toxicity by regulating cytokines and chemokines that amplify the activation of astrocytes. |
3.3. Mechanisms and Pathobiology of Alzheimer’s Disease Development Related to Lead Exposure
3.3.1. Lead Exposure Increases Tau Hyperphosphorylation, DNA Methylation, Amyloid Accumulation, Mitochondrial Dysfunction, Microglia Activation, and Neuronal Cell Death
3.3.2. Lead Exposure Increases DNA Damage, Causes Excessive mtROS Accumulation, and Alters Calcium and Iron Homeostasis and Apoptosis
3.3.3. Lead Exposure Disrupts Brain Cholesterol Metabolism, Increases Aβ Accumulation and Amyloid Plaque Deposition, and Impairs Cholesterol Homeostasis
3.3.4. Lead Exposure Disturbed Global Gene Expression Patterns in a Sex-Specific Manner
| SL | References | Study Objective | Study Type | Exposure | Key Findings |
|---|---|---|---|---|---|
| 1 | Rogers et al., 2016 [98] | Investigated the effect of Pb on iron homeostasis proteins in human neurons. | Human neuroblastoma SH-SY5Y cells—in vitro | Cells (100, 250, 500, 750, 1000 µM for 24 h) | Pb inhibits APP translation, raising cytosolic iron levels. Through the restoration of APP production, iron supplementation protects cells from Pb toxicity. |
| 2 | Wang et al., 2022 [88] | Investigated how Pb affected microRNAs (miRNAs), post-transcriptional regulators that may be involved in the pathophysiology of AD. | Mice—animal model | Mice—drinking water (0.2% Pb acetate solution for 3 months) | Pb exposure modulates miR-124-3p/BACE1 pathway, upregulating BACE1 and impacting amyloidogenic processing resembling AD. |
| 3 | Bandaru et al., 2022 [99] | Investigated the mitophagy marker proteins, including PINK1 and Parkin, in differentiated SH-SY5Y cells to examine the impact of Pb exposure on the PINK1/Parkin dependent pathway. | SH-SY5Y cells | Cells (5 µM for 24 h) | Pb exposure decreases mitochondrial mass, elevates MPTP opening, depolarizes membrane potential, and increases ROS generation, inducing neurotoxicity through the PINK1/Parkin-mediated mitophagy. |
| 4 | Eid et al., 2016 [90] | Investigated how early life exposure to Pb can cause epigenetic modifications and late-life changes. | Mice model | Mice—drinking water (0.2% Pb-acetate from PND 1 to PND 20) | Prenatal Pb exposure alters epigenetic regulators, modifying AD-related gene expression via histone/DNA methylation changes. |
| 5 | Xie et al., 2023 [100] | Investigated the effects of Pb exposure on AD-like pathogenesis in human cortical neurons. | Human iPSC-derived cortical neurons as a model system | Neurons (15, 50 ppb for 25, 45 days) | Pb exposure disrupts Ca regulation, induces epigenetic changes, and promotes AD-related tau and Aβ pathology. |
| 6 | vonderEmbse et al., 2017 [91] | Investigated the association between early toxicant exposure and systematic microglia activation, possibly reversing the pathological severity of AD. | Mouse model | Mouse—gavage technique (100 ppm for 3–6 months) | Pb exposure activates microglia and increases amyloid buildup. Females might be more vulnerable to AD as a result of early-life microglial injury. |
| 7 | Masoud et al., 2016 [92] | Investigated the early postnatal Pb exposure and its effect on the expression of select miRNAs, targeting AD-related protein. | Mice model | Mouse—mother’s milk (0.2% Pb acetate from Postnatal Day 1 (PND 1, first 24 h after birth) to PND 20) | Pb exposure triggers changes in miRNA expression targeting AD-related proteins. |
| 8 | Lee and Freeman, 2016 [105] | Investigated the connection between latent neurological changes and embryonic Pb exposure utilizing the brains of adult male and female zebrafish. | Zebrafish brain | Aquaria water (10 µg/L Pb [~0.048 µM] until 12 months of age) | Pb exposure in zebrafish leads to neurodegenerative gene expression changes in a sex-specific manner. |
| 9 | Wu et al., 2020 [93] | Examined how Pb exposure exacerbates the development of AD in mice by compromising the blood–brain barrier (BBB). | Mice model | Mice—drinking water (200 mg/L and 500 mg/L Pb acetate, until the age of 7-months) | Pb exposure results in aberrant alterations in BBB junction proteins and hastens the deposition of Aβ1–42 in the brains of APP/PS1 mice including rise of p-tau expression in APP/PS1 and C57BL/6J mice. |
| 10 | Gu et al., 2020, [94] | Investigated long-term Pb exposure effect on the BBB’s permeability using the Dynamic Contrast-Enhanced Computerized Tomography (DCE-CT) technique. | Mice model | Mice—oral gavage (13.5 or 27 mg/kg Pb) for 4 weeks) | Pb exposure increases the permeability surface area product, significantly induces brain perfusion, disrupts the brain vasculature and damages the BBB system. |
| 11 | Bandaru et al., 2023 [101] | Examined how Pb induces AD, via mitochondrial damage, using human neuronal cells. | Human neuronal cells (SH-SY5Y) | Human neuronal cells (5 µM for 24 h) | Pb exposure increases oxidative stress, reduces GSH levels, and antioxidant-related gene expressions, such as SOD2 (MnSOD) and Gpx4; mitochondrial dysfunction resembling features of AD. |
| 12 | Lee and Freeman, 2020 [106] | Investigated the relation between neurotoxic Pb exposure and de novo copy number alterations (CNAs) using zebrafish fibroblast cells. | Zebrafish cells | Cells (0.24, 2.4, or 24 μM Pb for 72 h) | Pb exposure causes de novo CNAs, which may be a mechanism causing neurological diseases. Amyloid precursor protein (APP), a crucial molecular target linked to the AD pathophysiology is connected to nearly every gene in a molecular network. |
| 13 | Liu et al., 2023 [104] | Investigated long-term Pb exposure effect on Aβ buildup in cerebral capillaries and the expression of a vital Aβ transporter, low-density lipoprotein receptor protein-1 (LRP1), in brain parenchyma and capillaries in Sprague-Dawley rats. | Rat model | Rat—oral gavage (14, 27 mg/kg for 4 or 8 weeks.) | Pb exposure increases Aβ buildup in the cerebral vasculature both in vitro and in vivo, including reduction of LRP1 expression in the studied brain region and fractions. |
| 14 | Eid et al., 2018 [95] | Investigated early life Pb exposure and latent over expression of AD-related gene regulation histone activation pathways. | Mice model | Drinking water (0.2% Pb acetate from PND 1 to PND 20) | Pb exposure produces a global gene repression profile via DNA methylation and histone modification pathways except in the genes linked to AD. |
| 15 | Bihaqi et al., 2014 [89] | Investigated infantile postnatal Pb exposure on the expression of tau in the aged mice’s brain. | Mice model | Drinking water (0.2% Pb acetate from PND 1 to PND 20) | Pb exposure increases tau protein and tau mRNA levels, serine/threonine phosphatase activity, and a changed p35/p25 protein ratio. |
| 16 | Bihaqi et al., 2018 [96] | Examined developmental Pb exposure effects on α-Syn pathways in tau-knockout mice and SHSY5Y cells. | Mice and SHSY5Y cells | Drinking water (0.2% Pb acetate from PND 1 to PND 20 and 5, 50, 100 µM for 48 h for cells) | Pb exposure upregulates α-Syn, Caspase-3, glycogen synthase kinase 3β (GSK-3β), and α-Syn and its phosphorylated forms via epigenetic mechanisms. |
| 17 | Huang et al., 2024 [97] | Investigated how Pb exposure aggravates AD progression and the role of microglial activation using APP/PS1 mice and Aβ1-42-treated BV-2 cells. | Mice model and BV-2 microglial cells | Drinking water (100 ppm until 4 months of age) | Chronic Pb exposure exacerbates memory and learning deficits in APP/PS1 mice, activating microglia via the mitochondrial dysfunction and excessive mtROS accumulation. |
| 18 | Zhou et al., 2018 [44] | Investigated the role of cholesterol metabolism in Pb-induced premature AD-like pathology in rats. | Male Sprague-Dawley rats | Drinking water (0.5–2% Pb acetate for 4 weeks) | Pb exposure disrupts brain cholesterol metabolism, triggering SREBP2-BACE1, reducing HMG-CR and LDL-R, and increasing ABCA1 and LXR-α, causing AD-like pathology. |
| 19 | Ayyalasomayajula et al., 2019 [102] | Examined epigallocatechin gallate’s (EGCG) role in reducing oxidative stress, apoptosis in neural cells exposed to Pb, β-APs. | SH-SY5Y cells | Cells (5 μM for 24 h) | Pb exposure increases oxidative stress, annexin V, Caspase-3, and apoptosis through the alteration of expression levels of Bax and Bcl2. |
| 20 | Lee et al., 2017 [107] | Investigated embryonic Pb exposure effect on AD related genes via sex-specific alterations in sorl1 expression in adult zebrafish. | Zebrafish | Aquaria water (100 ppb for 72 hpf) | Females are more prone to the onset of AD and SORL1, an AD genetic risk factor plays a critical role for this phenomenon. |
| 21 | Lokesh et al., 2024 [103] | Examined Pb and Aβ peptides’ (1–40 and 25–35) interaction with CDK5/p25 to understand Pb-induced neurotoxicity in neurons. | Human SH-SY5Y cells | Cells (5 μM for 24 and 48 h) | Pb exposure alters calcium levels, reduces antioxidants, increases oxidative damage, and disrupts CDK5-p25 signaling, affecting DNA repair and metabolism. |
3.4. Mechanisms and Pathobiology of Alzheimer’s Disease Development Related to Cadmium Exposure
3.4.1. Cadmium Exposure Increased Anxiety-like Behavior, Spatial Reference Memory Damage, Aβ Plaque Deposition, and Microglial Activation
3.4.2. Cadmium Exposure Induces Neuronal Cell Death Through Apoptosis and Autophagy
| SL | References | Study Objective | Study Type | Exposure | Key Findings |
|---|---|---|---|---|---|
| 1 | Liu, et al., 2023 [111] | Examined how low-dose environmental Cd exposure contributes to AD development. | Mice model | Mice—drinking water (1, or 10 mg/L for until 6 months of age) | Cd exposure causes a rise in anxiety-like behavior and disorderly movement, disruption to spatial reference memory, Aβ plaque formation in mice brains, an increase in microglia expression in the brain, and elevates IL-6 levels in the cortex and serum. |
| 2 | Zhang et al., 2021 [43] | Investigated Cd interactions with ApoE4 gene variants to modify the gut–liver axis in mice. | Mouse model | Mice—drinking water (0.6 and 3 mg/L for 14 weeks) | Cd exposure disrupts the gut–liver axis, increasing microbial AD biomarkers and inflammatory hepatic pathways. |
| 3 | Wang et al., 2022 [112] | Investigated Cd-ApoE effects on the transcriptome alterations in the livers and brains of ApoE3/ApoE4 transgenic mice. | Mice model | Mice—drinking water (0.6 mg/L for 6 weeks) | Cd dysregulates drug-processing genes in brain and liver, varying by sex and ApoE genotype. |
| 4 | Matsushita et al., 2023 [113] | Explored rescuing of Cd-induced cognitive impairment in ApoE4-KI mice via genetic and neurogenesis activation. | Mice model | Drinking water (0.6 mg/L) | Selective and conditional stimulation of adult neurogenesis restores Cd-induced impairments in hippocampus-dependent short-term spatial memory. |
| 5 | del Pino et al., 2016 [117] | Investigated how Cd induces cell death in basal forebrain cholinergic neurons. | SN56 Cell lines | SN56 cells (1, 10, 100 µM for 24 h) | Cd exposure induces cell death in cholinergic neurons, by blocking the M1 receptor, overexpressing AChE-S and GSK-3β, downregulating AChE-R, and raising the levels of Aβ, total, and phosphorylated tau proteins. |
| 6 | Deng et al., 2024 [116] | Investigated the underlying mechanism and impact of autophagy on the development of AD caused by environmental Cd. | Mouse neuroblastoma cells (Neuro-2a cells) | Cells (1, 2, and 4 μM for 24, 48 and 72 h) | Cd exposure disrupts autophagy, causing APP buildup and neuronal death. |
| 7 | Arab et al., 2023 [118] | Examined topiramate’s potential to counter Cd-induced cognitive deficits via hippocampal oxidative stress, apoptosis, and autophagy. | Rats model | Rat—oral gavage (5 mg/kg/day for eight weeks) | Cd exposure impairs retention memory, deteriorates the recognition memory, triggers hippocampal neuronal degeneration and signals as well as induces hippocampal apoptotic and autophagic cell death. |
| 8 | Zhang et al., 2020 [114] | Examined ApoE-e4 and Cd exposure interactions on cognition using an Alzheimer’s mouse model expressing human ApoE-e3 (ApoE3-KI [knock-in]) or ApoE-e4 (ApoE4-KI). | Mouse model | Mouse—drinking water (0.6 mg/L for 14 weeks) | Cd exposure accelerates cognitive decline, likely via reduced hippocampal neurogenesis. |
| 9 | Qian et al., 2024 [115] | Examined cellular senescence in AD by exploring Cd exposure effects on neuron senescence in vivo/vitro. | Mice and SHSY5Y cells | Mice—drinking water (10 mg/L for 7 weeks) | Cd induces neural senescence via p53/p21/Rb activation and SEL1L/HRD1-mediated SigmaR1 degradation, suggesting SigmaR1 as a neuroprotective biomarker. |
4. Discussion
5. Conclusions and Future Directions
5.1. Future Directions
5.1.1. Longitudinal and Epidemiological Studies
5.1.2. Mechanistic Studies
5.1.3. Therapeutic Development
5.1.4. Intervention Studies
5.1.5. Environmental Policy and Public Health
5.1.6. Advanced Models
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Islam, F.; Shohag, S.; Akhter, S.; Islam, M.R.; Sultana, S.; Mitra, S.; Chandran, D.; Khandaker, M.U.; Ashraf, G.M.; Idris, A.M.; et al. Exposure of metal toxicity in Alzheimer’s disease: An extensive review. Front. Pharmacol. 2022, 13, 903099. [Google Scholar] [CrossRef]
- Rahman, M.A.; Hannan, M.A.; Uddin, M.J.; Rahman, M.S.; Rashid, M.M.; Kim, B. Exposure to Environmental Arsenic and Emerging Risk of Alzheimer’s Disease: Perspective Mechanisms, Management Strategy, and Future Directions. Toxics 2021, 9, 188. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2016 Alzheimer’s disease facts and figures. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2016, 12, 459–509. [Google Scholar] [CrossRef]
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 40. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef]
- Tyczyńska, M.; Gędek, M.; Brachet, A.; Stręk, W.; Flieger, J.; Teresiński, G.; Baj, J. Trace Elements in Alzheimer’s Disease and Dementia: The Current State of Knowledge. J. Clin. Med. 2024, 13, 2381. [Google Scholar] [CrossRef]
- GBD 2019 Dementia Forecasting Collaborators. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: An analysis for the Global Burden of Disease Study 2019. Lancet Public Health 2022, 7, e105–e125. [Google Scholar] [CrossRef]
- Li, X.; Feng, X.; Sun, X.; Hou, N.; Han, F.; Liu, Y. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2019. Front. Aging Neurosci. 2022, 14, 937486. [Google Scholar] [CrossRef]
- Rostagno, A.A. Pathogenesis of Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 24, 107. [Google Scholar] [CrossRef]
- Blacker, D.; Tanzi, R.E. The genetics of Alzheimer disease: Current status and future prospects. Arch. Neurol. 1998, 55, 294–296. [Google Scholar] [CrossRef]
- Lanoiselée, H.M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. collaborators of the CNR-MAJ project APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017, 14, e1002270. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Santiago, J.A.; Bernstein, M.; Potashkin, J.A. Diet and lifestyle impact the development and progression of Alzheimer’s dementia. Front. Nutr. 2023, 10, 1213223. [Google Scholar] [CrossRef] [PubMed]
- Bellenguez, C.; Küçükali, F.; Jansen, I.E.; Kleineidam, L.; Moreno-Grau, S.; Amin, N.; Naj, A.C.; Campos-Martin, R.; Grenier-Boley, B.; Andrade, V.; et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 2022, 54, 412–436. [Google Scholar] [CrossRef]
- Bird, T.D. Genetic aspects of Alzheimer disease. Genet. Med. 2008, 10, 231–239. [Google Scholar] [CrossRef]
- Lau, V.; Ramer, L.; Tremblay, M.È. An aging, pathology burden, and glial senescence build-up hypothesis for late onset Alzheimer’s disease. Nat. Commun. 2023, 14, 1670. [Google Scholar] [CrossRef]
- Rahman, M.A.; Rahman, M.S.; Uddin, M.J.; Mamum-Or-Rashid, A.N.M.; Pang, M.G.; Rhim, H. Emerging risk of environmental factors: Insight mechanisms of Alzheimer’s diseases. Environ. Sci. Pollut. Res. Int. 2020, 27, 44659–44672. [Google Scholar] [CrossRef]
- Xia, X.; Jiang, Q.; McDermott, J.; Han, J.J. Aging and Alzheimer’s disease: Comparison and associations from molecular to system level. Aging Cell 2018, 17, e12802. [Google Scholar] [CrossRef]
- Murphy, M.P.; LeVine, H., III. Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimer’s Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef]
- Medeiros, R.; Baglietto-Vargas, D.; LaFerla, F.M. The role of tau in Alzheimer’s disease and related disorders. CNS Neurosci. Ther. 2011, 17, 514–524. [Google Scholar] [CrossRef]
- Yu, Y.; Chen, R.; Mao, K.; Deng, M.; Li, Z. The Role of Glial Cells in Synaptic Dysfunction: Insights into Alzheimer’s Disease Mechanisms. Aging Dis. 2024, 15, 459–479. [Google Scholar] [CrossRef]
- Ashleigh, T.; Swerdlow, R.H.; Beal, M.F. The role of mitochondrial dysfunction in Alzheimer’s disease pathogenesis. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2023, 19, 333–342. [Google Scholar] [CrossRef]
- Dhapola, R.; Beura, S.K.; Sharma, P.; Singh, S.K.; HariKrishnaReddy, D. Oxidative stress in Alzheimer’s disease: Current knowledge of signaling pathways and therapeutics. Mol. Biol. Rep. 2024, 51, 48. [Google Scholar] [CrossRef]
- Gella, A.; Durany, N. Oxidative stress in Alzheimer disease. Cell Adhes. Migr. 2009, 3, 88–93. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet. Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Joshi, M.; Joshi, S.; Khambete, M.; Degani, M. Role of calcium dysregulation in Alzheimer’s disease and its therapeutic implications. Chem. Biol. Drug Des. 2023, 101, 453–468. [Google Scholar] [CrossRef]
- Yu, J.T.; Chang, R.C.; Tan, L. Calcium dysregulation in Alzheimer’s disease: From mechanisms to therapeutic opportunities. Prog. Neurobiol. 2009, 89, 240–255. [Google Scholar] [CrossRef]
- Dosunmu, R.; Wu, J.; Basha, M.R.; Zawia, N.H. Environmental and dietary risk factors in Alzheimer’s disease. Expert Rev. Neurother. 2007, 7, 887–900. [Google Scholar] [CrossRef]
- Huat, T.J.; Camats-Perna, J.; Newcombe, E.A.; Valmas, N.; Kitazawa, M.; Medeiros, R. Metal Toxicity Links to Alzheimer’s Disease and Neuroinflammation. J. Mol. Biol. 2019, 431, 1843–1868. [Google Scholar] [CrossRef]
- Suresh, S.; Singh S, A.; Rushendran, R.; Vellapandian, C.; Prajapati, B. Alzheimer’s disease: The role of extrinsic factors in its development, an investigation of the environmental enigma. Front. Neurol. 2023, 14, 1303111. [Google Scholar] [CrossRef]
- Yegambaram, M.; Manivannan, B.; Beach, T.G.; Halden, R.U. Role of environmental contaminants in the etiology of Alzheimer’s disease: A review. Curr. Alzheimer Res. 2015, 12, 116–146. [Google Scholar] [CrossRef]
- Sheppard, O.; Coleman, M. Alzheimer’s Disease: Etiology, Neuropathology and Pathogenesis. In Alzheimer’s Disease: Drug Discovery; Huang, X., Ed.; Exon Publications: Brisbane, Australia, 2020. [Google Scholar]
- Henderson, A.S. The risk factors for Alzheimer’s disease: A review and a hypothesis. Acta Psychiatr. Scand. 1988, 78, 257–275. [Google Scholar] [CrossRef] [PubMed]
- Angon, P.B.; Islam, M.S.; Kc, S.; Das, A.; Anjum, N.; Poudel, A.; Suchi, S.A. Sources, effects and present perspectives of heavy metals contamination: Soil, plants and human food chain. Heliyon 2024, 10, e28357. [Google Scholar] [CrossRef] [PubMed]
- Tchounwou, P.B.; Yedjou, C.G.; Patlolla, A.K.; Sutton, D.J. Heavy metal toxicity and the environment. Exp. Suppl. 2012, 101, 133–164. [Google Scholar] [CrossRef]
- Bakulski, K.M.; Seo, Y.A.; Hickman, R.C.; Brandt, D.; Vadari, H.S.; Hu, H.; Park, S.K. Heavy Metals Exposure and Alzheimer’s Disease and Related Dementias. J. Alzheimer’s Dis. 2020, 76, 1215–1242. [Google Scholar] [CrossRef]
- Chin-Chan, M.; Navarro-Yepes, J.; Quintanilla-Vega, B. Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front. Cell. Neurosci. 2015, 9, 124. [Google Scholar] [CrossRef]
- Ijomone, O.M.; Ijomone, O.K.; Iroegbu, J.D.; Ifenatuoha, C.W.; Olung, N.F.; Aschner, M. Epigenetic influence of environmentally neurotoxic metals. Neurotoxicology 2020, 81, 51–65. [Google Scholar] [CrossRef]
- Horton, C.J.; Weng, H.Y.; Wells, E.M. Association between blood lead level and subsequent Alzheimer’s disease mortality. Environ. Epidemiol. 2019, 3, e045. [Google Scholar] [CrossRef]
- Min, J.Y.; Min, K.B. Blood cadmium levels and Alzheimer’s disease mortality risk in older US adults. Environ. Health A Glob. Access Sci. Source 2016, 15, 69. [Google Scholar] [CrossRef]
- Spitznagel, B.D.; Buchanan, R.A.; Consoli, D.C.; Thibert, M.K.; Bowman, A.B.; Nobis, W.P.; Harrison, F.E. Acute manganese exposure impairs glutamatergic function in a young mouse model of Alzheimer’s disease. Neurotoxicology 2023, 95, 1–11. [Google Scholar] [CrossRef]
- Tripathi, M.K.; Kartawy, M.; Ginzburg, S.; Amal, H. Arsenic alters nitric oxide signaling similar to autism spectrum disorder and Alzheimer’s disease-associated mutations. Transl. Psychiatry 2022, 12, 127. [Google Scholar] [CrossRef]
- Wang, X.; Huang, X.; Zhou, L.; Chen, J.; Zhang, X.; Xu, K.; Huang, Z.; He, M.; Shen, M.; Chen, X.; et al. Association of arsenic exposure and cognitive impairment: A population-based cross-sectional study in China. Neurotoxicology 2021, 82, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Matsushita, M.; Zhang, L.; Wang, H.; Shi, X.; Gu, H.; Xia, Z.; Cui, J.Y. Cadmium exposure modulates the gut-liver axis in an Alzheimer’s disease mouse model. Commun. Biol. 2021, 4, 1398. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.C.; Gao, Z.Y.; Wang, J.; Wu, M.Q.; Hu, S.; Chen, F.; Liu, J.X.; Pan, H.; Yan, C.H. Lead exposure induces Alzheimers’s disease (AD)-like pathology and disturbes cholesterol metabolism in the young rat brain. Toxicol. Lett. 2018, 296, 173–183. [Google Scholar] [CrossRef]
- Peres, T.V.; Schettinger, M.R.; Chen, P.; Carvalho, F.; Avila, D.S.; Bowman, A.B.; Aschner, M. Manganese-induced neurotoxicity: A review of its behavioral consequences and neuroprotective strategies. BMC Pharmacol. Toxicol. 2016, 17, 57. [Google Scholar] [CrossRef]
- Martin, K.; Huggins, T.; King, C.; Carroll, M.A.; Catapane, E.J. The neurotoxic effects of manganese on the dopaminergic innervation of the gill of the bivalve mollusc, Crassostrea virginica. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2008, 148, 152–159. [Google Scholar] [CrossRef]
- Martins, A.C.; Morcillo, P., Jr.; Ijomone, O.M.; Venkataramani, V.; Harrison, F.E.; Lee, E.; Bowman, A.B.; Aschner, M. New Insights on the Role of Manganese in Alzheimer’s Disease and Parkinson’s Disease. Int. J. Environ. Res. Public Health 2019, 16, 3546. [Google Scholar] [CrossRef]
- Tong, Y.; Yang, H.; Tian, X.; Wang, H.; Zhou, T.; Zhang, S.; Yu, J.; Zhang, T.; Fan, D.; Guo, X.; et al. High manganese, a risk for Alzheimer’s disease: High manganese induces amyloid-β related cognitive impairment. J. Alzheimer’s Dis. 2014, 42, 865–878. [Google Scholar] [CrossRef]
- Canfield, R.L.; Jusko, T.A.; Kordas, K. Environmental lead exposure and children’s cognitive function. Riv. Ital. Di Pediatr. Ital. J. Pediatr. 2005, 31, 293–300. [Google Scholar]
- Neuwirth, L.S.; Lopez, O.E.; Schneider, J.S.; Markowitz, M.E. Low-level Lead Exposure Impairs Fronto-executive Functions: A Call to Update the DSM-5 with Lead Poisoning as a Neurodevelopmental Disorder. Psychol. Neurosci. 2020, 13, 299–325. [Google Scholar] [CrossRef]
- Reuben, A. Childhood Lead Exposure and Adult Neurodegenerative Disease. J. Alzheimer’s Dis. 2018, 64, 17–42. [Google Scholar] [CrossRef]
- Sanders, T.; Liu, Y.; Buchner, V.; Tchounwou, P.B. Neurotoxic effects and biomarkers of lead exposure: A review. Rev. Environ. Health 2009, 24, 15–45. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.H.; Ge, G.; Gao, K.; Pang, Y.; Chai, R.C.; Jia, X.H.; Kong, J.G.; Yu, A.C. Calcium Signaling Involvement in Cadmium-Induced Astrocyte Cytotoxicity and Cell Death Through Activation of MAPK and PI3K/Akt Signaling Pathways. Neurochem. Res. 2015, 40, 1929–1944. [Google Scholar] [CrossRef] [PubMed]
- Branca, J.J.V.; Fiorillo, C.; Carrino, D.; Paternostro, F.; Taddei, N.; Gulisano, M.; Pacini, A.; Becatti, M. Cadmium-Induced Oxidative Stress: Focus on the Central Nervous System. Antioxidants 2020, 9, 492. [Google Scholar] [CrossRef]
- Li, C.X.; Talukder, M.; Xu, Y.R.; Zhu, S.Y.; Wang, Y.X.; Li, J.L. Cadmium causes cerebral mitochondrial dysfunction through regulating mitochondrial HSF1. Environ. Pollut. 2024, 360, 124677. [Google Scholar] [CrossRef]
- Ali, T.; Khan, A.; Alam, S.I.; Ahmad, S.; Ikram, M.; Park, J.S.; Lee, H.J.; Kim, M.O. Cadmium, an Environmental Contaminant, Exacerbates Alzheimer’s Pathology in the Aged Mice’s Brain. Front. Aging Neurosci. 2021, 13, 650930. [Google Scholar] [CrossRef]
- Arruebarrena, M.A.; Hawe, C.T.; Lee, Y.M.; Branco, R.C. Mechanisms of Cadmium Neurotoxicity. Int. J. Mol. Sci. 2023, 24, 16558. [Google Scholar] [CrossRef]
- Peng, Q.; Bakulski, K.M.; Nan, B.; Park, S.K. Cadmium and Alzheimer’s disease mortality in U.S. adults: Updated evidence with a urinary biomarker and extended follow-up time. Environ. Res. 2017, 157, 44–51. [Google Scholar] [CrossRef]
- Hippius, H.; Neundörfer, G. The discovery of Alzheimer’s disease. Dialogues Clin. Neurosci. 2003, 5, 101–108. [Google Scholar] [CrossRef]
- Andrade, V.M.; Mateus, M.L.; Batoréu, M.C.; Aschner, M.; Marreilha dos Santos, A.P. Lead, Arsenic, and Manganese Metal Mixture Exposures: Focus on Biomarkers of Effect. Biol. Trace Elem. Res. 2015, 166, 13–23. [Google Scholar] [CrossRef]
- Rodríguez-Barranco, M.; Lacasaña, M.; Aguilar-Garduño, C.; Alguacil, J.; Gil, F.; González-Alzaga, B.; Rojas-García, A. Association of arsenic, cadmium and manganese exposure with neurodevelopment and behavioural disorders in children: A systematic review and meta-analysis. Sci. Total Environ. 2013, 454–455, 562–577. [Google Scholar] [CrossRef]
- Sanders, A.P.; Desrosiers, T.A.; Warren, J.L.; Herring, A.H.; Enright, D.; Olshan, A.F.; Meyer, R.E.; Fry, R.C. Association between arsenic, cadmium, manganese, and lead levels in private wells and birth defects prevalence in North Carolina: A semi-ecologic study. BMC Public Health 2014, 14, 955. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.E.; Shah, P.; Pollock, B.G.; Gerretsen, P.; Graff-Guerrero, A. Lead (Pb) in Alzheimer’s Dementia: A Systematic Review of Human Case- Control Studies. Curr. Alzheimer Res. 2019, 16, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Chib, S.; Singh, S. Manganese and related neurotoxic pathways: A potential therapeutic target in neurodegenerative diseases. Neurotoxicol. Teratol. 2022, 94, 107124. [Google Scholar] [CrossRef] [PubMed]
- Chin-Chan, M.; Cobos-Puc, L.; Alvarado-Cruz, I.; Bayar, M.; Ermolaeva, M. Early-life Pb exposure as a potential risk factor for Alzheimer’s disease: Are there hazards for the Mexican population? JBIC J. Biol. Inorg. Chem. 2019, 24, 1285–1303. [Google Scholar] [CrossRef]
- Gong, G.; O’Bryant, S.E. The arsenic exposure hypothesis for Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2010, 24, 311–316. [Google Scholar] [CrossRef]
- Rahaman, M.S.; Rahman, M.M.; Mise, N.; Sikder, M.T.; Ichihara, G.; Uddin, M.K.; Kurasaki, M.; Ichihara, S. Environmental arsenic exposure and its contribution to human diseases, toxicity mechanism and management. Environ. Pollut. 2021, 289, 117940. [Google Scholar] [CrossRef]
- Rahaman, M.S.; Mise, N.; Ichihara, S. Arsenic contamination in food chain in Bangladesh: A review on health hazards, socioeconomic impacts and implications. Hyg. Environ. Health Adv. 2022, 2, 100004. [Google Scholar] [CrossRef]
- Tyler, C.R.; Allan, A.M. The Effects of Arsenic Exposure on Neurological and Cognitive Dysfunction in Human and Rodent Studies: A Review. Curr. Environ. Health Rep. 2014, 1, 132–147. [Google Scholar] [CrossRef]
- Pakzad, D.; Akbari, V.; Sepand, M.R.; Aliomrani, M. Risk of neurodegenerative disease due to tau phosphorylation changes and arsenic exposure via drinking water. Toxicol. Res. 2021, 10, 325–333. [Google Scholar] [CrossRef]
- Niño, S.A.; Morales-Martínez, A.; Chi-Ahumada, E.; Carrizales, L.; Salgado-Delgado, R.; Pérez-Severiano, F.; Díaz-Cintra, S.; Jiménez-Capdeville, M.E.; Zarazúa, S. Arsenic Exposure Contributes to the Bioenergetic Damage in an Alzheimer’s Disease Model. ACS Chem. Neurosci. 2018, 10, 323–336. [Google Scholar] [CrossRef]
- Niño, S.A.; Vázquez-Hernández, N.; Arevalo-Villalobos, J.; Chi-Ahumada, E.; Martín-Amaya-Barajas, F.L.; Díaz-Cintra, S.; Martel-Gallegos, G.; González-Burgos, I.; Jiménez-Capdeville, M.E. Cortical Synaptic Reorganization Under Chronic Arsenic Exposure. Neurotox. Res. 2021, 39, 1970–1980. [Google Scholar] [CrossRef]
- Niño, S.A.; Martel-Gallegos, G.; Castro-Zavala, A.; Ortega-Berlanga, B.; Delgado, J.M.; Hernández-Mendoza, H.; Romero-Guzmán, E.; Ríos-Lugo, J.; Rosales-Mendoza, S.; Jiménez-Capdeville, M.E.; et al. Chronic Arsenic Exposure Increases Aβ (1–42) Production and Receptor for Advanced Glycation End Products Expression in Rat Brain. Chem. Res. Toxicol. 2018, 31, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Wisessaowapak, C.; Visitnonthachai, D.; Watcharasit, P.; Satayavivad, J. Prolonged arsenic exposure increases tau phosphorylation in differentiated SH-SY5Y cells: The contribution of GSK3 and ERK1/2. Environ. Toxicol. Pharmacol. 2021, 84, 103626. [Google Scholar] [CrossRef] [PubMed]
- Rizor, A.; Pajarillo, E.; Nyarko-Danquah, I.; Digman, A.; Mooneyham, L.; Son, D.S.; Aschner, M.; Lee, E. Manganese-induced reactive oxygen species activate IκB kinase to upregulate YY1 and impair glutamate transporter EAAT2 function in human astrocytes in vitro. Neurotoxicology 2021, 86, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Mattison, D.R.; Momoli, F.; Alyanak, C.; Aschner, M.; Baker, M.; Cashman, N.; Dydak, U.; Farhat, N.; Guilarte, T.R.; Karyakina, N.; et al. Diagnosis of manganism and manganese neurotoxicity: A workshop report. Med. Int. 2024, 4, 11. [Google Scholar] [CrossRef]
- Ye, Q.; Park, J.E.; Gugnani, K.; Betharia, S.; Pino-Figueroa, A.; Kim, J. Influence of iron metabolism on manganese transport and toxicity. Met. Integr. Biometal Sci. 2017, 9, 1028–1046. [Google Scholar] [CrossRef]
- Xu, Y.; Wei, L.; Tang, S.; Shi, Q.; Wu, B.; Yang, X.; Zou, Y.; Wang, X.; Ao, Q.; Meng, L.; et al. Regulation PP2Ac methylation ameliorating autophagy dysfunction caused by Mn is associated with mTORC1/ULK1 pathway. Food Chem. Toxicol. 2021, 156, 112441. [Google Scholar] [CrossRef]
- Pajarillo, E.; Rizor, A.; Son, D.S.; Aschner, M.; Lee, E. The transcription factor REST up-regulates tyrosine hydroxylase and antiapoptotic genes and protects dopaminergic neurons against manganese toxicity. J. Biol. Chem. 2020, 295, 3040–3054. [Google Scholar] [CrossRef]
- Lu, C.; Meng, Z.; He, Y.; Xiao, D.; Cai, H.; Xu, Y.; Liu, X.; Wang, X.; Mo, L.; Liang, Z.; et al. Involvement of gap junctions in astrocyte impairment induced by manganese exposure. Brain Res. Bull. 2018, 140, 107–113. [Google Scholar] [CrossRef]
- Kirkley, K.S.; Popichak, K.A.; Afzali, M.F.; Legare, M.E.; Tjalkens, R.B. Microglia amplify inflammatory activation of astrocytes in manganese neurotoxicity. J. Neuroinflamm. 2017, 14, 99. [Google Scholar] [CrossRef]
- Fan, R.Z.; Sportelli, C.; Lai, Y.; Salehe, S.S.; Pinnell, J.R.; Brown, H.J.; Richardson, J.R.; Luo, S.; Tieu, K. A partial Drp1 knockout improves autophagy flux independent of mitochondrial function. Mol. Neurodegener. 2024, 19, 26. [Google Scholar] [CrossRef]
- Pajarillo, E.; Demayo, M.; Digman, A.; Nyarko-Danquah, I.; Son, D.S.; Aschner, M.; Lee, E. Deletion of RE1-silencing transcription factor in striatal astrocytes exacerbates manganese-induced neurotoxicity in mice. Glia 2022, 70, 1886–1901. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, J.; Yang, X.; Li, Z.; Wang, J.; Lu, C.; Nan, A.; Zou, Y. Dysregulated APP expression and α-secretase processing of APP is involved in manganese-induced cognitive impairment. Ecotoxicol. Environ. Saf. 2021, 220, 112365. [Google Scholar] [CrossRef] [PubMed]
- Bakulski, K.M.; Hu, H.; Park, S.K. Lead, cadmium and Alzheimer’s disease. Genet. Neurol. Behav. Diet Dement. 2020, 2, 813–830. [Google Scholar] [CrossRef]
- WHO. Exposure to Lead: A Major Public Health Concern. Preventing Disease Through Healthy Environments; World Health Organization: Geneva, Switzerland, 2010. [Google Scholar]
- Grandjean, P.; Bellanger, M. Calculation of the disease burden associated with environmental chemical exposures: Application of toxicological information in health economic estimation. Environ. Health A Glob. Access Sci. Source 2017, 16, 123. [Google Scholar] [CrossRef]
- Wang, R.; Wu, Z.; Liu, R.; Bai, L.; Lin, Y.; Ba, Y.; Huang, H. Age-related miRNAs dysregulation and abnormal BACE1 expression following Pb exposure in adolescent mice. Environ. Toxicol. 2022, 37, 1902–1913. [Google Scholar] [CrossRef]
- Bihaqi, S.W.; Bahmani, A.; Adem, A.; Zawia, N.H. Infantile postnatal exposure to lead (Pb) enhances tau expression in the cerebral cortex of aged mice: Relevance to AD. Neurotoxicology 2014, 44, 114–120. [Google Scholar] [CrossRef]
- Eid, A.; Bihaqi, S.W.; Renehan, W.E.; Zawia, N.H. Developmental lead exposure and lifespan alterations in epigenetic regulators and their correspondence to biomarkers of Alzheimer’s disease. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2016, 2, 123–131. [Google Scholar] [CrossRef]
- vonderEmbse, A.N.; Hu, Q.; DeWitt, J.C. Developmental toxicant exposure in a mouse model of Alzheimer’s disease induces differential sex-associated microglial activation and increased susceptibility to amyloid accumulation. J. Dev. Orig. Health Dis. 2017, 8, 493–501. [Google Scholar] [CrossRef]
- Masoud, A.M.; Bihaqi, S.W.; Machan, J.T.; Zawia, N.H.; Renehan, W.E. Early-Life Exposure to Lead (Pb) Alters the Expression of microRNA that Target Proteins Associated with Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 51, 1257–1264. [Google Scholar] [CrossRef]
- Wu, S.; Liu, H.; Zhao, H.; Wang, X.; Chen, J.; Xia, D.; Xiao, C.; Cheng, J.; Zhao, Z.; He, Y. Environmental lead exposure aggravates the progression of Alzheimer’s disease in mice by targeting on blood brain barrier. Toxicol. Lett. 2020, 319, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Territo, P.R.; Persohn, S.A.; Bedwell, A.A.; Eldridge, K.; Speedy, R.; Chen, Z.; Zheng, W.; Du, Y. Evaluation of chronic lead effects in the blood brain barrier system by DCE-CT. J. Trace Elem. Med. Biol. 2020, 62, 126648. [Google Scholar] [CrossRef]
- Eid, A.; Bihaqi, S.W.; Hemme, C.; Gaspar, J.M.; Hart, R.P.; Zawia, N.H. Histone acetylation maps in aged mice developmentally exposed to lead: Epigenetic drift and Alzheimer-related genes. Epigenomics 2018, 10, 573–583. [Google Scholar] [CrossRef]
- Bihaqi, S.W.; Alansi, B.; Masoud, A.M.; Mushtaq, F.; Subaiea, G.M.; Zawia, N.H. Influence of Early Life Lead (Pb) Exposure on α-Synuclein, GSK-3β and Caspase-3 Mediated Tauopathy: Implications on Alzheimer’s Disease. Curr. Alzheimer Res. 2018, 15, 1114–1122. [Google Scholar] [CrossRef]
- Huang, D.; Chen, L.; Ji, Q.; Xiang, Y.; Zhou, Q.; Chen, K.; Zhang, X.; Zou, F.; Zhang, X.; Zhao, Z.; et al. Lead aggravates Alzheimer’s disease pathology via mitochondrial copper accumulation regulated by COX17. Redox Biol. 2024, 69, 102990. [Google Scholar] [CrossRef]
- Rogers, J.T.; Venkataramani, V.; Washburn, C.; Liu, Y.; Tummala, V.; Jiang, H.; Smith, A.; Cahill, C.M. A role for amyloid precursor protein translation to restore iron homeostasis and ameliorate lead (Pb) neurotoxicity. J. Neurochem. 2016, 138, 479–494. [Google Scholar] [CrossRef]
- Bandaru, L.J.M.; Ayyalasomayajula, N.; Murumulla, L.; Dixit, P.K.; Challa, S. Defective mitophagy and induction of apoptosis by the depleted levels of PINK1 and Parkin in Pb and β-amyloid peptide induced toxicity. Toxicol. Mech. Methods 2022, 32, 559–568. [Google Scholar] [CrossRef]
- Xie, J.; Wu, S.; Szadowski, H.; Min, S.; Yang, Y.; Bowman, A.B.; Rochet, J.C.; Freeman, J.L.; Yuan, C. Developmental Pb exposure increases AD risk via altered intracellular Ca2+ homeostasis in hiPSC-derived cortical neurons. J. Biol. Chem. 2023, 299, 105023. [Google Scholar] [CrossRef]
- Bandaru, L.J.M.; Murumulla, L.; Challa, S. Exposure of combination of environmental pollutant, lead (Pb) and β-amyloid peptides causes mitochondrial dysfunction and oxidative stress in human neuronal cells. J. Bioenerg. Biomembr. 2023, 55, 79–89. [Google Scholar] [CrossRef]
- Ayyalasomayajula, N.; Ajumeera, R.; Chellu, C.S.; Challa, S. Mitigative effects of epigallocatechin gallate in terms of diminishing apoptosis and oxidative stress generated by the combination of lead and amyloid peptides in human neuronal cells. J. Biochem. Mol. Toxicol. 2019, 33, e22393. [Google Scholar] [CrossRef]
- Lokesh, M.; Bandaru, L.J.M.; Rajanna, A.; Rao, J.S.; Challa, S. Unveiling Potential Neurotoxic Mechansisms: Pb-Induced Activation of CDK5-p25 Signaling Axis in Alzheimer’s Disease Development, Emphasizing CDK5 Inhibition and Formation of Toxic p25 Species. Mol. Neurobiol. 2024, 61, 3090–3103. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.L.; Shen, X.; Gu, H.; Zhao, G.; Du, Y.; Zheng, W. High affinity of β-amyloid proteins to cerebral capillaries: Implications in chronic lead exposure-induced neurotoxicity in rats. Fluids Barriers CNS 2023, 20, 32. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Freeman, J.L. Embryonic exposure to 10 μg L−1 lead results in female-specific expression changes in genes associated with nervous system development and function and Alzheimer’s disease in aged adult zebrafish brain. Met. Integr. Biometal Sci. 2016, 8, 589–596. [Google Scholar] [CrossRef]
- Lee, J.; Freeman, J.L. Exposure to the Heavy-Metal Lead Induces DNA Copy Number Alterations in Zebrafish Cells. Chem. Res. Toxicol. 2020, 33, 2047–2053. [Google Scholar] [CrossRef]
- Lee, J.; Peterson, S.M.; Freeman, J.L. Sex-specific characterization and evaluation of the Alzheimer’s disease genetic risk factor sorl1 in zebrafish during aging and in the adult brain following a 100 ppb embryonic lead exposure. J. Appl. Toxicol. 2017, 37, 400–407. [Google Scholar] [CrossRef]
- ATSDR. Toxicological Profile for Cadmium. 2012. Available online: https://www.atsdr.cdc.gov/toxprofiles/tp5.pdf (accessed on 12 December 2024).
- Straif, K.; Benbrahim-Tallaa, L.; Baan, R.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Bouvard, V.; ∙Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogensepart C: Metals, arsenic, dusts, and fibres. Lancet Oncol. 2009, 10, 453–454. [Google Scholar]
- Satarug, S.; Garrett, S.H.; Sens, M.A.; Sens, D.A. Cadmium, environmental exposure, and health outcomes. Environ. Health Perspect. 2010, 118, 182–190. [Google Scholar] [CrossRef]
- Liu, J.; Xie, Y.; Lu, Y.; Zhao, Z.; Zhuang, Z.; Yang, L.; Huang, H.; Li, H.; Mao, Z.; Pi, S.; et al. APP/PS1 Gene-Environmental Cadmium Interaction Aggravates the Progression of Alzheimer’s Disease in Mice via the Blood-Brain Barrier, Amyloid-β, and Inflammation. J. Alzheimer’s Dis. 2023, 94, 115–136. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, L.; Xia, Z.; Cui, J.Y. Effect of Chronic Cadmium Exposure on Brain and Liver Transporters and Drug-Metabolizing Enzymes in Male and Female Mice Genetically Predisposed to Alzheimer’s Disease. Drug Metab. Dispos. Biol. Fate Chem. 2022, 50, 1414–1428. [Google Scholar] [CrossRef]
- Matsushita, M.T.; Wang, H.; Abel, G.M.; Xia, Z. Inducible and Conditional Activation of Adult Neurogenesis Rescues Cadmium-Induced Hippocampus-Dependent Memory Deficits in ApoE4-KI Mice. Int. J. Mol. Sci. 2023, 24, 9118. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, H.; Abel, G.M.; Storm, D.R.; Xia, Z. The Effects of Gene-Environment Interactions Between Cadmium Exposure and Apolipoprotein E4 on Memory in a Mouse Model of Alzheimer’s Disease. Toxicol. Sci. 2020, 173, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.; Li, T.Y.; Zheng, Z.X.; Zhang, H.Y.; Xu, W.Q.; Mo, S.M.; Cui, J.J.; Chen, W.J.; Lin, Y.C.; Lin, Z.N. The involvement of SigmaR1K142 degradation mediated by ERAD in neural senescence linked with CdCl2 exposure. J. Hazard. Mater. 2024, 472, 134466. [Google Scholar] [CrossRef]
- Deng, P.; Fan, T.; Gao, P.; Peng, Y.; Li, M.; Li, J.; Qin, M.; Hao, R.; Wang, L.; Li, M.; et al. SIRT5-Mediated Desuccinylation of RAB7A Protects Against Cadmium-Induced Alzheimer’s Disease-Like Pathology by Restoring Autophagic Flux. Adv. Sci. 2024, 11, e2402030. [Google Scholar] [CrossRef]
- del Pino, J.; Zeballos, G.; Anadón, M.J.; Moyano, P.; Díaz, M.J.; García, J.M.; Frejo, M.T. Cadmium-induced cell death of basal forebrain cholinergic neurons mediated by muscarinic M1 receptor blockade, increase in GSK-3β enzyme, β-amyloid and tau protein levels. Arch. Toxicol. 2016, 90, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Arab, H.H.; Eid, A.H.; Yahia, R.; Alsufyani, S.E.; Ashour, A.M.; El-Sheikh, A.A.K.; Darwish, H.W.; Saad, M.A.; Al-Shorbagy, M.Y.; Masoud, M.A. Targeting Autophagy, Apoptosis, and SIRT1/Nrf2 Axis with Topiramate Underlies Its Neuroprotective Effect against Cadmium-Evoked Cognitive Deficits in Rats. Pharmaceuticals 2023, 16, 1214. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, G.; Rahaman, M.S.; Perez, E.; Khan, K.M. Associations of Environmental Exposure to Arsenic, Manganese, Lead, and Cadmium with Alzheimer’s Disease: A Review of Recent Evidence from Mechanistic Studies. J. Xenobiot. 2025, 15, 47. https://doi.org/10.3390/jox15020047
Ahmed G, Rahaman MS, Perez E, Khan KM. Associations of Environmental Exposure to Arsenic, Manganese, Lead, and Cadmium with Alzheimer’s Disease: A Review of Recent Evidence from Mechanistic Studies. Journal of Xenobiotics. 2025; 15(2):47. https://doi.org/10.3390/jox15020047
Chicago/Turabian StyleAhmed, Giasuddin, Md. Shiblur Rahaman, Enrique Perez, and Khalid M. Khan. 2025. "Associations of Environmental Exposure to Arsenic, Manganese, Lead, and Cadmium with Alzheimer’s Disease: A Review of Recent Evidence from Mechanistic Studies" Journal of Xenobiotics 15, no. 2: 47. https://doi.org/10.3390/jox15020047
APA StyleAhmed, G., Rahaman, M. S., Perez, E., & Khan, K. M. (2025). Associations of Environmental Exposure to Arsenic, Manganese, Lead, and Cadmium with Alzheimer’s Disease: A Review of Recent Evidence from Mechanistic Studies. Journal of Xenobiotics, 15(2), 47. https://doi.org/10.3390/jox15020047