
Bisphenol A (BPA) and Cardiovascular or Cardiometabolic Diseases


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Relationship between Blood or Urinary BPA Levels and Cardiovascular or Cardiometabolic Diseases
2.1. BPA Levels and Diabetes
2.2. BPA Levels and Obesity
2.3. BPA Levels and Hypertension
2.4. BPA Levels and Kidney Diseases
2.5. BPA Levels and Cardiovascular Diseases
2.6. BPA Levels and Liver Diseases
3. Effects of BPA on Cardiovascular or Cardiometabolic Organs
3.1. Effects of BPA on Skeletal Muscles
3.2. Effects of BPA on Adipocytes and Adipose Tissues
3.2.1. BPA-Mediated Adipogenesis
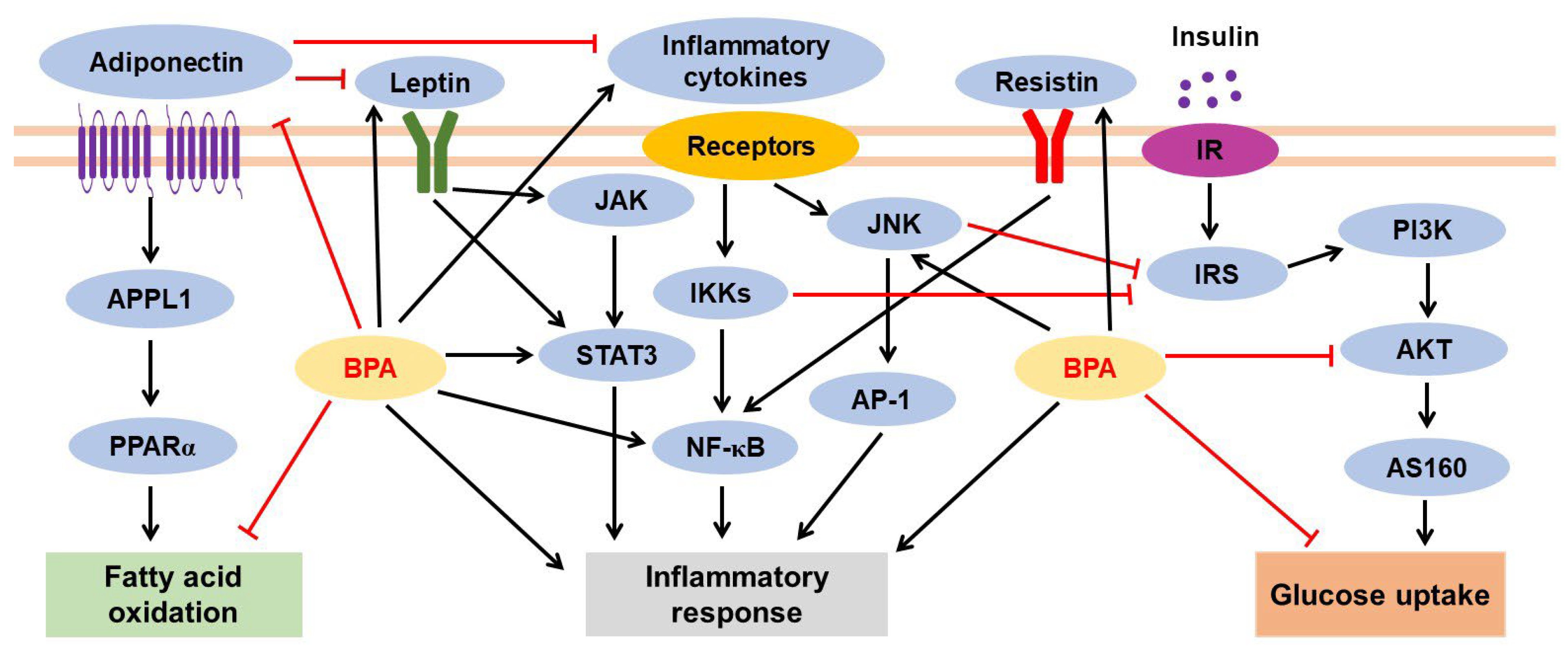
3.2.2. BPA-Mediated Inflammatory Response and Adipokine Dysregulation
3.2.3. BPA-Induced Insulin Resistance
3.2.4. BPA Levels in Adipose Tissues
3.3. Effects of BPA on Pancreatic Tissues
3.4. Effects of BPA on Liver Tissues
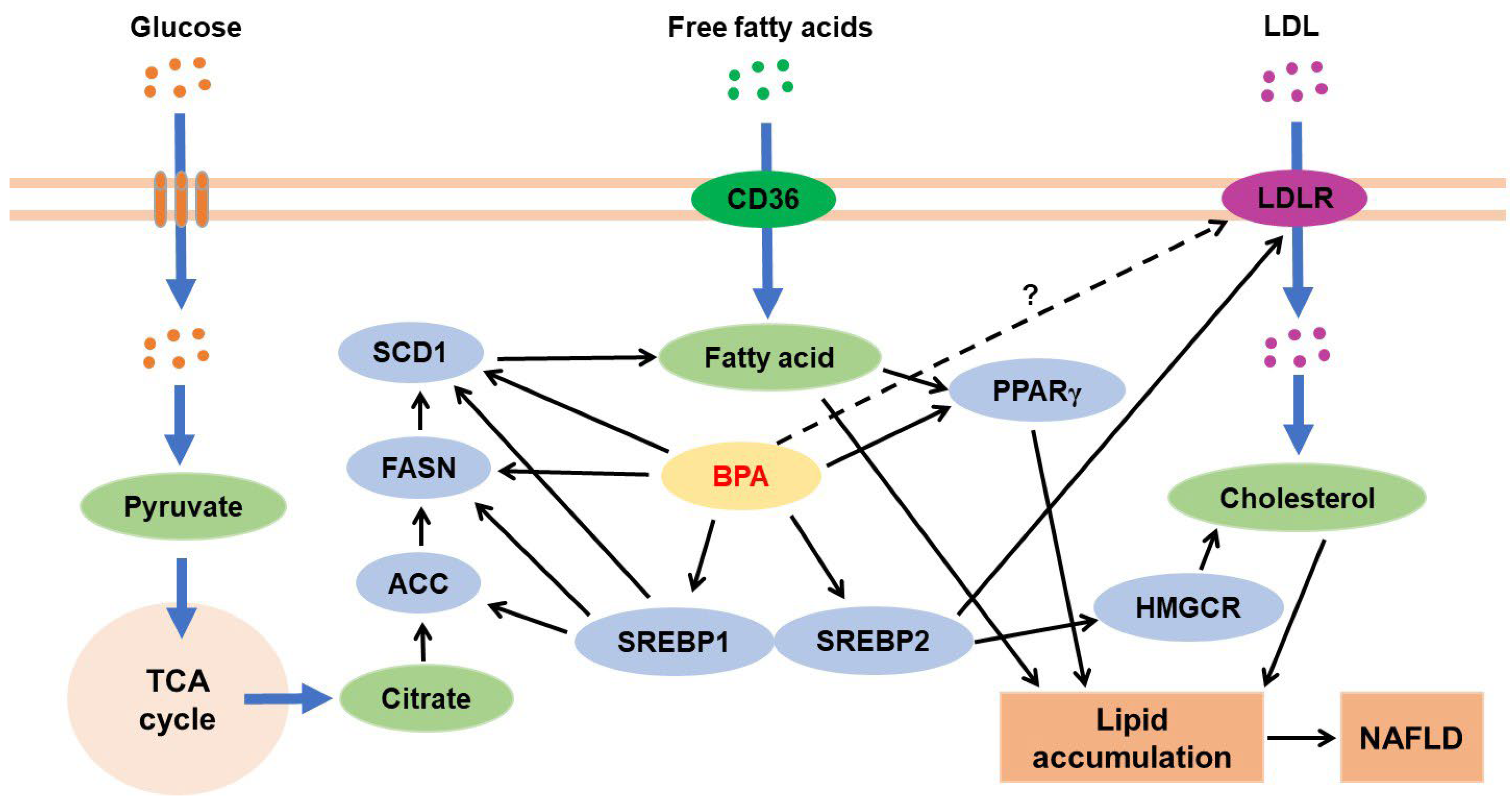
3.4.1. BPA and Hepatic Lipid Metabolism

3.4.2. BPA and Hepatic Glucose Metabolism
3.4.3. BPA and Liver Injury
3.5. Effects of BPA on the Cardiovascular System
3.5.1. Effects of BPA on Heart Tissues
3.5.2. Effects of BPA on Blood Vessels
3.6. Effects of BPA on Kidney Tissues
3.7. Effects of BPA on Intestinal and Gut Microbiota
3.8. BPA and Exercises
4. BPA Exposure and Alternation of Lipid Compositions
5. Factors Influencing BPA-Mediated Disorders in Cardiovascular or Cardiometabolic Systems
6. Effects of BPA on the Efficacy of Therapeutic Drugs for Cardiovascular or Cardiometabolic Diseases
7. Summary and Overall Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Corrales, J.; Kristofco, L.A.; Steele, W.B.; Yates, B.S.; Breed, C.S.; Williams, E.S.; Brooks, B.W. Global assessment of bisphenol A in the environment: Review and analysis of its occurrence and bioaccumulation. Dose Response 2015, 13, 1559325815598308. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Kondo, F.; Katayama, Y. Human exposure to bisphenol A. Toxicology 2006, 226, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Vilarinho, F.; Sendón, R.; van der Kellen, A.; Vaz, M.F.; SanchesSilva, A. Bisphenol A in food as a result of its migration from food packaging. Trends. Food Sci. Technol. 2019, 91, 33–65. [Google Scholar] [CrossRef]
- Kang, J.H.; Katayama, Y.; Kondo, F. Biodegradation or metabolism of bisphenol A: From microorganisms to mammals. Toxicology 2006, 217, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Asai, D.; Katayama, Y. Bisphenol A in the aquatic environment and its endocrine-disruptive effects on aquatic organisms. Crit. Rev. Toxicol. 2007, 37, 607–625. [Google Scholar] [CrossRef] [PubMed]
- Murata, M.; Kang, J.H. Bisphenol A (BPA) and cell signaling pathways. Biotechnol. Adv. 2018, 36, 311–327. [Google Scholar] [CrossRef] [PubMed]
- Almeida, S.; Raposo, A.; Almeida-González, M.; Carrascosa, C. Bisphenol A: Food exposure and impact on human health. Compr. Rev. Food Sci. Food Saf. 2018, 17, 1503–1517. [Google Scholar] [CrossRef]
- Colorado-Yohar, S.M.; Castillo-González, A.C.; Sánchez-Meca, J.; Rubio-Aparicio, M.; Sánchez-Rodríguez, D.; Salamanca-Fernández, E.; Ardanaz, E.; Amiano, P.; Fernández, M.F.; Mendiola, J.; et al. Concentrations of bisphenol-A in adults from the general population: A systematic review and meta-analysis. Sci. Total Environ. 2021, 775, 145755. [Google Scholar] [CrossRef]
- Wang, X.; Wang, X.; Chen, Q.; Luo, Z.C.; Zhao, S.; Wang, W.; Zhang, H.J.; Zhang, J.; Ouyang, F. Urinary bisphenol A concentration and gestational diabetes mellitus in Chinese women. Epidemiology 2017, 28 (Suppl. S1), S41–S47. [Google Scholar] [CrossRef]
- Wang, B.; Li, M.; Zhao, Z.; Lu, J.; Chen, Y.; Xu, Y.; Xu, M.; Wang, W.; Wang, T.; Bi, Y.; et al. Urinary bisphenol A concentration and glucose homeostasis in non-diabetic adults: A repeated-measures, longitudinal study. Diabetologia 2019, 62, 1591–1600. [Google Scholar] [CrossRef]
- Rancière, F.; Botton, J.; Slama, R.; Lacroix, M.Z.; Debrauwer, L.; Charles, M.A.; Roussel, R.; Balkau, B.; Magliano, D.J.; DESIR Study Group. Exposure to bisphenol A and bisphenol S and incident type 2 diabetes: A Case-Cohort Study in the French Cohort DESIR. Environ. Health Perspect. 2019, 127, 107013. [Google Scholar] [CrossRef] [PubMed]
- Ahmadkhaniha, R.; Mansouri, M.; Yunesian, M.; Omidfar, K.; Jeddi, M.Z.; Larijani, B.; Mesdaghinia, A.; Rastkari, N. Association of urinary bisphenol A concentration with type-2 diabetes mellitus. J. Environ. Health Sci. Eng. 2014, 12, 64. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Park, Y.J.; Kim, M.J.; Kim, S.; Choi, S.; Park, J.; Cho, Y.H.; Hong, S.; Yoo, J.; Park, H.; et al. Associations of urinary concentrations of phthalate metabolites, bisphenol A, and parabens with obesity and diabetes mellitus in a Korean adult population: Korean National Environmental Health Survey (KoNEHS) 2015–2017. Environ. Int. 2021, 146, 106227. [Google Scholar] [CrossRef] [PubMed]
- Murphy, L.; Mérida-Ortega, Á.; Cebrián, M.E.; Hernández-Garciadiego, L.; Gómez-Ruiz, H.; Gamboa-Loira, B.; López-Carrillo, L. Exposure to bisphenol A and diabetes risk in Mexican women. Environ. Sci. Pollut. Res. Int. 2019, 26, 26332–26338. [Google Scholar] [CrossRef] [PubMed]
- Li, A.J.; Xue, J.; Lin, S.; Al-Malki, A.L.; Al-Ghamdi, M.A.; Kumosani, T.A.; Kannan, K. Urinary concentrations of environmental phenols and their association with type 2 diabetes in a population in Jeddah, Saudi Arabia. Environ. Res. 2018, 166, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Haq, M.E.U.; Akash, M.S.H.; Sabir, S.; Mahmood, M.H.; Rehman, K. Human exposure to bisphenol A through dietary sources and development of diabetes mellitus: A cross-sectional study in Pakistani population. Environ. Sci. Pollut. Res. Int. 2020, 27, 26262–26275. [Google Scholar] [CrossRef] [PubMed]
- Aekplakorn, W.; Chailurkit, L.O.; Ongphiphadhanakul, B. Relationship of serum bisphenol A with diabetes in the Thai population, National Health Examination Survey IV, 2009. J. Diabetes 2015, 7, 240–249. [Google Scholar] [CrossRef]
- Tosirisuk, N.; Sakorn, N.; Jantarat, C.; Nosoongnoen, W.; Aroonpakmongkol, S.; Supornsilchai, V. Increased bisphenol A levels in Thai children and adolescents with type 1 diabetes mellitus. Pediatr. Int. 2021, 64, e14944. [Google Scholar] [CrossRef]
- Carlsson, A.; Sørensen, K.; Andersson, A.M.; Frederiksen, H.; Juul, A. Bisphenol A, phthalate metabolites and glucose homeostasis in healthy normal-weight children. Endocr. Connect. 2018, 7, 232–238. [Google Scholar] [CrossRef]
- Lang, I.A.; Galloway, T.S.; Scarlett, A.; Henley, W.E.; Depledge, M.; Wallace, R.B.; Melzer, D. Association of urinary bisphenol A concentration with medical disorders and laboratory abnormalities in adults. JAMA 2008, 300, 1303–1310. [Google Scholar] [CrossRef]
- Shankar, A.; Teppala, S. Urinary bisphenol A and hypertension in a multiethnic sample of US adults. J. Environ. Public Health 2012, 2012, 481641. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Cornelis, M.C.; Townsend, M.K.; Tobias, D.K.; Eliassen, A.H.; Franke, A.A.; Hauser, R.; Hu, F.B. Association of urinary concentrations of bisphenol A and phthalate metabolites with risk of type 2 diabetes: A prospective investigation in the Nurses’ Health Study (NHS) and NHSII cohorts. Environ. Health Perspect. 2014, 122, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Pastor-Belda, M.; Bastida, D.; Campillo, N.; Pérez-Cárceles, M.D.; Motas, M.; Viñas, P. A study of the influence on diabetes of free and conjugated bisphenol A concentrations in urine: Development of a simple microextraction procedure using gas chromatography-mass spectrometry. J. Pharm. Biomed. Anal. 2016, 129, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Turgut, F.; Sungur, S.; Okur, R.; Yaprak, M.; Ozsan, M.; Ustun, I.; Gokce, C. Higher serum bisphenol A levels in diabetic hemodialysis patients. Blood Purif. 2016, 42, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Sabanayagam, C.; Teppala, S.; Shankar, A. Relationship between urinary bisphenol A levels and prediabetes among subjects free of diabetes. Acta Diabetol. 2013, 50, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Soundararajan, A.; Prabu, P.; Mohan, V.; Gibert, Y.; Balasubramanyam, M. Novel insights of elevated systemic levels of bisphenol-A (BPA) linked to poor glycemic control, accelerated cellular senescence and insulin resistance in patients with type 2 diabetes. Mol. Cell Biochem. 2019, 458, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.; Liang, J.; Liao, Q.; Huang, H.; Guo, X.; Lin, M.; Liu, B.; Wei, B.; Zeng, X.; Liu, S.; et al. Associations of bisphenol exposure with the risk of gestational diabetes mellitus: A nested case-control study in Guangxi, China. Environ. Sci. Pollut. Res. Int. 2023, 30, 25170–25180. [Google Scholar] [CrossRef]
- Yang, J.; Wang, H.; Du, H.; Xu, L.; Liu, S.; Yi, J.; Chen, Y.; Jiang, Q.; He, G. Serum bisphenol A, glucose homeostasis, and gestational diabetes mellitus in Chinese pregnant women: A prospective study. Environ. Sci. Pollut. Res. Int. 2021, 28, 12546–12554. [Google Scholar] [CrossRef]
- Menale, C.; Grandone, A.; Nicolucci, C.; Cirillo, G.; Crispi, S.; Di Sessa, A.; Marzuillo, P.; Rossi, S.; Mita, D.G.; Perrone, L.; et al. Bisphenol A is associated with insulin resistance and modulates adiponectin and resistin gene expression in obese children. Pediatr. Obes. 2017, 12, 380–387. [Google Scholar] [CrossRef]
- İnce, T.; Balcı, A.; Yalçın, S.S.; Özkemahlı, G.; Erkekoglu, P.; Kocer-Gumusel, B.; Yurdakök, K. Urinary bisphenol-A levels in children with type 1 diabetes mellitus. J. Pediatr. Endocrinol. Metab. 2018, 31, 829–836. [Google Scholar] [CrossRef]
- Silver, M.K.; O’Neill, M.S.; Sowers, M.R.; Park, S.K. Urinary bisphenol A and type-2 diabetes in U.S. adults: Data from NHANES 2003-2008. PLoS ONE 2011, 6, e26868. [Google Scholar] [CrossRef] [PubMed]
- Tai, X.; Chen, Y. Urinary bisphenol A concentrations positively associated with glycated hemoglobin and other indicators of diabetes in Canadian men. Environ. Res. 2016, 147, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.; Ridpath, A.; Carpenter, J.; Kieszak, S.; Sircar, K.; Espinosa-Bode, A.; Nelson, D.; Martin, C. Urine bisphenol A and arsenic levels in residents of the Cheyenne River Sioux Tribe, South Dakota, with and without diabetes. J. Med. Toxicol. 2020, 16, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Park, H. Association between urinary concentrations of bisphenol A and type 2 diabetes in Korean adults: A population-based cross-sectional study. Int. J. Hyg. Environ. Health. 2013, 216, 467–471. [Google Scholar] [CrossRef] [PubMed]
- LaKind, J.S.; Goodman, M.; Naiman, D.Q. Use of NHANES data to link chemical exposures to chronic diseases: A cautionary tale. PLoS ONE 2012, 7, e51086. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Tang, S.; Peng, C.; Gao, R.; Yang, S.; Luo, T.; Cheng, Q.; Wang, Y.; Wang, Z.; Zhen, Q.; et al. Bisphenol A is not associated with a 5-year incidence of type 2 diabetes: A prospective nested case-control study. Acta Diabetol. 2018, 55, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Hauser, R.; Hu, F.B.; Franke, A.A.; Liu, S.; Sun, Q. Urinary concentrations of bisphenol A and phthalate metabolites and weight change: A prospective investigation in US women. Int. J. Obes. 2014, 38, 1532–1537. [Google Scholar] [CrossRef]
- Hao, M.; Ding, L.; Xuan, L.; Wang, T.; Li, M.; Zhao, Z.; Lu, J.; Xu, Y.; Chen, Y.; Wang, W.; et al. Urinary bisphenol A concentration and the risk of central obesity in Chinese adults: A prospective study. J. Diabetes 2018, 10, 442–448. [Google Scholar] [CrossRef]
- Moon, S.; Seo, M.Y.; Choi, K.; Chang, Y.S.; Kim, S.H.; Park, M.J. Urinary bisphenol A concentrations and the risk of obesity in Korean adults. Sci. Rep. 2021, 11, 1603. [Google Scholar] [CrossRef]
- Choi, J.Y.; Lee, J.; Huh, D.A.; Moon, K.W. Urinary bisphenol concentrations and its association with metabolic disorders in the US and Korean populations. Environ. Pollut. 2022, 295, 118679. [Google Scholar] [CrossRef]
- Do, M.T.; Chang, V.C.; Mendez, M.A.; de Groh, M. Urinary bisphenol A and obesity in adults: Results from the Canadian Health Measures Survey. Health Promot. Chronic Dis. Prev. Can. 2017, 37, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Lehmler, H.J.; Sun, Y.; Xu, G.; Sun, Q.; Snetselaar, L.G.; Wallace, R.B.; Bao, W. Association of bisphenol A and its substitutes, bisphenol F and bisphenol S, with obesity in United States children and adolescents. Diabetes Metab. J. 2019, 43, 59–75. [Google Scholar] [CrossRef] [PubMed]
- Aktağ, E.; Yurdakök, K.; Yalçın, S.S.; Kandemir, N. Urinary bisphenol A levels in prepubertal children with exogenous obesity according to presence of metabolic syndrome. J. Pediatr. Endocrinol. Metab. 2021, 34, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Okubo, Y.; Handa, A.; Belin, T. Serial cross-sectional study for the association between urinary bisphenol A and paediatric obesity: Recent updates using NHANES 2003-2014. Pediatr. Obes. 2019, 14, e12566. [Google Scholar] [CrossRef] [PubMed]
- Mustieles, V.; Casas, M.; Ferrando-Marco, P.; Ocón-Hernández, O.; Reina-Pérez, I.; Rodríguez-Carrillo, A.; Vela-Soria, F.; Pérez-Lobato, R.; Navarrete-Muñoz, E.M.; Freire, C.; et al. Bisphenol A and adiposity measures in peripubertal boys from the INMA-Granada cohort. Environ. Res. 2019, 173, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zhang, J.; Wu, C.; Xiao, H.; Lv, S.; Lu, D.; Qi, X.; Feng, C.; Liang, W.; Chang, X.; et al. Urinary bisphenol A concentrations and adiposity measures at age 7 years in a prospective birth cohort. Chemosphere 2020, 251, 126340. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.M.; Li, N.; Arbuckle, T.E.; Dodds, L.; Massarelli, I.; Fraser, W.D.; Lanphear, B.P.; Muckle, G. Association between gestational urinary bisphenol a concentrations and adiposity in young children: The MIREC study. Environ. Res. 2019, 172, 454–461. [Google Scholar] [CrossRef]
- Xue, J.; Wu, Q.; Sakthivel, S.; Pavithran, P.V.; Vasukutty, J.R.; Kannan, K. Urinary levels of endocrine-disrupting chemicals, including bisphenols, bisphenol A diglycidyl ethers, benzophenones, parabens, and triclosan in obese and non-obese Indian children. Environ. Res. 2015, 137, 120–128. [Google Scholar] [CrossRef]
- Gajjar, P.; Liu, Y.; Li, N.; Buckley, J.P.; Chen, A.; Lanphear, B.P.; Kalkwarf, H.J.; Cecil, K.M.; Yolton, K.; Braun, J.M. Associations of mid-childhood bisphenol A and bisphenol S exposure with mid-childhood and adolescent obesity. Environ. Epidemiol. 2021, 6, e187. [Google Scholar] [CrossRef]
- Jacobson, M.H.; Woodward, M.; Bao, W.; Liu, B.; Trasande, L. Urinary bisphenols and obesity prevalence among U.S. children and adolescents. J. Endocr. Soc. 2019, 3, 1715–1726. [Google Scholar] [CrossRef]
- Shankar, A.; Teppala, S. Relationship between urinary bisphenol A levels and diabetes mellitus. J. Clin. Endocrinol. Metab. 2012, 96, 3822–3826. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Hong, Y.C. Exposure to bisphenol A from drinking canned beverages increases blood pressure: Randomized crossover trial. Hypertension 2015, 65, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Kim, J.H.; Lim, Y.H.; Park, H.Y.; Hong, Y.C. Associations of bisphenol A exposure with heart rate variability and blood pressure. Hypertension 2012, 60, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Liu, H.; Zhou, S.; Zhang, X.; Peng, C.; Zhou, H.; Tong, Y.; Lu, Q. Association of bisphenol A and its alternatives bisphenol S and F exposure with hypertension and blood pressure: A cross-sectional study in China. Environ. Pollut. 2020, 257, 113639. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Yang, G.; Zhou, S.; Zhang, X.; Peng, C.; Lu, Q. Bisphenol A, S, and F exposure, ESR1/2, CAT, and eNOS genetic polymorphisms, and the risk of hypertension. Ecotoxicol. Environ. Saf. 2021, 224, 112684. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Cho, Y.H.; Hong, Y.C. MicroRNA expression in response to bisphenol A is associated with high blood pressure. Environ. Int. 2020, 141, 105791. [Google Scholar] [CrossRef]
- Aekplakorn, W.; Chailurkit, L.O.; Ongphiphadhanakul, B. Association of serum bisphenol a with hypertension in Thai population. Int. J. Hypertens. 2015, 2015, 594189. [Google Scholar] [CrossRef]
- Wang, T.; Xu, M.; Xu, Y.; Lu, J.; Li, M.; Chen, Y.; Wang, W.; Lai, S.; Bi, Y.; Ning, G. Association of bisphenol A exposure with hypertension and early macrovascular diseases in Chinese adults: A cross-sectional study. Medicine 2015, 94, e1814. [Google Scholar] [CrossRef]
- Camara, L.R.; Arbuckle, T.E.; Trottier, H.; Fraser, W.D. Associations between maternal exposure to bisphenol A or triclosan and gestational hypertension and preeclampsia: The MIREC Study. Am. J. Perinatol. 2019, 36, 1127–1135. [Google Scholar] [CrossRef]
- Chawla, L.S.; Bellomo, R.; Bihorac, A.; Goldstein, S.L.; Siew, E.D.; Bagshaw, S.M.; Bittleman, D.; Cruz, D.; Endre, Z.; Fitzgerald, R.L.; et al. Acute Disease Quality Initiative Workgroup 16. Acute kidney disease and renal recovery: Consensus report of the Acute Disease Quality Initiative (ADQI) 16 Workgroup. Nat. Rev. Nephrol. 2017, 13, 241–257. [Google Scholar] [CrossRef]
- Lameire, N.H.; Levin, A.; Kellum, J.A.; Cheung, M.; Jadoul, M.; Winkelmayer, W.C.; Stevens, P.E.; Conference Participants. Harmonizing acute and chronic kidney disease definition and classification: Report of a Kidney Disease: Improving Global Outcomes (KDIGO) Consensus Conference. Kidney Int. 2021, 100, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Lee, J.P.; Choi, K. Exposure to phthalates and environmental phenols in association with chronic kidney disease (CKD) among the general US population participating in multi-cycle NHANES (2005–2016). Sci. Total Environ. 2021, 791, 148343. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.H.; Wu, Y.; Liu, M.; Attina, T.M.; Naidu, M.; Karthikraj, R.; Kannan, K.; Warady, B.A.; Furth, S.; Vento, S.; et al. Serially assessed bisphenol A and phthalate exposure and association with kidney function in children with chronic kidney disease in the US and Canada: A longitudinal cohort study. PLoS Med. 2020, 17, e1003384. [Google Scholar] [CrossRef] [PubMed]
- Malits, J.; Attina, T.M.; Karthikraj, R.; Kannan, K.; Naidu, M.; Furth, S.; Warady, B.A.; Vento, S.; Trachtman, H.; Trasande, L. Renal function and exposure to bisphenol A and phthalates in children with chronic kidney disease. Environ. Res. 2018, 167, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Park, J.Y.; Kim, S.; An, J.N.; Lee, J.; Park, H.; Jung, S.K.; Kim, S.Y.; Lee, J.P.; Choi, K. Association of exposure to phthalates and environmental phenolics with markers of kidney function: Korean National Environmental Health Survey (KoNEHS) 2015–2017. Environ. Int. 2020, 143, 105877. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.B. Concentrations of bisphenol A and its associations with urinary albumin creatinine ratios across the various stages of renal function. Environ. Sci. Pollut. Res. Int. 2021, 28, 9946–9953. [Google Scholar] [CrossRef] [PubMed]
- Nie, H.; Wang, F.; Zhang, Y.; Zhang, S.; Han, X.; Zhang, X.; Guo, H.; He, M. Associations of serum bisphenol A levels with incident chronic kidney disease risk. Sci. Total Environ. 2021, 771, 145401. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Liu, T.; Shi, Y.; Zhuang, F.; Lu, J.; Zhu, Q.; Ding, F. Bisphenol A analogs in patients with chronic kidney disease and dialysis therapy. Ecotoxicol. Environ. Saf. 2019, 185, 109684. [Google Scholar] [CrossRef]
- Hu, J.; Wang, Y.; Xiang, X.; Peng, C.; Gao, R.; Goswami, R.; Zhou, H.; Zhang, Y.; Zhen, Q.; Cheng, Q.; et al. Serum bisphenol A as a predictor of chronic kidney disease progression in primary hypertension: A 6-year prospective study. J. Hypertens. 2016, 34, 332–337. [Google Scholar] [CrossRef]
- Hu, J.; Yang, S.; Wang, Y.; Goswami, R.; Peng, C.; Gao, R.; Zhou, H.; Zhang, Y.; Cheng, Q.; Zhen, Q.; et al. Serum bisphenol A and progression of type 2 diabetic nephropathy: A 6-year prospective study. Acta Diabetol. 2015, 52, 1135–1141. [Google Scholar] [CrossRef]
- Bosch-Panadero, E.; Mas, S.; Sanchez-Ospina, D.; Camarero, V.; Pérez-Gómez, M.V.; Saez-Calero, I.; Abaigar, P.; Ortiz, A.; Egido, J.; González-Parra, E. The choice of hemodialysis membrane affects bisphenol A levels in blood. J. Am. Soc. Nephrol. 2016, 27, 1566–1574. [Google Scholar] [CrossRef] [PubMed]
- Mas, S.; Bosch-Panadero, E.; Abaigar, P.; Camarero, V.; Mahillo, I.; Civantos, E.; Sanchez-Ospina, D.; Ruiz-Priego, A.; Egido, J.; Ortiz, A.; et al. Influence of dialysis membrane composition on plasma bisphenol A levels during online hemodiafiltration. PLoS ONE 2018, 13, e0193288. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Ohashi, A.; Hori, H.; Hibiya, M.; Shoji, Y.; Kunisaki, M.; Akita, M.; Yagi, A.; Sugiyama, K.; Shimozato, S.; et al. Accumulation of bisphenol A in hemodialysis patients. Blood Purif. 2007, 25, 290–394. [Google Scholar] [CrossRef] [PubMed]
- Badding, M.A.; Vargas, J.R.; Fortney, J.; Cheng, Q.J.; Ho, C.H. Toxicological risk assessment of bisphenol a released from dialyzers under simulated-use and exaggerated extraction conditions. Regul. Toxicol. Pharmacol. 2020, 118, 104787. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Liu, B.; Rong, S.; Dai, S.Y.; Trasande, L.; Lehmler, H.J. Association between bisphenol A exposure and risk of all-cause and cause-specific mortality in US adults. JAMA Netw. Open 2020, 3, e2011620. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Rao, X.; Ye, J.; Ling, Y.; Mi, S.; Chen, H.; Fan, C.; Li, Y. Relationship between urinary bisphenol a levels and cardiovascular diseases in the U.S. adult population, 2003-2014. Ecotoxicol. Environ. Saf. 2020, 192, 110300. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; He, J.; Shi, W. Association between urinary environmental phenols and the prevalence of cardiovascular diseases in US adults. Environ. Sci. Pollut. Res. Int. 2022, 29, 42947–42954. [Google Scholar] [CrossRef]
- Melzer, D.; Gates, P.; Osborne, N.J.; Henley, W.E.; Cipelli, R.; Young, A.; Money, C.; McCormack, P.; Schofield, P.; Mosedale, D.; et al. Urinary bisphenol A concentration and angiography-defined coronary artery stenosis. PLoS ONE 2012, 7, e43378. [Google Scholar] [CrossRef]
- Melzer, D.; Osborne, N.J.; Henley, W.E.; Cipelli, R.; Young, A.; Money, C.; McCormack, P.; Luben, R.; Khaw, K.T.; Wareham, N.J.; et al. Urinary bisphenol A concentration and risk of future coronary artery disease in apparently healthy men and women. Circulation 2012, 125, 1482–1490. [Google Scholar] [CrossRef]
- Li, R.; Yang, S.; Gao, R.; Deng, Y.; Liu, J.; Yuan, C.; Yao, Q.; Lv, X.; Wang, K.; Ye, X.; et al. Relationship between the environmental endocrine disruptor bisphenol A and dyslipidemia: A five-year prospective study. Endocr. Pract. 2020, 26, 399–406. [Google Scholar] [CrossRef]
- Tsen, C.M.; Liu, J.H.; Yang, D.P.; Chao, H.R.; Chen, J.L.; Chou, W.C.; Ho, Y.C.; Chuang, C.Y. Study on the correlation of bisphenol A exposure, pro-inflammatory gene expression, and C-reactive protein with potential cardiovascular disease symptoms in young adults. Environ. Sci. Pollut. Res. Int. 2021, 28, 32580–32591. [Google Scholar] [CrossRef] [PubMed]
- Chu, P.L.; Lin, C.Y.; Sung, F.C.; Su, T.C. Apoptotic microparticles mediate the association between bisphenol A and subclinical atherosclerosis in a young population: A population-based study. Ecotoxicol. Environ. Saf. 2021, 224, 112663. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Q.; Liu, X.; Shen, Y.; Yu, P.; Chen, S.; Hu, J.; Yu, J.; Li, J.; Wang, H.S.; Cheng, X.; et al. Elevated serum bisphenol A level in patients with dilated cardiomyopathy. Int. J. Environ. Res. Public Health 2015, 12, 5329–5337. [Google Scholar] [CrossRef] [PubMed]
- Lind, P.M.; Lind, L. Circulating levels of bisphenol A and phthalates are related to carotid atherosclerosis in the elderly. Atherosclerosis 2011, 218, 207–213. [Google Scholar] [CrossRef] [PubMed]
- An, S.J.; Yang, E.J.; Oh, S.; Park, K.J.; Kim, T.; Hong, Y.P.; Yang, Y.J. The association between urinary bisphenol A levels and nonalcoholic fatty liver disease in Korean adults: Korean National Environmental Health Survey (KoNEHS) 2015–2017. Environ. Health Prev. Med. 2021, 26, 91. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Yoo, E.R.; Li, A.A.; Cholankeril, G.; Tighe, S.P.; Kim, W.; Harrison, S.A.; Ahmed, A. Elevated urinary bisphenol A levels are associated with non-alcoholic fatty liver disease among adults in the United States. Liver Int. 2019, 39, 1335–1342. [Google Scholar] [CrossRef]
- Verstraete, S.G.; Wojcicki, J.M.; Perito, E.R.; Rosenthal, P. Bisphenol a increases risk for presumed non-alcoholic fatty liver disease in Hispanic adolescents in NHANES 2003–2010. Environ. Health. 2018, 17, 12. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.R.; Park, H.; Bae, S.; Lim, Y.H.; Kim, J.H.; Cho, S.H.; Hong, Y.C. Urinary bisphenol A concentrations are associated with abnormal liver function in the elderly: A repeated panel study. J. Epidemiol. Community Health 2014, 68, 312–317. [Google Scholar] [CrossRef]
- Lee, S.; Lee, H.A.; Park, B.; Han, H.; Park, B.H.; Oh, S.Y.; Hong, Y.S.; Ha, E.H.; Park, H. A prospective cohort study of the association between bisphenol A exposure and the serum levels of liver enzymes in children. Environ. Res. 2018, 161, 195–201. [Google Scholar] [CrossRef]
- Fu, X.; He, J.; Zheng, D.; Yang, X.; Wang, P.; Tuo, F.; Wang, L.; Li, S.; Xu, J.; Yu, J. Association of endocrine disrupting chemicals levels in serum, environmental risk factors, and hepatic function among 5- to 14-year-old children. Toxicology 2022, 465, 153011. [Google Scholar] [CrossRef]
- Ko, J.H.; Kang, J.H.; Park, C.; Shin, D.W.; Kim, J.H.; Kim, H.; Han, S.B. Effect of bisphenol A on insulin-mediated glucose metabolism in vivo and in vitro. Mol. Cell. Toxicol. 2008, 4, 348–354. [Google Scholar]
- Ahmed, F.; Chehadé, L.; Garneau, L.; Caron, A.; Aguer, C. The effects of acute BPA exposure on skeletal muscle mitochondrial function and glucose metabolism. Mol. Cell Endocrinol. 2020, 499, 110580. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Magdalena, P.; Vieira, E.; Soriano, S.; Menes, L.; Burks, D.; Quesada, I.; Nadal, A. Bisphenol A exposure during pregnancy disrupts glucose homeostasis in mothers and adult male offspring. Environ. Health Perspect. 2010, 118, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Indumathi, D.; Jayashree, S.; Selvaraj, J.; Sathish, S.; Mayilvanan, C.; Akilavalli, N.; Balasubramanian, K. Effect of bisphenol-A on insulin signal transduction and glucose oxidation in skeletal muscle of adult male albino rat. Hum. Exp. Toxicol. 2013, 32, 960–971. [Google Scholar] [CrossRef] [PubMed]
- Mullainadhan, V.; Viswanathan, M.P.; Karundevi, B. Effect of bisphenol-A (BPA) on insulin signal transduction and GLUT4 translocation in gastrocnemius muscle of adult male albino rat. Int. J. Biochem. Cell Biol. 2017, 90, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Batista, T.M.; Alonso-Magdalena, P.; Vieira, E.; Amaral, M.E.; Cederroth, C.R.; Nef, S.; Quesada, I.; Carneiro, E.M.; Nadal, A. Short-term treatment with bisphenol-A leads to metabolic abnormalities in adult male mice. PLoS ONE 2012, 7, e33814. [Google Scholar] [CrossRef]
- Moon, M.K.; Jeong, I.K.; Jung Oh, T.; Ahn, H.Y.; Kim, H.H.; Park, Y.J.; Jang, H.C.; Park, K.S. Long-term oral exposure to bisphenol A induces glucose intolerance and insulin resistance. J. Endocrinol. 2015, 226, 35–42. [Google Scholar] [CrossRef]
- Galyon, K.D.; Farshidi, F.; Han, G.; Ross, M.G.; Desai, M.; Jellyman, J.K. Maternal bisphenol A exposure alters rat offspring hepatic and skeletal muscle insulin signaling protein abundance. Am. J. Obstet. Gynecol. 2017, 216, 290.e1–290.e9. [Google Scholar] [CrossRef]
- Karlsson, H.K.; Zierath, J.R.; Kane, S.; Krook, A.; Lienhard, G.E.; Wallberg-Henriksson, H. Insulin-stimulated phosphorylation of the Akt substrate AS160 is impaired in skeletal muscle of type 2 diabetic subjects. Diabetes 2005, 54, 1692–1697. [Google Scholar] [CrossRef]
- Zhang, R.; Pessah, I.N. Divergent mechanisms leading to signaling dysfunction in embryonic muscle by bisphenol A and tetrabromobisphenol A. Mol. Pharmacol. 2017, 91, 428–436. [Google Scholar] [CrossRef]
- Wade, M.; Delawder, V.; Reneau, P.; Dos Santos, J.M. The effect of BPA exposure on insulin resistance and type 2 diabetes-The impact of muscle contraction. Med. Hypotheses 2020, 140, 109675. [Google Scholar] [CrossRef] [PubMed]
- Cohen, I.C.; Cohenour, E.R.; Harnett, K.G.; Schuh, S.M. BPA, BPAF and TMBPF alter adipogenesis and fat accumulation in human mesenchymal stem cells, with implications for obesity. Int. J. Mol. Sci. 2021, 22, 5363. [Google Scholar] [CrossRef] [PubMed]
- Ohlstein, J.F.; Strong, A.L.; McLachlan, J.A.; Gimble, J.M.; Burow, M.E.; Bunnell, B.A. Bisphenol A enhances adipogenic differentiation of human adipose stromal/stem cells. J. Mol. Endocrinol. 2014, 53, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Schaffert, A.; Krieg, L.; Weiner, J.; Schlichting, R.; Ueberham, E.; Karkossa, I.; Bauer, M.; Landgraf, K.; Junge, K.M.; Wabitsch, M.; et al. Alternatives for the worse: Molecular insights into adverse effects of bisphenol A and substitutes during human adipocyte differentiation. Environ. Int. 2021, 156, 106730. [Google Scholar] [CrossRef] [PubMed]
- Wabitsch, M.; Brenner, R.E.; Melzner, I.; Braun, M.; Möller, P.; Heinze, E.; Debatin, K.M.; Hauner, H. Characterization of a human preadipocyte cell strain with high capacity for adipose differentiation. Int. J. Obes. Relat. Metab. Disord. 2001, 25, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Salehpour, A.; Shidfar, F.; Hedayati, M.; Neshatbini Tehrani, A.; Farshad, A.A.; Mohammadi, S. Bisphenol A enhances adipogenic signaling pathways in human mesenchymal stem cells. Genes Environ. 2020, 42, 13. [Google Scholar] [CrossRef] [PubMed]
- Atlas, E.; Pope, L.; Wade, M.G.; Kawata, A.; Boudreau, A.; Boucher, J.G. Bisphenol A increases aP2 expression in 3T3L1 by enhancing the transcriptional activity of nuclear receptors at the promoter. Adipocyte 2014, 3, 170–179. [Google Scholar] [CrossRef]
- Biasiotto, G.; Zanella, I.; Masserdotti, A.; Pedrazzani, R.; Papa, M.; Caimi, L.; Di Lorenzo, D. Municipal wastewater affects adipose deposition in male mice and increases 3T3-L1 cell differentiation. Toxicol. Appl. Pharmacol. 2016, 297, 32–40. [Google Scholar] [CrossRef]
- Ariemma, F.; D’Esposito, V.; Liguoro, D.; Oriente, F.; Cabaro, S.; Liotti, A.; Cimmino, I.; Longo, M.; Beguinot, F.; Formisano, P.; et al. Low-dose bisphenol-A impairs adipogenesis and generates dysfunctional 3T3-L1 adipocytes. PLoS ONE 2016, 11, e0150762. [Google Scholar] [CrossRef]
- Longo, M.; Zatterale, F.; Naderi, J.; Nigro, C.; Oriente, F.; Formisano, P.; Miele, C.; Beguinot, F. Low-dose bisphenol-A promotes epigenetic changes at Pparγ promoter in adipose precursor cells. Nutrients 2020, 12, 3498. [Google Scholar] [CrossRef]
- De Filippis, E.; Li, T.; Rosen, E.D. Exposure of adipocytes to bisphenol-A in vitro interferes with insulin action without enhancing adipogenesis. PLoS ONE 2018, 13, e0201122. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.; Ferrini, M.G.; Jellyman, J.K.; Han, G.; Ross, M.G. In vivo and in vitro bisphenol A exposure effects on adiposity. J. Dev. Orig. Health Dis. 2018, 9, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Dunder, L.; Halin Lejonklou, M.; Lind, L.; Risérus, U.; Lind, P.M. Low-dose developmental bisphenol A exposure alters fatty acid metabolism in Fischer 344 rat offspring. Environ. Res. 2018, 166, 117–129. [Google Scholar] [CrossRef]
- Angle, B.M.; Do, R.P.; Ponzi, D.; Stahlhut, R.W.; Drury, B.E.; Nagel, S.C.; Welshons, W.V.; Besch-Williford, C.L.; Palanza, P.; Parmigiani, S.; et al. Metabolic disruption in male mice due to fetal exposure to low but not high doses of bisphenol A (BPA): Evidence for effects on body weight, food intake, adipocytes, leptin, adiponectin, insulin and glucose regulation. Reprod. Toxicol. 2013, 42, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.A.; Shioda, K.; Mitsunaga, S.; Yawata, S.; Angle, B.M.; Nagel, S.C.; Vom Saal, F.S.; Shioda, T. Prenatal exposure to bisphenol A disrupts naturally occurring bimodal DNA methylation at proximal promoter of fggy, an obesity-relevant gene encoding a carbohydrate kinase, in gonadal white adipose tissues of CD-1 mice. Endocrinology 2018, 159, 779–794. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Chen, M.; Wang, J.; Xu, M.; Sun, J.; Ding, L.; Lv, X.; Ma, Q.; Bi, Y.; Liu, R.; et al. Bisphenol A promotes adiposity and inflammation in a nonmonotonic dose-response way in 5-week-old male and female C57BL/6J mice fed a low-calorie diet. Endocrinology 2016, 157, 2333–2345. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, J.W.; Walker, E.A.; Bujalska, I.J.; Draper, N.; Lavery, G.G.; Cooper, M.S.; Hewison, M.; Stewart, P.M. 11β-hydroxysteroid dehydrogenase type 1: A tissue-specific regulator of glucocorticoid response. Endocr. Rev. 2004, 25, 831–866. [Google Scholar] [CrossRef]
- Wang, J.; Sun, B.; Hou, M.; Pan, X.; Li, X. The environmental obesogen bisphenol A promotes adipogenesis by increasing the amount of 11β-hydroxysteroid dehydrogenase type 1 in the adipose tissue of children. Int. J. Obes. 2013, 37, 999–1005. [Google Scholar] [CrossRef]
- Heine, P.A.; Taylor, J.A.; Iwamoto, G.A.; Lubahn, D.B.; Cooke, P.S. Increased adipose tissue in male and female estrogen receptor-α knockout mice. Proc. Natl. Acad. Sci. USA 2000, 97, 12729–12734. [Google Scholar] [CrossRef]
- Davis, K.E.; Neinast, D.M.; Sun, K.; Skiles, M.W.; Bills, D.J.; Zehr, A.J.; Zeve, D.; Hahner, D.L.; Cox, W.D.; Gent, M.L.; et al. The sexually dimorphic role of adipose and adipocyte estrogen receptors in modulating adipose tissue expansion, inflammation, and fibrosis. Mol. Metab. 2013, 2, 227–242. [Google Scholar] [CrossRef]
- Menale, C.; Piccolo, M.T.; Cirillo, G.; Calogero, R.A.; Papparella, A.; Mita, L.; Del Giudice, E.M.; Diano, N.; Crispi, S.; Mita, D.G. Bisphenol A effects on gene expression in adipocytes from children: Association with metabolic disorders. J. Mol. Endocrinol. 2015, 54, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, I.; Oriente, F.; D’Esposito, V.; Liguoro, D.; Liguoro, P.; Ambrosio, M.R.; Cabaro, S.; D’Andrea, F.; Beguinot, F.; Formisano, P.; et al. Low-dose bisphenol-A regulates inflammatory cytokines through GPR30 in mammary adipose cells. J. Mol. Endocrinol. 2019, 63, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, F.; Sarsenbayeva, A.; Katsogiannos, P.; Aguer, C.; Pereira, M.J. The effects of bisphenol A and bisphenol S on adipokine expression and glucose metabolism in human adipose tissue. Toxicology 2020, 445, 152600. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Valentino, R.; D’Esposito, V.; Passaretti, F.; Liotti, A.; Cabaro, S.; Longo, M.; Perruolo, G.; Oriente, F.; Beguinot, F.; Formisano, P. Bisphenol-A impairs insulin action and up-regulates inflammatory pathways in human subcutaneous adipocytes and 3T3-L1 cells. PLoS ONE 2013, 8, e82099. [Google Scholar] [CrossRef] [PubMed]
- Pu, Y.; Gingrich, J.D.; Steibel, J.P.; Veiga-Lopez, A. Sex-specific modulation of fetal adipogenesis by gestational bisphenol A and bisphenol S exposure. Endocrinology 2017, 158, 3844–3858. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Savastano, S.; Tarantino, G.; D’Esposito, V.; Passaretti, F.; Cabaro, S.; Liotti, A.; Liguoro, D.; Perruolo, G.; Ariemma, F.; Finelli, C.; et al. Bisphenol-A plasma levels are related to inflammatory markers, visceral obesity and insulin-resistance: A cross-sectional study on adult male population. J. Transl. Med. 2015, 13, 169. [Google Scholar] [CrossRef] [PubMed]
- Rönn, M.; Lind, L.; Örberg, J.; Kullberg, J.; Söderberg, S.; Larsson, A.; Johansson, L.; Ahlström, H.; Lind, P.M. Bisphenol A is related to circulating levels of adiponectin, leptin and ghrelin, but not to fat mass or fat distribution in humans. Chemosphere 2014, 112, 42–48. [Google Scholar] [CrossRef]
- Ampem, G.; Junginger, A.; Yu, H.; Balogh, L.; Thuróczy, J.; Schneider, M.E.; Röszer, T. The environmental obesogen bisphenol A increases macrophage self-renewal. Cell Tissue Res. 2019, 378, 81–96. [Google Scholar] [CrossRef]
- Dai, Y.E.; Chen, W.; Qi, H.; Liu, Q.Q. Effect of bisphenol A on SOCS-3 and insulin signaling transduction in 3T3-L1 adipocytes. Mol. Med. Rep. 2016, 14, 331–336. [Google Scholar] [CrossRef]
- Puttabyatappa, M.; Martin, J.D.; Andriessen, V.; Stevenson, M.; Zeng, L.; Pennathur, S.; Padmanabhan, V. Developmental programming: Changes in mediators of insulin sensitivity in prenatal bisphenol A-treated female sheep. Reprod. Toxicol. 2019, 85, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Doerge, D.R.; Twaddle, N.C.; Vanlandingham, M.; Fisher, J.W. Pharmacokinetics of bisphenol A in serum and adipose tissue following intravenous administration to adult female CD-1 mice. Toxicol. Lett. 2012, 211, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Kito, K.; Kondo, F. Factors influencing the migration of bisphenol A from cans. J. Food Protect. 2003, 66, 1444–1447. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Asimakopoulos, A.G.; Kannan, K. Accumulation of 19 environmental phenolic and xenobiotic heterocyclic aromatic compounds in human adipose tissue. Environ. Int. 2015, 78, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.F.; Arrebola, J.P.; Taoufiki, J.; Navalón, A.; Ballesteros, O.; Pulgar, R.; Vilchez, J.L.; Olea, N. Bisphenol-A and chlorinated derivatives in adipose tissue of women. Reprod. Toxicol. 2007, 24, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Geens, T.; Neels, H.; Covaci, A. Distribution of bisphenol-A, triclosan and n-nonylphenol in human adipose tissue, liver and brain. Chemosphere 2012, 87, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Keshavarz-Maleki, R.; Kaviani, A.; Omranipour, R.; Gholami, M.; Khoshayand, M.R.; Ostad, S.N.; Sabzevari, O. Bisphenol-A in biological samples of breast cancer mastectomy and mammoplasty patients and correlation with levels measured in urine and tissue. Sci. Rep. 2021, 11, 18411. [Google Scholar] [CrossRef]
- Campbell, J.E.; Newgard, C.B. Mechanisms controlling pancreatic islet cell function in insulin secretion. Nat. Rev. Mol. Cell Biol. 2021, 22, 142–158. [Google Scholar] [CrossRef]
- Alonso-Magdalena, P.; Laribi, O.; Ropero, A.B.; Fuentes, E.; Ripoll, C.; Soria, B.; Nadal, A. Low doses of bisphenol A and diethylstilbestrol impair Ca2+ signals in pancreatic α-cells through a nonclassical membrane estrogen receptor within intact islets of Langerhans. Environ. Health Perspect. 2005, 113, 969–977. [Google Scholar] [CrossRef]
- Makaji, E.; Raha, S.; Wade, M.G.; Holloway, A.C. Effect of environmental contaminants on Beta cell function. Int. J. Toxicol. 2011, 30, 410–418. [Google Scholar] [CrossRef]
- Xin, F.; Jiang, L.; Liu, X.; Geng, C.; Wang, W.; Zhong, L.; Yang, G.; Chen, M. Bisphenol A induces oxidative stress-associated DNA damage in INS-1 cells. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2014, 769, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Weldingh, N.M.; Jørgensen-Kaur, L.; Becher, R.; Holme, J.A.; Bodin, J.; Nygaard, U.C.; Bølling, A.K. Bisphenol A is more potent than phthalate metabolites in reducing pancreatic β-cell function. Biomed. Res. Int. 2017, 2017, 4614379. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Xia, W.; Zhou, Z.; Li, Y.; Lin, Y.; Wei, J.; Wei, Z.; Xu, B.; Shen, J.; Li, W.; et al. Low-level phenolic estrogen pollutants impair islet morphology and β-cell function in isolated rat islets. J. Endocrinol. 2012, 215, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Manukyan, L.; Dunder, L.; Lind, P.M.; Bergsten, P.; Lejonklou, M.H. Developmental exposure to a very low dose of bisphenol A induces persistent islet insulin hypersecretion in Fischer 344 rat offspring. Environ. Res. 2019, 172, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Rashid, C.; Xin, F.; Li, C.; Polyak, E.; Duemler, A.; van der Meer, T.; Stefaniak, M.; Wajid, S.; Doliba, N.; et al. Sex- and dose-specific effects of maternal bisphenol A exposure on pancreatic islets of first- and second-generation adult mice offspring. Environ. Health Perspect. 2017, 125, 097022. [Google Scholar] [CrossRef] [PubMed]
- García-Arévalo, M.; Alonso-Magdalena, P.; Servitja, J.M.; Boronat-Belda, T.; Merino, B.; Villar-Pazos, S.; Medina-Gómez, G.; Novials, A.; Quesada, I.; Nadal, A. Maternal Exposure to bisphenol-A during pregnancy increases pancreatic β-cell growth during early life in male mice offspring. Endocrinology 2016, 157, 4158–4171. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Xia, W.; Chang, H.; Huo, W.; Li, Y.; Xu, S. Paternal BPA exposure in early life alters Igf2 epigenetic status in sperm and induces pancreatic impairment in rat offspring. Toxicol. Lett. 2015, 238, 30–38. [Google Scholar] [CrossRef]
- Mao, Z.; Xia, W.; Huo, W.; Zheng, T.; Bassig, B.A.; Chang, H.; Chen, T.; Li, F.; Pan, Y.; Peng, Y.; et al. Pancreatic impairment and Igf2 hypermethylation induced by developmental exposure to bisphenol A can be counteracted by maternal folate supplementation. J. Appl. Toxicol. 2017, 37, 825–835. [Google Scholar] [CrossRef]
- Chang, H.; Wang, D.; Xia, W.; Pan, X.; Huo, W.; Xu, S.; Li, Y. Epigenetic disruption and glucose homeostasis changes following low-dose maternal bisphenol A exposure. Toxicol. Res. 2016, 5, 1400–1409. [Google Scholar] [CrossRef]
- Whitehead, R.; Guan, H.; Arany, E.; Cernea, M.; Yang, K. Prenatal exposure to bisphenol A alters mouse fetal pancreatic morphology and islet composition. Horm. Mol. Biol. Clin. Investig. 2016, 25, 171–179. [Google Scholar] [CrossRef]
- Soriano, S.; Alonso-Magdalena, P.; García-Arévalo, M.; Novials, A.; Muhammed, S.J.; Salehi, A.; Gustafsson, J.A.; Quesada, I.; Nadal, A. Rapid insulinotropic action of low doses of bisphenol-A on mouse and human islets of Langerhans: Role of estrogen receptor β. PLoS ONE 2012, 7, e31109. [Google Scholar] [CrossRef]
- Villar-Pazos, S.; Martinez-Pinna, J.; Castellano-Muñoz, M.; Alonso-Magdalena, P.; Marroqui, L.; Quesada, I.; Gustafsson, J.A.; Nadal, A. Molecular mechanisms involved in the non-monotonic effect of bisphenol-a on Ca2+ entry in mouse pancreatic β-cells. Sci. Rep. 2017, 7, 11770. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Pinna, J.; Marroqui, L.; Hmadcha, A.; Lopez-Beas, J.; Soriano, S.; Villar-Pazos, S.; Alonso-Magdalena, P.; Dos Santos, R.S.; Quesada, I.; Martin, F.; et al. Oestrogen receptor β mediates the actions of bisphenol-A on ion channel expression in mouse pancreatic beta cells. Diabetologia. 2019, 62, 1667–1680. [Google Scholar] [CrossRef]
- Alonso-Magdalena, P.; Ropero, A.B.; Carrera, M.P.; Cederroth, C.R.; Baquié, M.; Gauthier, B.R.; Nef, S.; Stefani, E.; Nadal, A. Pancreatic insulin content regulation by the estrogen receptor ERα. PLoS ONE 2008, 3, e2069. [Google Scholar] [CrossRef] [PubMed]
- Boronat-Belda, T.; Ferrero, H.; Al-Abdulla, R.; Quesada, I.; Gustafsson, J.A.; Nadal, Á.; Alonso-Magdalena, P. Bisphenol-A exposure during pregnancy alters pancreatic β-cell division and mass in male mice offspring: A role for ERβ. Food Chem. Toxicol. 2020, 145, 111681. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, K.M.; Figueiredo, L.S.; Araujo, T.R.; Freitas, I.N.; Silva, J.N.; Boschero, A.C.; Ribeiro, R.A. Prolonged bisphenol-A exposure decreases endocrine pancreatic proliferation in response to obesogenic diet in ovariectomized mice. Steroids 2020, 160, 108658. [Google Scholar] [CrossRef]
- Furman, B.L. Streptozotocin-induced diabetic models in mice and rats. Curr. Protoc. 2021, 1, e78. [Google Scholar] [CrossRef]
- Kang, H.S.; Yang, H.; Ahn, C.; Kang, H.Y.; Hong, E.J.; Jaung, E.B. Effects of xenoestrogens on streptozotocin-induced diabetic mice. J. Physiol. Pharmacol. 2014, 65, 273–282. [Google Scholar] [PubMed]
- Ahn, C.; Kang, H.S.; Lee, J.H.; Hong, E.J.; Jung, E.M.; Yoo, Y.M.; Jeung, E.B. Bisphenol A and octylphenol exacerbate type 1 diabetes mellitus by disrupting calcium homeostasis in mouse pancreas. Toxicol. Lett. 2018, 295, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Bodin, J.; Bølling, A.K.; Samuelsen, M.; Becher, R.; Løvik, M.; Nygaard, U.C. Long-term bisphenol A exposure accelerates insulitis development in diabetes-prone NOD mice. Immunopharmacol. Immunotoxicol. 2013, 35, 349–358. [Google Scholar] [CrossRef]
- Bodin, J.; Bølling, A.K.; Becher, R.; Kuper, F.; Løvik, M.; Nygaard, U.C. Transmaternal bisphenol A exposure accelerates diabetes type 1 development in NOD mice. Toxicol. Sci. 2014, 137, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Bodin, J.; Kocbach Bølling, A.; Wendt, A.; Eliasson, L.; Becher, R.; Kuper, F.; Løvik, M.; Nygaard, U.C. Exposure to bisphenol A, but not phthalates, increases spontaneous diabetes type 1 development in NOD mice. Toxicol. Rep. 2015, 2, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Cavaghan, M.K.; Ehrmann, D.A.; Polonsky, K.S. Interactions between insulin resistance and insulin secretion in the development of glucose intolerance. J. Clin. Investig. 2000, 106, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Nolan, C.J.; Prentki, M. Insulin resistance and insulin hypersecretion in the metabolic syndrome and type 2 diabetes: Time for a conceptual framework shift. Diab. Vasc. Dis. Res. 2019, 16, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Toita, R.; Murata, M. Liver cell-targeted delivery of therapeutic molecules. Crit. Rev. Biotechnol. 2016, 36, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.L.; Zhang, J.; Goodyer, C.G.; Hayward, S.; Cooke, G.M.; Curran, I.H. Bisphenol A in human placental and fetal liver tissues collected from Greater Montreal area (Quebec) during 1998–2008. Chemosphere 2012, 89, 505–511. [Google Scholar] [CrossRef]
- Nahar, M.S.; Liao, C.; Kannan, K.; Harris, C.; Dolinoy, D.C. In utero bisphenol A concentration, metabolism, and global DNA methylation across matched placenta, kidney, and liver in the human fetus. Chemosphere 2015, 124, 54–60. [Google Scholar] [CrossRef]
- Zhang, J.; Cooke, G.M.; Curran, I.H.; Goodyer, C.G.; Cao, X.L. GC-MS analysis of bisphenol A in human placental and fetal liver samples. J. Chromatogr. B 2011, 879, 209–214. [Google Scholar] [CrossRef]
- Lin, R.; Wu, D.; Wu, F.J.; Meng, Y.; Zhang, J.H.; Wang, X.G.; Jia, L.H. Non-alcoholic fatty liver disease induced by perinatal exposure to bisphenol A is associated with activated mTOR and TLR4/NF-κB signaling pathways in offspring rats. Front. Endocrinol. 2019, 10, 620. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, A.; Li, T.; Gao, R.; Peng, C.; Liu, L.; Cheng, Q.; Mei, M.; Song, Y.; Xiang, X.; et al. Dysregulated autophagy in hepatocytes promotes bisphenol A-induced hepatic lipid accumulation in male mice. Endocrinology 2017, 158, 2799–2812. [Google Scholar] [CrossRef]
- Shimpi, P.C.; More, V.R.; Paranjpe, M.; Donepudi, A.C.; Goodrich, J.M.; Dolinoy, D.C.; Rubin, B.; Slitt, A.L. Hepatic lipid accumulation and Nrf2 expression following perinatal and peripubertal exposure to bisphenol A in a mouse model of nonalcoholic liver disease. Environ. Health Perspect. 2017, 125, 087005. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.; Gao, R.; Peng, C.; Yi, J.; Liu, L.; Yang, S.; Li, D.; Hu, J.; Luo, T.; Mei, M.; et al. Bisphenol A promotes hepatic lipid deposition involving Kupffer cells M1 polarization in male mice. J. Endocrinol. 2017, 234, 143–154. [Google Scholar] [CrossRef]
- Huc, L.; Lemarié, A.; Guéraud, F.; Héliès-Toussaint, C. Low concentrations of bisphenol A induce lipid accumulation mediated by the production of reactive oxygen species in the mitochondria of HepG2 cells. Toxicol. Vitr. 2012, 26, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Xia, W.; Zhu, Y.; Li, X.; Wang, D.; Liu, J.; Chang, H.; Li, G.; Xu, B.; Chen, X.; et al. Mitochondrial dysfunction in early life resulted from perinatal bisphenol A exposure contributes to hepatic steatosis in rat offspring. Toxicol. Lett. 2014, 228, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Pirozzi, C.; Lama, A.; Annunziata, C.; Cavaliere, G.; Ruiz-Fernandez, C.; Monnolo, A.; Comella, F.; Gualillo, O.; Stornaiuolo, M.; Mollica, M.P.; et al. Oral bisphenol A worsens liver immune-metabolic and mitochondrial dysfunction induced by high-fat diet in adult mice: Cross-talk between oxidative stress and inflammasome pathway. Antioxidants 2020, 9, 1201. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, L.S.; Oliveira, K.M.; Freitas, I.N.; Silva, J.A., Jr.; Silva, J.N.; Favero-Santos, B.C.; Bonfleur, M.L.; Carneiro, E.M.; Ribeiro, R.A. Bisphenol-A exposure worsens hepatic steatosis in ovariectomized mice fed on a high-fat diet: Role of endoplasmic reticulum stress and fibrogenic pathways. Life Sci. 2020, 256, 118012. [Google Scholar] [CrossRef]
- Long, Z.; Fan, J.; Wu, G.; Liu, X.; Wu, H.; Liu, J.; Chen, Y.; Su, S.; Cheng, X.; Xu, Z.; et al. Gestational bisphenol A exposure induces fatty liver development in male offspring mice through the inhibition of HNF1b and upregulation of PPARγ. Cell Biol. Toxicol. 2021, 37, 65–84. [Google Scholar] [CrossRef]
- Marmugi, A.; Ducheix, S.; Lasserre, F.; Polizzi, A.; Paris, A.; Priymenko, N.; Bertrand-Michel, J.; Pineau, T.; Guillou, H.; Martin, P.G.; et al. Low doses of bisphenol A induce gene expression related to lipid synthesis and trigger triglyceride accumulation in adult mouse liver. Hepatology 2012, 55, 395–407. [Google Scholar] [CrossRef]
- Strakovsky, R.S.; Wang, H.; Engeseth, N.J.; Flaws, J.A.; Helferich, W.G.; Pan, Y.X.; Lezmi, S. Developmental bisphenol A (BPA) exposure leads to sex-specific modification of hepatic gene expression and epigenome at birth that may exacerbate high-fat diet-induced hepatic steatosis. Toxicol. Appl. Pharmacol. 2015, 284, 101–112. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, H.; Zou, J.; Feng, X.; Feng, D. Bisphenol A induces cholesterol biosynthesis in HepG2 cells via SREBP-2/HMGCR signaling pathway. J. Toxicol. Sci. 2019, 44, 481–491. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, H.; Zou, J.; Mai, H.; Su, D.; Feng, X.; Feng, D. Bisphenol A exposure induces cholesterol synthesis and hepatic steatosis in C57BL/6 mice by down-regulating the DNA methylation levels of SREBP-2. Food Chem. Toxicol. 2019, 133, 110786. [Google Scholar] [CrossRef] [PubMed]
- Tonini, C.; Segatto, M.; Bertoli, S.; Leone, A.; Mazzoli, A.; Cigliano, L.; Barberio, L.; Mandalà, M.; Pallottini, V. Prenatal exposure to BPA: The effects on hepatic lipid metabolism in male and female rat fetuses. Nutrients 2021, 13, 1970. [Google Scholar] [CrossRef] [PubMed]
- Madison, B.B. Srebp2: A master regulator of sterol and fatty acid synthesis. J. Lipid Res. 2016, 57, 333–335. [Google Scholar] [CrossRef] [PubMed]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [PubMed]
- Toita, R.; Kang, J.H. Long-term profile of serological biomarkers, hepatic inflammation, and fibrosis in a mouse model of non-alcoholic fatty liver disease. Toxicol. Lett. 2020, 332, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Toita, R.; Kawano, T.; Fujita, S.; Murata, M.; Kang, J.H. Increased hepatic inflammation in a normal-weight mouse after long-term high-fat diet feeding. J. Toxicol. Pathol. 2018, 31, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Marchlewicz, E.; McCabe, C.; Djuric, Z.; Hoenerhoff, M.; Barks, J.; Tang, L.; Song, P.X.; Peterson, K.; Padmanabhan, V.; Dolinoy, D.C. Gestational exposure to high fat diets and bisphenol A alters metabolic outcomes in dams and offspring but produces hepatic steatosis only in dams. Chemosphere 2022, 286, 131645. [Google Scholar] [CrossRef]
- Wei, J.; Sun, X.; Chen, Y.; Li, Y.; Song, L.; Zhou, Z.; Xu, B.; Lin, Y.; Xu, S. Perinatal exposure to bisphenol A exacerbates nonalcoholic steatohepatitis-like phenotype in male rat offspring fed on a high-fat diet. J. Endocrinol. 2014, 222, 313–325. [Google Scholar] [CrossRef]
- Perreault, L.; McCurdy, C.; Kerege, A.A.; Houck, J.; Færch, K.; Bergman, B.C. Bisphenol A impairs hepatic glucose sensing in C57BL/6 male mice. PLoS ONE 2013, 8, e69991. [Google Scholar] [CrossRef]
- Li, G.; Chang, H.; Xia, W.; Mao, Z.; Li, Y.; Xu, S. F0 maternal BPA exposure induced glucose intolerance of F2 generation through DNA methylation change in Gck. Toxicol. Lett. 2014, 228, 192–199. [Google Scholar] [CrossRef]
- Jayashree, S.; Indumathi, D.; Akilavalli, N.; Sathish, S.; Selvaraj, J.; Balasubramanian, K. Effect of Bisphenol-A on insulin signal transduction and glucose oxidation in liver of adult male albino rat. Environ Toxicol Pharmacol. 2013, 35, 300–310. [Google Scholar] [CrossRef]
- Han, J.; Wang, Y. mTORC1 signaling in hepatic lipid metabolism. Protein Cell. 2018, 9, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Yecies, J.L.; Zhang, H.H.; Menon, S.; Liu, S.; Yecies, D.; Lipovsky, A.I.; Gorgun, C.; Kwiatkowski, D.J.; Hotamisligil, G.S.; Lee, C.H.; et al. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab. 2011, 14, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Mun, G.I.; Choi, E.; Kim, M.; Jeong, J.S.; Kang, K.W.; Jee, S.; Lim, K.M.; Lee, Y.S. Submicromolar bisphenol A induces proliferation and DNA damage in human hepatocyte cell lines in vitro and in juvenile rats in vivo. Food Chem. Toxicol. 2018, 111, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Elswefy, S.E.; Abdallah, F.R.; Atteia, H.H.; Wahba, A.S.; Hasan, R.A. Inflammation, oxidative stress and apoptosis cascade implications in bisphenol A-induced liver fibrosis in male rats. Int. J. Exp. Pathol. 2016, 97, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Elswefy, S.E.; Abdallah, F.R.; Wahba, A.S.; Hasan, R.A.; Atteia, H.H. Antifibrotic effect of curcumin, N-acetyl cysteine and propolis extract against bisphenol A-induced hepatotoxicity in rats: Prophylaxis versus co-treatment. Life Sci. 2020, 260, 118245. [Google Scholar] [CrossRef] [PubMed]
- Kazemi, S.; Mousavi, S.N.; Aghapour, F.; Rezaee, B.; Sadeghi, F.; Moghadamnia, A.A. Induction effect of bisphenol A on gene expression involving hepatic oxidative stress in rat. Oxid. Med. Cell Longev. 2016, 2016, 6298515. [Google Scholar] [CrossRef] [PubMed]
- Shmarakov, I.O.; Borschovetska, V.L.; Blaner, W.S. Hepatic detoxification of bisphenol A is retinoid-dependent. Toxicol. Sci. 2017, 157, 141–155. [Google Scholar] [CrossRef]
- Xia, W.; Jiang, Y.; Li, Y.; Wan, Y.; Liu, J.; Ma, Y.; Mao, Z.; Chang, H.; Li, G.; Xu, B.; et al. Early-life exposure to bisphenol a induces liver injury in rats involvement of mitochondria-mediated apoptosis. PLoS ONE 2014, 9, e90443. [Google Scholar] [CrossRef]
- Weinhouse, C.; Anderson, O.S.; Bergin, I.L.; Vandenbergh, D.J.; Gyekis, J.P.; Dingman, M.A.; Yang, J.; Dolinoy, D.C. Dose-dependent incidence of hepatic tumors in adult mice following perinatal exposure to bisphenol A. Environ. Health Perspect. 2014, 122, 485–491. [Google Scholar] [CrossRef]
- Elliott, P.; McKenna, W.J. Hypertrophic cardiomyopathy. Lancet 2004, 363, 1881–1891. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, I.; Minamino, T. Physiological and pathological cardiac hypertrophy. J. Mol. Cell Cardiol. 2016, 97, 245–262. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.B.; Raad, M.; Sebag, I.A.; Chalifour, L.E. Sex-specific cardiovascular responses to control or high fat diet feeding in C57bl/6 mice chronically exposed to bisphenol A. Toxicol. Rep. 2015, 2, 1310–1318. [Google Scholar] [CrossRef] [PubMed]
- MohanKumar, S.M.; Rajendran, T.D.; Vyas, A.K.; Hoang, V.; Asirvatham-Jeyaraj, N.; Veiga-Lopez, A.; Olivier, N.B.; Padmanabhan, V.; MohanKumar, P.S. Effects of prenatal bisphenol-A exposure and postnatal overfeeding on cardiovascular function in female sheep. J. Dev. Orig. Health Dis. 2017, 8, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liao, M.; Huang, R.; You, Y.; Lin, X.; Yang, H.; Fan, L.; Zhong, Y.; Li, X.; Li, J.; et al. Perinatal combinational exposure to bisphenol A and a high-fat diet contributes to transgenerational dysregulation of cardiovascular and metabolic systems in mice. Front. Cell Dev. Biol. 2022, 10, 834346. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Yang, S.; Li, X.; Liang, F.; Zhou, R.; Wang, H.; Feng, Y.; Wang, Y. Low doses of BPA induced abnormal mitochondrial fission and hypertrophy in human embryonic stem cell-derived cardiomyocytes via the calcineurin-DRP1 signaling pathway: A comparison between XX and XY cardiomyocytes. Toxicol. Appl. Pharmacol. 2020, 388, 114850. [Google Scholar] [CrossRef] [PubMed]
- Belcher, S.M.; Gear, R.B.; Kendig, E.L. Bisphenol A alters autonomic tone and extracellular matrix structure and induces sex-specific effects on cardiovascular function in male and female CD-1 mice. Endocrinology 2015, 156, 882–895. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Xia, W.; Yang, J.; Zhu, Y.; Chang, H.; Liu, J.; Huo, W.; Xu, B.; Chen, X.; Li, Y.; et al. BPA-induced DNA hypermethylation of the master mitochondrial gene PGC-1α contributes to cardiomyopathy in male rats. Toxicology 2015, 329, 21–31. [Google Scholar] [CrossRef]
- Gear, R.; Kendziorski, J.A.; Belcher, S.M. Effects of bisphenol A on incidence and severity of cardiac lesions in the NCTR-Sprague-Dawley rat: A CLARITY-BPA study. Toxicol. Lett. 2017, 275, 123–135. [Google Scholar] [CrossRef]
- Bruno, K.A.; Mathews, J.E.; Yang, A.L.; Frisancho, J.A.; Scott, A.J.; Greyner, H.D.; Molina, F.A.; Greenaway, M.S.; Cooper, G.M.; Bucek, A.; et al. BPA alters estrogen receptor expression in the heart after viral infection activating cardiac mast cells and T cells leading to perimyocarditis and fibrosis. Front. Endocrinol. 2019, 10, 598. [Google Scholar] [CrossRef]
- Bruno, K.A.; Macomb, L.P.; Morales-Lara, A.C.; Mathews, J.E.; Frisancho, J.A.; Yang, A.L.; Di Florio, D.N.; Edenfield, B.H.; Whelan, E.R.; Salomon, G.R.; et al. Sex-specific effects of plastic caging in murine viral myocarditis. Int. J. Mol. Sci. 2021, 22, 8834. [Google Scholar] [CrossRef] [PubMed]
- Reventun, P.; Sanchez-Esteban, S.; Cook, A.; Cuadrado, I.; Roza, C.; Moreno-Gomez-Toledano, R.; Muñoz, C.; Zaragoza, C.; Bosch, R.J.; Saura, M. Bisphenol A induces coronary endothelial cell necroptosis by activating RIP3/CamKII dependent pathway. Sci. Rep. 2020, 10, 4190. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Liang, Q.; Chen, Y.; Wang, H.S. Molecular mechanisms underlying the rapid arrhythmogenic action of bisphenol A in female rat hearts. Endocrinology 2013, 154, 4607–4617. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Hong, K.; Wang, H.S. Progesterone protects against bisphenol A-induced arrhythmias in female rat cardiac myocytes via rapid signaling. Endocrinology 2017, 158, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Mao, N.; Gao, Q.; Hu, H.; Zhu, T.; Hao, L. BPA disrupts the cardioprotection by 17β-oestradiol against ischemia/reperfusion injury in isolated guinea pig hearts. Steroids 2019, 146, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Chen, Y.; Dong, M.; Song, W.; Belcher, S.M.; Wang, H.S. Bisphenol A and 17β-estradiol promote arrhythmia in the female heart via alteration of calcium handling. PLoS ONE 2011, 6, e25455. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Song, W.; Chen, Y.; Hong, K.; Rubinstein, J.; Wang, H.S. Low-dose bisphenol A and estrogen increase ventricular arrhythmias following ischemia-reperfusion in female rat hearts. Food Chem. Toxicol. 2013, 56, 75–80. [Google Scholar] [CrossRef]
- Hinderer, S.; Schenke-Layland, K. Cardiac fibrosis-A short review of causes and therapeutic strategies. Adv. Drug Deliv. Rev. 2019, 146, 77–82. [Google Scholar] [CrossRef]
- Amin, D.M. Role of copeptin as a novel biomarker of bisphenol A toxic effects on cardiac tissues: Biochemical, histological, immunohistological, and genotoxic study. Environ. Sci. Pollut. Res. Int. 2019, 26, 36037–36047. [Google Scholar] [CrossRef]
- García-Arévalo, M.; Lorza-Gil, E.; Cardoso, L.; Batista, T.M.; Araujo, T.R.; Ramos, L.A.F.; Areas, M.A.; Nadal, A.; Carneiro, E.M.; Davel, A.P. Ventricular fibrosis and coronary remodeling following short-term exposure of healthy and malnourished mice to bisphenol A. Front. Physiol. 2021, 12, 638506. [Google Scholar] [CrossRef]
- Rasdi, Z.; Kamaludin, R.; Ab Rahim, S.; Syed Ahmad Fuad, S.B.; Othman, M.H.D.; Siran, R.; Mohd Nor, N.S.; Abdul Hamid Hasani, N.; Sheikh Abdul Kadir, S.H. The impacts of intrauterine bisphenol A exposure on pregnancy and expression of miRNAs related to heart development and diseases in animal model. Sci. Rep. 2020, 10, 5882. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhang, L.; Wu, X.; Hou, L.; Li, Z.; Ju, J.; Li, Q.; Qin, W.; Li, J.; Zhang, Q.; et al. Bisphenol A, an environmental estrogen-like toxic chemical, induces cardiac fibrosis by activating the ERK1/2 pathway. Toxicol. Lett. 2016, 250–251, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Xia, M.; Zhang, L.; Cheng, W.; Yan, J.; Sun, Y.; Wang, Y.; Jiang, H. Individual and combined effects of BPA, BPS and BPAF on the cardiomyocyte differentiation of embryonic stem cells. Ecotoxicol. Environ. Saf. 2021, 220, 112366. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Huang, Y.; Zhang, L.; Liu, W. Involvement of estrogen receptor and GPER in bisphenol A induced proliferation of vascular smooth muscle cells. Toxicol. Vitr. 2019, 56, 156–162. [Google Scholar] [CrossRef]
- Saura, M.; Marquez, S.; Reventun, P.; Olea-Herrero, N.; Arenas, M.I.; Moreno-Gómez-Toledano, R.; Gómez-Parrizas, M.; Muñóz-Moreno, C.; González-Santander, M.; Zaragoza, C.; et al. Oral administration of bisphenol A induces high blood pressure through angiotensin II/CaMKII-dependent uncoupling of eNOS. FASEB J. 2014, 28, 4719–4728. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, M.; Raja, B.; Manivannan, J. Exposure to a “safe” dose of environmental pollutant bisphenol A elevates oxidative stress and modulates vasoactive system in hypertensive rats. Hum. Exp. Toxicol. 2021, 40 (Suppl. S12), S654–S665. [Google Scholar] [CrossRef] [PubMed]
- Paz Ocaranza, M.; Riquelme, J.A.; García, L.; Jalil, J.E.; Chiong, M.; Santos, R.A.S.; Lavandero, S. Counter-regulatory renin-angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 116–129. [Google Scholar] [CrossRef]
- Friques, A.G.F.; Santos, F.D.N.; Angeli, D.B.; Silva, F.A.C.; Dias, A.T.; Aires, R.; Leal, M.A.S.; Nogueira, B.V.; Amorim, F.G.; Campagnaro, B.P.; et al. Bisphenol A contamination in infant rats: Molecular, structural, and physiological cardiovascular changes and the protective role of kefir. J. Nutr. Biochem. 2020, 75, 108254. [Google Scholar] [CrossRef]
- Kim, M.J.; Moon, M.K.; Kang, G.H.; Lee, K.J.; Choi, S.H.; Lim, S.; Oh, B.C.; Park, D.J.; Park, K.S.; Jang, H.C.; et al. Chronic exposure to bisphenol A can accelerate atherosclerosis in high-fat-fed apolipoprotein E knockout mice. Cardiovasc. Toxicol. 2014, 14, 120–128. [Google Scholar] [CrossRef]
- Sui, Y.; Park, S.H.; Helsley, R.N.; Sunkara, M.; Gonzalez, F.J.; Morris, A.J.; Zhou, C. Bisphenol A increases atherosclerosis in pregnane X receptor-humanized ApoE deficient mice. J. Am. Heart Assoc. 2014, 3, e000492. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, C.; Yang, J.; Yuan, F.; Cheng, R.; Chen, R.; Shen, Y.; Huang, L. Impairment of sirtuin 1-mediated DNA repair is involved in bisphenol A-induced aggravation of macrophage inflammation and atherosclerosis. Chemosphere 2021, 265, 128997. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Ning, B.; Waqar, A.B.; Niimi, M.; Li, S.; Satoh, K.; Shiomi, M.; Ye, T.; Dong, S.; Fan, J. Bisphenol A exposure enhances atherosclerosis in WHHL rabbits. PLoS ONE 2014, 9, e110977. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Ning, B.; Waqar, A.B.; Niimi, M.; Li, S.; Satoh, K.; Shiomi, M.; Ye, T.; Dong, S.; Fan, J. Bisphenol A exposure induces metabolic disorders and enhances atherosclerosis in hyperlipidemic rabbits. J. Appl. Toxicol. 2015, 35, 1058–1070. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Toita, R.; Asai, D.; Yamaoka, T.; Murata, M. Reduction of inorganic phosphate-induced human smooth muscle cells calcification by inhibition of protein kinase A and p38 mitogen-activated protein kinase. Heart Vessel. 2014, 29, 718–722. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, G.; Iacobini, C.; Blasetti Fantauzzi, C.; Menini, S. The dark and bright side of atherosclerotic calcification. Atherosclerosis 2015, 238, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Toita, R.; Otani, K.; Kawano, T.; Fujita, S.; Murata, M.; Kang, J.H. Protein kinase A (PKA) inhibition reduces human aortic smooth muscle cell calcification stimulated by inflammatory response and inorganic phosphate. Life Sci. 2018, 209, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Bourne, L.E.; Wheeler-Jones, C.P.; Orriss, I.R. Regulation of mineralisation in bone and vascular tissue: A comparative review. J. Endocrinol. 2021, 248, R51–R65. [Google Scholar] [CrossRef]
- Lee, S.J.; Lee, I.K.; Jeon, J.H. Vascular calcification-new insights into its mechanism. Int. J. Mol. Sci. 2020, 21, 2685. [Google Scholar] [CrossRef]
- Kanno, S.; Hirano, S.; Kayama, F. Effects of phytoestrogens and environmental estrogens on osteoblastic differentiation in MC3T3-E1 cells. Toxicology 2004, 196, 137–145. [Google Scholar] [CrossRef]
- Thent, Z.C.; Froemming, G.R.A.; Ismail, A.B.M.; Fuad, S.B.S.A.; Muid, S. Employing different types of phytoestrogens improve bone mineralization in bisphenol A stimulated osteoblast. Life Sci. 2018, 210, 214–223. [Google Scholar] [CrossRef]
- Kopp, J.B.; Anders, H.J.; Susztak, K.; Podestà, M.A.; Remuzzi, G.; Hildebrandt, F.; Romagnani, P. Podocytopathies. Nat. Rev. Dis. Primers 2020, 6, 68. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Gómez-Toledano, R.; Arenas, M.I.; González-Martínez, C.; Olea-Herrero, N.; Reventún, P.; Di Nunzio, M.; Sánchez-Esteban, S.; Arilla-Ferreiro, E.; Saura, M.; Bosch, R.J. Bisphenol A impaired cell adhesion by altering the expression of adhesion and cytoskeleton proteins on human podocytes. Sci. Rep. 2020, 10, 16638. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Yang, S.; Li, T.; Gao, R.; Hu, J.; Luo, T.; Qing, H.; Zhen, Q.; Hu, R.; Li, X.; et al. Role of neutrophil extracellular traps in chronic kidney injury induced by bisphenol-A. J. Endocrinol. 2019, 241, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Bosch-Panadero, E.; Mas, S.; Civantos, E.; Abaigar, P.; Camarero, V.; Ruiz-Priego, A.; Ortiz, A.; Egido, J.; González-Parra, E. Bisphenol A is an exogenous toxin that promotes mitochondrial injury and death in tubular cells. Environ. Toxicol. 2018, 33, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Kong, Y.; Ommati, M.M.; Tang, Z.; Li, H.; Li, L.; Zhao, C.; Shi, Z.; Wang, J. Bisphenol A-induced apoptosis, oxidative stress and DNA damage in cultured rhesus monkey embryo renal epithelial Marc-145 cells. Chemosphere 2019, 234, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Gómez-Toledano, R.; Arenas, M.I.; Muñoz-Moreno, C.; Olea-Herrero, N.; Reventun, P.; Izquierdo-Lahuerta, A.; Antón-Cornejo, A.; González-Santander, M.; Zaragoza, C.; Saura, M.; et al. Comparison of the renal effects of bisphenol A in mice with and without experimental diabetes. Role of sexual dimorphism. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166296. [Google Scholar] [CrossRef] [PubMed]
- Peerapanyasut, W.; Kobroob, A.; Palee, S.; Chattipakorn, N.; Wongmekiat, O. Bisphenol A aggravates renal ischemia-reperfusion injury by disrupting mitochondrial homeostasis and N-acetylcysteine mitigates the injurious outcomes. IUBMB Life 2020, 72, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Liu, Z.; Liu, T. The antagonistic effect of bisphenol A and nonylphenol on liver and kidney injury in rats. Immunopharmacol. Immunotoxicol. 2021, 43, 527–535. [Google Scholar] [CrossRef]
- Priego, A.R.; Parra, E.G.; Mas, S.; Morgado-Pascual, J.L.; Ruiz-Ortega, M.; Rayego-Mateos, S. Bisphenol A modulates autophagy and exacerbates chronic kidney damage in mice. Int. J. Mol. Sci. 2021, 22, 7189. [Google Scholar] [CrossRef]
- Dong, Y.; Zhang, Z.; Liu, H.; Jia, L.; Qin, M.; Wang, X. Exacerbating lupus nephritis following BPA exposure is associated with abnormal autophagy in MRL/lpr mice. Am. J. Transl. Res. 2020, 12, 649–659. [Google Scholar]
- Nuñez, P.; Fernandez, T.; García-Arévalo, M.; Alonso-Magdalena, P.; Nadal, A.; Perillan, C.; Arguelles, J. Effects of bisphenol A treatment during pregnancy on kidney development in mice: A stereological and histopathological study. J. Dev. Orig. Health Dis. 2018, 9, 208–214. [Google Scholar] [CrossRef]
- Bao, L.; Zhao, C.; Feng, L.; Zhao, Y.; Duan, S.; Qiu, M.; Wu, K.; Zhang, N.; Hu, X.; Fu, Y. Ferritinophagy is involved in bisphenol A-induced ferroptosis of renal tubular epithelial cells through the activation of the AMPK-mTOR-ULK1 pathway. Food Chem. Toxicol. 2022, 163, 112909. [Google Scholar] [CrossRef] [PubMed]
- Diamante, G.; Cely, I.; Zamora, Z.; Ding, J.; Blencowe, M.; Lang, J.; Bline, A.; Singh, M.; Lusis, A.J.; Yang, X. Systems toxicogenomics of prenatal low-dose BPA exposure on liver metabolic pathways, gut microbiota, and metabolic health in mice. Environ. Int. 2021, 146, 106260. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Zhang, H.; Jiang, X.; Zou, J.; Li, Q.; Mai, H.; Su, D.; Ling, W.; Feng, X. Bisphenol A exposure induces gut microbiota dysbiosis and consequent activation of gut-liver axis leading to hepatic steatosis in CD-1 mice. Environ. Pollut. 2020, 265 Pt A, 114880. [Google Scholar] [CrossRef]
- Ni, Y.; Hu, L.; Yang, S.; Ni, L.; Ma, L.; Zhao, Y.; Zheng, A.; Jin, Y.; Fu, Z. Bisphenol A impairs cognitive function and 5-HT metabolism in adult male mice by modulating the microbiota-gut-brain axis. Chemosphere 2021, 282, 130952. [Google Scholar] [CrossRef] [PubMed]
- Reddivari, L.; Veeramachaneni, D.N.R.; Walters, W.A.; Lozupone, C.; Palmer, J.; Hewage, M.K.K.; Bhatnagar, R.; Amir, A.; Kennett, M.J.; Knight, R.; et al. Perinatal bisphenol A exposure induces chronic inflammation in rabbit offspring via modulation of gut bacteria and their metabolites. mSystems 2017, 2, e00093-17. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Sarma, S.J.; Marshall, B.L.; Liu, Y.; Kinkade, J.A.; Bellamy, M.M.; Mao, J.; Helferich, W.G.; Schenk, A.K.; Bivens, N.J.; et al. Developmental exposure of California mice to endocrine disrupting chemicals and potential effects on the microbiome-gut-brain axis at adulthood. Sci. Rep. 2020, 10, 10902. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Chen, S.; Zhang, L.; Qu, W.; Chen, Z. Bisphenol A increases intestinal permeability through disrupting intestinal barrier function in mice. Environ. Pollut. 2019, 254 Pt A, 112960. [Google Scholar] [CrossRef]
- Wang, K.; Qiu, L.; Zhu, J.; Sun, Q.; Qu, W.; Yu, Y.; Zhao, Z.; Yu, Y.; Shao, G. Environmental contaminant BPA causes intestinal damage by disrupting cellular repair and injury homeostasis in vivo and in vitro. Biomed. Pharmacother. 2021, 137, 111270. [Google Scholar] [CrossRef] [PubMed]
- Misme-Aucouturier, B.; De Carvalho, M.; Delage, E.; Dijoux, E.; Klein, M.; Brosseau, C.; Bodinier, M.; Guzylack-Piriou, L.; Bouchaud, G. Oral exposure to bisphenol A exacerbates allergic inflammation in a mouse model of food allergy. Toxicology 2022, 472, 153188. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Kondo, F. Bisphenol A degradation by bacteria isolated from river water. Arch. Environ. Contam. Toxicol. 2002, 43, 265–269. [Google Scholar] [CrossRef] [PubMed]
- López-Moreno, A.; Torres-Sánchez, A.; Acuña, I.; Suárez, A.; Aguilera, M. Representative Bacillus sp. AM1 from gut microbiota harbor versatile molecular pathways for bisphenol A biodegradation. Int. J. Mol. Sci. 2021, 22, 4952. [Google Scholar] [CrossRef] [PubMed]
- López-Moreno, A.; Ruiz-Moreno, Á.; Pardo-Cacho, J.; Cerk, K.; Torres-Sánchez, A.; Ortiz, P.; Úbeda, M.; Aguilera, M. Culturing and molecular approaches for identifying microbiota taxa impacting children’s obesogenic phenotypes related to xenobiotic dietary exposure. Nutrients 2022, 14, 241. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Rui, M.; Nie, Y.; Lu, G. Influence of gastrointestinal tract on metabolism of bisphenol A as determined by in vitro simulated system. J. Hazard Mater. 2018, 355, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Gao, H.; Du, Z.; Liu, H.; Cheng, Q.; Zhang, F.; Ye, J.; Wang, A.; Dou, Y.; Ma, B.; et al. A new approach for reducing pollutants level: A longitudinal cohort study of physical exercises in young people. BMC Public Health 2022, 22, 223. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.A.; Painter, M.S.; Javurek, A.B.; Ellersieck, M.R.; Wiedmeyer, C.E.; Thyfault, J.P.; Rosenfeld, C.S. Sex-dependent effects of developmental exposure to bisphenol A and ethinyl estradiol on metabolic parameters and voluntary physical activity. J. Dev. Orig. Health Dis. 2015, 6, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Lin, Y.; Qiu, X.; Liu, J.; Zhu, T.; Araujo, J.A.; Fiehn, O.; Zhu, Y. Triglyceride profiles are associated with subacute exposure to bisphenol A in healthy young adults. Sci. Total Environ. 2022, 825, 153991. [Google Scholar] [CrossRef] [PubMed]
- Altamirano, G.A.; Muñoz-de-Toro, M.; Luque, E.H.; Gómez, A.L.; Delconte, M.B.; Kass, L. Milk lipid composition is modified by perinatal exposure to bisphenol A. Mol. Cell Endocrinol. 2015, 411, 258–267. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Li, L.; Eguchi, A.; Kannan, K.; Kim, E.Y.; Iwata, H. Effects on the liver lipidome of rat offspring prenatally exposed to bisphenol A. Sci. Total Environ. 2021, 759, 143466. [Google Scholar] [CrossRef]
- Blaauwendraad, S.M.; Voerman, E.; Trasande, L.; Kannan, K.; Santos, S.; Ruijter, G.J.G.; Sol, C.M.; Marchioro, L.; Shokry, E.; Koletzko, B.; et al. Associations of maternal bisphenol urine concentrations during pregnancy with neonatal metabolomic profiles. Metabolomics 2021, 17, 84. [Google Scholar] [CrossRef]
- Zhao, C.; Xie, P.; Yang, T.; Wang, H.; Chung, A.C.K.; Cai, Z. Identification of glycerophospholipid fatty acid remodeling by using mass spectrometry imaging in bisphenol S induced mouse liver. Chin. Chem. Lett. 2018, 29, 1281–1283. [Google Scholar] [CrossRef]
- Zhao, C.; Yong, T.; Zhang, Y.; Jin, Y.; Xiao, Y.; Wang, H.; Zhao, B.; Cai, Z. Evaluation of the splenic injury following exposure of mice to bisphenol S: A mass spectrometry-based lipidomics and imaging analysis. Environ. Int. 2020, 135, 105378. [Google Scholar] [CrossRef] [PubMed]
- Asai, D.; Kawano, T.; Murata, M.; Nakashima, H.; Toita, R.; Kang, J.H. Effect of fetal bovine serum concentration on lysophosphatidylcholine-mediated proliferation and apoptosis of human aortic smooth muscle cells. J. Oleo Sci. 2020, 69, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.T.; Ramesh, T.; Toh, X.R.; Nguyen, L.N. Emerging roles of lysophospholipids in health and disease. Prog. Lipid Res. 2020, 80, 101068. [Google Scholar] [CrossRef] [PubMed]
- Toita, R.; Asai, D.; Otani, K.; Kawano, T.; Murata, M.; Kang, J.H. Suppression of lysophosphatidylcholine-induced human aortic smooth muscle cell calcification by protein kinase A inhibition. Lipids 2019, 54, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Hanioka, N.; Isobe, T.; Tanaka-Kagawa, T.; Jinno, H.; Ohkawara, S. In vitro glucuronidation of bisphenol A in liver and intestinal microsomes: Interspecies differences in humans and laboratory animals. Drug Chem. Toxicol. 2022, 45, 1565–1569. [Google Scholar] [CrossRef] [PubMed]
- Pritchett, J.J.; Kuester, R.K.; Sipes, I.G. Metabolism of bisphenol a in primary cultured hepatocytes from mice, rats, and humans. Drug Metab. Dispos. 2002, 30, 1180–1185. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, F.; Zhang, G.H.; Du, K.; Shen, L.; Ma, R.; Wang, X.; Wang, X.; Zhang, J. Maternal prenatal urinary bisphenol A level and child cardio-metabolic risk factors: A prospective cohort study. Environ. Pollut. 2020, 265 Pt A, 115008. [Google Scholar] [CrossRef]
- Liu, J.; Yu, P.; Qian, W.; Li, Y.; Zhao, J.; Huan, F.; Wang, J.; Xiao, H. Perinatal bisphenol A exposure and adult glucose homeostasis: Identifying critical windows of exposure. PLoS ONE 2013, 8, e64143. [Google Scholar] [CrossRef]
- Zhou, R.; Cheng, W.; Feng, Y.; Wang, W.; Liang, F.; Luo, F.; Yang, S.; Wang, Y. Combined effects of BPA and PFOS on fetal cardiac development: In vitro and in vivo experiments. Environ. Toxicol. Pharmacol. 2020, 80, 103434. [Google Scholar] [CrossRef]
- Dökmeci, A.H.; Karaboğa, İ.; Güzel, S.; Erboğa, Z.F.; Yılmaz, A. Toxicological assessment of low-dose bisphenol A, lead and endosulfan combination: Chronic toxicity study in male rats. Environ. Sci. Pollut. Res. Int. 2022, 29, 10558–10574. [Google Scholar] [CrossRef]
- Wang, D.; Zhu, W.; Yan, S.; Meng, Z.; Yan, J.; Teng, M.; Jia, M.; Li, R.; Zhou, Z. Impaired lipid and glucose homeostasis in male mice offspring after combined exposure to low-dose bisphenol A and arsenic during the second half of gestation. Chemosphere 2018, 210, 998–1005. [Google Scholar] [CrossRef]
- Troisi, R.; Titus, L.; Hatch, E.E.; Palmer, J.R.; Huo, D.; Strohsnitter, W.C.; Adam, E.; Ricker, W.; Hyer, M.; Hoover, R.N. A prospective cohort study of prenatal DES exposure and cardiovascular disease risk. J. Clin. Endocrinol. Metab. 2018, 103, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Haddad, R.; Kasneci, A.; Sebag, I.A.; Chalifour, L.E. Cardiac structure/function, protein expression, and DNA methylation are changed in adult female mice exposed to diethylstilbestrol in utero. Can. J. Physiol. Pharmacol. 2013, 91, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.F.; Canário, A.V.M.; Power, D.M.; Campinho, M.A. Ioxynil and diethylstilbestrol disrupt vascular and heart development in zebrafish. Environ Int. 2019, 124, 511–520. [Google Scholar] [CrossRef]
- Hafezi, S.A.; Abdel-Rahman, W.M. The endocrine disruptor bisphenol A (BPA) exerts a wide range of effects in carcinogenesis and response to therapy. Curr. Mol. Pharmacol. 2019, 12, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Lagunas-Rangel, F.A.; Liu, W.; Schiöth, H.B. Can exposure to environmental pollutants be associated with less effective chemotherapy in cancer patients? Int. J. Environ. Res. Public Health 2022, 19, 2064. [Google Scholar] [CrossRef] [PubMed]
- Palmisano, B.T.; Zhu, L.; Stafford, J.M. Role of estrogens in the regulation of liver lipid metabolism. Adv. Exp. Med. Biol. 2017, 1043, 227–256. [Google Scholar] [PubMed]
- Zhang, Y.; Shi, Y.; Li, Z.; Sun, L.; Zhang, M.; Yu, L.; Wu, S. BPA disrupts 17-estradiol-mediated hepatic protection against ischemia/reperfusion injury in rat liver by upregulating the Ang II/AT1R signaling pathway. Mol. Med. Rep. 2020, 22, 416–422. [Google Scholar] [CrossRef]
- Iorga, A.; Cunningham, C.M.; Moazeni, S.; Ruffenach, G.; Umar, S.; Eghbali, M. The protective role of estrogen and estrogen receptors in cardiovascular disease and the controversial use of estrogen therapy. Biol. Sex Differ. 2017, 8, 33. [Google Scholar] [CrossRef]
- Kiuchi, S.; Ikeda, T. Management of hypertension associated with cardiovascular failure. J. Cardiol. 2022, 79, 698–702. [Google Scholar] [CrossRef]
- Strippoli, G.F.; Bonifati, C.; Craig, M.; Navaneethan, S.D.; Craig, J.C. Angiotensin converting enzyme inhibitors and angiotensin II receptor antagonists for preventing the progression of diabetic kidney disease. Cochrane. Database Syst. Rev. 2006, 2006, CD006257. [Google Scholar]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, J.-H.; Asai, D.; Toita, R. Bisphenol A (BPA) and Cardiovascular or Cardiometabolic Diseases. J. Xenobiot. 2023, 13, 775-810. https://doi.org/10.3390/jox13040049
Kang J-H, Asai D, Toita R. Bisphenol A (BPA) and Cardiovascular or Cardiometabolic Diseases. Journal of Xenobiotics. 2023; 13(4):775-810. https://doi.org/10.3390/jox13040049
Chicago/Turabian StyleKang, Jeong-Hun, Daisuke Asai, and Riki Toita. 2023. "Bisphenol A (BPA) and Cardiovascular or Cardiometabolic Diseases" Journal of Xenobiotics 13, no. 4: 775-810. https://doi.org/10.3390/jox13040049
APA StyleKang, J.-H., Asai, D., & Toita, R. (2023). Bisphenol A (BPA) and Cardiovascular or Cardiometabolic Diseases. Journal of Xenobiotics, 13(4), 775-810. https://doi.org/10.3390/jox13040049