Abstract
The current scenario of male infertility is not yet fully elucidated; however, there is increasing evidence that it is associated with the widespread exposure to endocrine-disrupting chemicals (EDCs), and in particular to obesogens. These compounds interfere with hormones involved in the regulation of metabolism and are associated with weight gain, being also able to change the functioning of the male reproductive axis and, consequently, the testicular physiology and metabolism that are pivotal for spermatogenesis. The disruption of these tightly regulated metabolic pathways leads to adverse reproductive outcomes. The permanent exposure to obesogens has raised serious health concerns. Evidence suggests that obesogens are one of the leading causes of the marked decline of male fertility and key players in shaping the future health outcomes not only for those who are directly exposed but also for upcoming generations. In addition to the changes that lead to inefficient functioning of the male gametes, obesogens induce alterations that are “imprinted” on the genes of the male gametes, establishing a link between generations and contributing to the transmission of defects. Unveiling the molecular mechanisms by which obesogens induce toxicity that may end-up in epigenetic modifications is imperative. This review describes and discusses the suggested molecular targets and potential mechanisms for obesogenic–disrupting chemicals and the subsequent effects on male reproductive health.
1. Introduction
Today, we are exposed to a cocktail of man-made chemicals, some of which have the potential to disrupt our hormonal system. Any compound or mixture of compounds, which interferes with any aspect of the endocrine system is classified by the Endocrine Society as an endocrine-disrupting chemical (EDC) [1]. These chemicals are ubiqutious in consumer products, being widely detected in the indoor environment (e.g., homes, workplaces, schools, gyms, transportation) and in diet samples. Generally, their occurrence in the indoor environment is a consequence of their widespread use in construction materials and consumer products, including, for example, house furnishings, electronic devices, wallpaper, textiles, kitchen utensils, cleaning products, and cosmetics, amongst many others. Indoor activities such as the use of biocides, deodorisers, the burning of incense and candles, smoking and cooking are also responsible for the release of EDCs into the indoor environment. Furthermore, all materials carried from outdoors on our shoes or clothes also increase our exposure. EDCs are also present in food items due to food production (e.g., pesticides), processing (antimicrobials, preservatives) and storage (plasticizers), which makes diet a significant route of EDCs. Such continuous and widespread exposure has raised concerns not only within the scientific community but also to the health authorities and policymakers due to the adverse effects on human health. As the endocrine system is at the interface with the environment, EDCs disturb the homeostatic mechanisms of the body. The endocrine system plays a central role in regulating critical biological functions as metabolism, development, reproduction, and behavior. Understanding how these chemicals affect physiologic processes and initiate pathological conditions is essential to dissect the etiology of hormone-related diseases. Scientific evidence has demonstrated the relationship between EDCs and neurobehavioral and neurodevelopmental changes, immune disorders, cancer development, reproductive health problems and metabolic diseases in humans [1,2,3]. All these findings are in line with what was reported from animal models [1,4,5]. The group of compounds classified as EDCs is extremely heterogeneous and includes industrially produced chemicals, such as polychlorinated biphenyls (PCBs), bisphenol A (BPA), phthalates, pesticides, fungicides, and pharmaceutical drugs [1]. Likewise, natural chemicals found in human and animal diets (e.g., phytoestrogens, such as genistein and daidzein, or even mycotoxins) can also act as EDCs [1]. Furthermore, several of these compounds are lipophilic which make them particularly relevant from a physiologic point of view. EDCs act by different mechanisms and elicit several responses. One recent concern about the adverse effects of EDCs is the ability that some of them have to induce weight gain [6]. Today these chemicals are classified as obesogens, a term coined by Blumberg and Grun in 2006 [7]. Obesogens are a limited group of EDCs capable of inducing adipogenesis and altering the mechanisms that regulate appetite, satiety and energy metabolism. Though the relationship between lifestyle habits of modern societies and the obesity rates has been established, obesogens have been considered major contributors to the obesity epidemic [8,9,10]. The mechanisms by which obesogens exert their effects are much broader than was initially postulated, see reviews [11,12,13,14,15]. In Europe, exposures to EDCs have contributed substantially to obesity [8,16,17,18,19] and albeit obesogens are only part of the problem, it is necessary to become aware of the risks of permanent exposure to these chemicals. Reducing our exposure to these chemicals is therefore important to decrease the chances of becoming more prone to develop obesity. In parallel to the adverse effects of obesogens on whole body metabolism, another issue of concern is male reproductive health problems resulting from exposure to EDCs, whether they are obesogenic or not. In the last decades, male fertility has decreased significantly. Today, 15% of the population is infertile, and one in seven couples is infertile at reproductive age [20]. About 50% of the cases are due to the male factor, and in many of them, it occurs due to idiopathic infertility [20]. The etiology of this adverse trend remains a subject of intense debate, but there has been a consensual awareness regarding the role of external factors, such as lifestyle habits and its associated factors, especially the permanent exposure to EDCs. The decline of male fertility has also been concurrent with the overall increase in exposure to EDCs, leading to these contaminants being singled out as an important cause of male infertility [21]. Treatment is usually associated with assisted reproductive technologies, which are applied to increase the chances of conception. Infertility is not a life-threatening disease, but it is vital to society, and the high costs associated with the infertility treatments and medically assisted reproduction techniques make these alternatives almost inaccessible to most infertile couples. The male reproductive function is highly susceptible to the effects of EDCs [21,22], and most of these chemicals are lipophilic, mimicking naturally occurring hormones. Male reproduction is highly dependent on endocrine regulation, especially the reproductive events, such as steroidogenesis and spermatogenesis that are dependent on Leydig (LCs) and Sertoli cells (SCs), respectively [23]. The majority of EDCs interfere with the receptors of endogenous hormones, impairing genome and non-genomic responses [24], which is extremely relevant since most of the effects are exerted through disturbance of estrogen-, anti-estrogen, androgen- and anti-androgen-mediated processes [25,26]. However, some EDCs can modify hormone bioavailability by interfering with its secretion and transport or by disrupting the enzymatic pathways involved in hormone synthesis and metabolism [27,28]. It is essential to understand how EDCs, especially obesogens, induce male infertility namely in the deregulation of the mechanisms that govern the function of the reproductive axis, sperm formation, and its functioning (Figure 1).
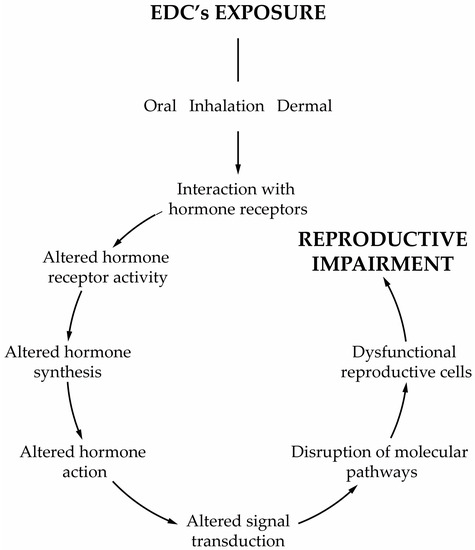
Figure 1.
Schematic representation of the most important characteristics of obesogens’ effects.
Information regarding human data is scarce due to ethical issues in performing human studies to unveil the adverse effects of EDCs. Furthermore, most of the available data demonstrated the effects of EDCs alone, and the understanding of the potential human health risks requires the study of the complex mixtures. In this context, animal studies have suggested that environmental cues play a significant role in sperm quality and thus, in male fertility [29,30,31]. However, there is a need to converge information from basic knowledge about EDCs’ mechanisms, and epidemiological studies to predict and assess potential effects in humans. Furthermore, EDCs can also act on multiple generations, through small modifications induced in the DNA of male gametes and contributing to a transgenerational amplification of reproductive defects, but also other adverse health outcomes.
This chapter will present and discuss the impacts of EDCs, especially obesogens, on male reproductive health (Figure 2). The putative effects of obesogenic EDCs on sperm function and sperm-related events and the subsequent consequences to overall male fertility potential will also be explored.
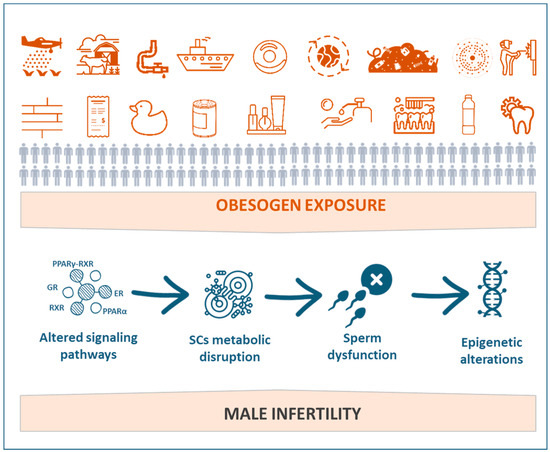
Figure 2.
Schematic representation of the implications of exposure to obesogens on male reproductive impairment.
2. Brief History of Endocrine-Disrupting Chemicals
In the beginning of the 20th century, pig farmers in the United States became concerned about the reproductive complications in their swine herds fed with a moldy grain [32]. Later in the 1940s, sheep farmers in Western Australia also observed reproductive problems in their sheep after grazing on specific fields of clover [33]. These were the first evidence of the effects of EDCs on animals. The idea that EDCs could find their way into the animals began to gain strength when Gassner and collaborators [34] described that the presence of certain chemicals in livestock feedlots could increase exposure. However, it was only in 1962, after the “Silent Spring” book, that the scientific community awakened to this issue. Rachel Carlson gave an important warning message of how the indiscriminate use of pesticides can leave “our fingerprint” in ecosystems, ultimately affecting ecosystems and human health. This wake-up call came to join the previous one by Bane-Jones, “The Advancement of Medical Research and Education through the Department of Health, Education and Welfare” [35] for the creation of an agency specifically committed to studying the effects of environmental exposure in human health. So, in 1966, the National Institute of Environmental Health Sciences (NIEHS) was created and four years later, together with the United States Environmental Protection Agency (US-EPA), they implemented regulations allowing the protection of the environment and human health. By that time, several studies provided clues on how chemicals caused endocrine disruption; for instance, evidence from minks showing reproductive complications and increased mortality rates, which were ascribed to fish from Great Lakes contaminated with PCBs [36,37]. By the same time, Gilbertson and Reynolds [38] observed low birth rates and high proportions of eggs failing to hatch in terns feeding from fish contaminated with hexachlorobenzene in the sea harbor. However, none could imagine that, around the 1970s, one of the most important medical breakthroughs would take place when the effects of diethylstilbestrol (DES) were discovered. DES is a synthetic estrogen, that had been used since 1930 for several indications, such as to reduce miscarriage or hormone therapy for menopausal women’s symptoms, or even estrogen deficiency. DES was later shown to be related to serious health consequences like rare cancers, reproductive malformations, and other conditions alerting the long-term effects of DES that were only noticeable after one or even more generations [39,40]. After that, EDCs’ research focused on the understanding of how certain chemicals accumulated and degraded in organisms [41]. The interest in EDCs was increasing, and the importance of their study was evidenced by the increasing number of scientific meetings dedicated to this topic. In 1991, one of the most important conferences, the Wingspread Conference, took place, where the concepts of “endocrine disruption” and “endocrine disruptors” were established [42]. From that date, the classical view of hormone function was revised, and it was apparent that there was more than one “key” for the same “lock”. The 1990s were prolific in demonstrating the harmful effects of EDCs. In 1992, Carlsen and collaborators [43] attracted the attention of the scientific community when they associated the falling sperm counts with the exposures to EDCs over the past five decades. In 1995, the US-EPA sponsored a workshop to gather interested parties with the aim to identify gaps regarding environmental influences on humans and wildlife. The workshop group felt the need to develop further investigations specifically regarding the health-effects of carcinogenesis, reproductive toxicity, neurotoxicity, immunotoxicity, risk assessment paradigm-hazard identification, dose-response assessment, exposure assessment, and risk characterization. In Europe, researchers assembled at the first European Workshop dedicated to the impact of EDCs, the Weybridge meeting [44], while similar events took place around the world [45]. In 1998, the US-EPA announced the “Endocrine Disruption Screening Program,” which was mandated under the Food Quality Protection Act and Safe Drinking Water Act to establish a framework for priority setting, screening, and testing of more than 85,000 chemicals in commerce. The idea behind the program was that prioritization would be based on existing information about chemical uses, production volume, structure-activity, and toxicity. Through the Registration, Evaluation, Authorization, and Restriction of Chemicals legislation, usually known as REACH, which became law in the European Union in 2007, chemicals, in which potential EDCs are included, require a portfolio of safety information before being released into the environment, rather than waiting for problems ahead, a fundamental aspect of the precautionary principle. With the growing scientific concerns and public debate about the potential adverse effects of EDCs exposure, the World Health Organization (WHO) published the global assessment of the state-of-the-science of EDCs with the purpose of providing an objective, global assessment of the current state-of-the-science relative to environmental endocrine disruption in humans, experimental studies, and wildlife species [46]. Eighteen years after Wingspread, the Endocrine Society of USA published a scientific statement showing how experimental and epidemiological data converge to implicate EDCs as one of the major concerns to human health [47]. In 2013, the WHO and the United Nations Environment Program released the most comprehensive study on EDCs [48], calling for more research to fully explore the association between EDCs and the risks to the health of human and animal life. In 2014, a multidisciplinary group of scientists, from basic to clinical settings, gathered in Parma for a workshop aiming to examine the relationship between EDC exposure and the increased incidence of metabolic disorders worldwide [49]. In 2015, the Endocrine Society of USA reviewed the latest science and updated the first statement on the recent knowledge and existing gaps [1]. There is a general and conclusive agreement about the EDCs’ hazards. The benefits of “chemical revolution” have shown us the “other side of the coin” where the subsequent environmental contamination is related to a wide range of behavioral, endocrine, and neurobiological complications. The research in this field has advanced rapidly; nevertheless, the strategy of health and politic authorities involved in the study of EDCs must be updated and revised in a continuum.
3. Endocrine Disruptors as Obesogens: Evidence from Basic and Clinical Studies
The endocrine system is known to play an essential role in energy balance, fat deposition, and fat distribution in the body because many endocrine organs and hormones work together to regulate metabolism and body weight. A significant advance in our understanding of how the environment can influence health, particularly by affecting developmental programming, came from the recognition that chemicals could disrupt the function of the endocrine system. However, the interaction of multiple factors across the lifespan and generations makes it difficult to determine what extent obesogenic EDCs contribute to the obesity pandemic in humans, despite its great importance and increasing interest from not only scientists, but also by health authorities, the general public and policy makers. Obesogens act as metabolic disrupters, promoting adipogenesis, increasing energy storage in fat tissue, and interfering with the hormonal control of appetite and satiety. There are nearly 50 known obesogens (Figure 3, Table 1), or chemicals supposed to exert obesogenic action (for review see [12,50]), and though some works are purely correlative [51], others have deepened understanding regarding the molecular mechanisms on how obesogens stimulate adipogenesis [52,53]. Obesogens interact with peroxisome proliferator-activated receptors (PPARs), particularly the peroxisome proliferator-activated receptor γ (PPARγ), the key regulator of lipid metabolism, adipocyte function and differentiation [54]. Studies have allowed us to understand the relationship between obesogens and weight gain better. One of the first studies demonstrated that tributyltin (TBT), a human-made chemical introduced into the environment mainly due to its widespread use as an antifouling agent in ships and other submerged structures, acts as a nanomolar-affinity ligand for PPARγ and its heterodimeric partner, 9-cis retinoic acid receptor (RXR) [7,20,55,56]. TBT is considered the obesogen model and belongs to the organotins which comprises of several families sharing structural similarities [57,58] (including e.g., butyltins, phenyltins and octyltins), being used in diverse applications including the aforementioned biocidal activities, in the production of polyurethane foams and silicones, and in the stabilization of plastics [59]. TBT is lipophilic so it may accumulate in our bodies [60,61,62,63,64,65]. It has been proposed that TBT commit cells to the adipocyte development by altering the chromatin landscape through activation of RXR, which decreases trimethylation in the lysine 27 of the histone 3 (H3K27me3), leading to increased expression of genes involved in adipogenesis [66]. Additionally, mesenchymal stem cells (MSCs) from mice exposed to TBT in the womb were biased towards the adipose lineage at the expense of the osteoblast lineage. This was confirmed under in vitro conditions where MSCs exposed to TBT were shunted toward the adipose fate in a PPARγ-dependent pathway. The activation of RXR seemed to be enough for adipogenic commitment, and the rexinoids acting through RXR activated it and altered the expression of the enhancer of zeste homolog 2, leading to H3K27me3, and thus promoting adipose commitment and programming subsequent differentiation [66]. Furthermore, in vitro studies have shown that TBT also increases basal glucose uptake in adipocytes exposed to 50 nM of TBT [67]. Similar to organotins, phthalates have also been classified as obesogens. Phthalates are esters of phthalic acid; high molecular weight phthalates are used mainly as plasticizers to increase the flexibility, transparency, and durability of plastic materials and can be found in several products of our daily life such as packaging, children’s toys, electronics, flooring and medical equipment. Low molecular weight phthalates are used as solvents in personal care products including cosmetics, air fresheners, food products, varnishes and coatings, pharmaceuticals and textiles. In vitro studies demonstrated that bis(2-ethylhexyl) phthalate (DEHP) and its metabolite mono-(2-ethylhexyl)-phthalate (MEHP) enhance differentiation of 3T3-L1 preadipocytes into adipocytes and murine MSCs in a dose-dependent manner through activation of the PPARγ pathway [68,69]. However, phthalates also stimulate adipogenesis by exerting antiandrogenic effects through PPARα. Another high production chemical widely used in polycarbonate plastics, can linings and thermal paper such as the one used in currency and in cash register receipts is bisphenol A (BPA), which had already revealed obesogenic action by predisposing murine preadipocyte differentiation, not only acting through nuclear estrogen receptors [70], but also via nongenomic mechanisms acting through cell membrane-associated estrogen receptors [71]. This is possible because BPA’s structure enables it to fit into the binding site of the estrogen receptor, which allows the compound to activate both nuclear and cell membrane-localized estrogen receptors [72]. This action directly modulates the insulin dependent phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT) pathway and regulates glucose uptake [73]. Other chemicals have been identified as potential obesogens by promoting adipogenic differentiation; for instance, parabens which are used as preservatives in personal care products, foods, pharmaceutical products, and paper products. The cell line 3T3-L1 exposed to 100 µM of ethyl- propyl-, and butylparaben demonstrated increased adipogenesis, showed lipid accumulation and an up-regulated activity of PPARγ [74], as well as an up-regulation in the transcription of PPARγ in C3H10T1/2 multipotent stem cells [75]. Moreover, it exhibited butyl- and benzylparaben-induced adipogenesis, though in a dose-dependent manner [76]. Parabens act as PPARγ agonists, but they can mediate their obesogenic effects through other pathways, such as the case of the glucocorticoid receptor [77] or through endocannabinoids as demonstrated by [76].
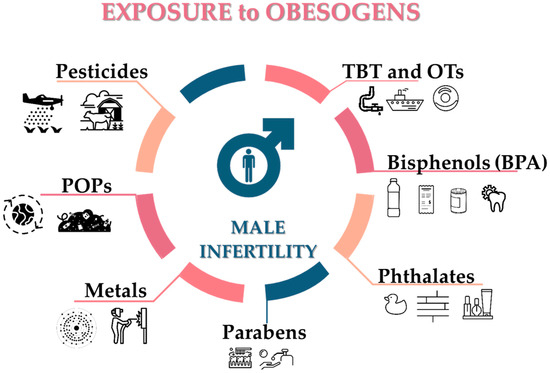
Figure 3.
Potential sources of obesogens known to affect the male reproductive system. TBT: tributyltin; OTs: organotin compounds; BPA: bisphenol-A; POPs: persistent organic pollutants.

Table 1.
Summary of proposed obesogens and possible sources of exposure.
Several classes of persistent organic pollutants (POPs) were already associated with obesity development. Polychlorinated biphenyls (PCBs), for example, are POPs that were used in electrical and industrial equipment. PCB101, PCB153, and PCB180 impair leptin signaling in differentiated 3T3-L1 adipocytes with associated increased lipid content and reduction of the AMP-activated protein kinase/acetyl-CoA carboxylase pathway, which is directly involved in the fatty acid oxidation [78]. Organochlorine compounds (OCs), also POPs, and widely used as pesticides, for example, also alter increased basal free fatty acid uptake by mature 3T3-L1 adipocytes. This increase was followed by augmentation of adipokine release, namely adiponectin and resistin [79]. Other environmental contaminants, such as the metals lead, cadmium and mercury and some phytoestrogens (genistein and daidzein), also exhibit obesogenic effects. One should note that the effects vary with the time of the exposure, since EDCs present non-monotonic dose–response curves, with results for higher doses often being opposite from what was observed for lower doses. Genistein, for example, and in contrast to its effects in adults [80], may produce obesogenic effects in animals exposed at critical periods of development. Interestingly, mice exposed to lower doses of phytoestrogen during gestation and lactation present a lower birth weight, but develop obesity, high leptin, and 17β-estradiol (E2) levels, and impaired glucose metabolism [81]. In vitro studies have enlightened the molecular mechanisms by which obesogens enhance adipogenesis at environmentally relevant doses. Importantly, some of these studies have been performed using cell lines, and thus further studies to evaluate in detail the obesity risk assessment for humans from exposure to obesogens alone or in a mixture are required. Therefore, epidemiological surveys alongside animal models have a crucial role in this context, so their use must be heavily weighed in the assessment paradigms. Many obesogens alter the function of gene expression and the hormone secretion of the white adipose tissue, the number and volume of adipocytes, or the body weight of the animal after exposure. Zuo and collaborators [82] orally administered TBT (0.5, 5 and 50 µg/Kg) once every three days, and after 45 days of treatment, the authors observed a significant increase of the weight in the animals treated with the intermediate levels (5 µg/Kg) without changing food consumption. This was concurrent with an altered hormonal profile of metabolic hormones, where the levels of leptin and resistin were significantly increased, while the release of adiponectin was decreased. Increased levels of leptin and resistin are linked with obesity [83,84], and this may result from mechanisms such as those previously described [66]. Nevertheless, it is possible that TBT may exert estrogenic activity as reported by Penza and collaborators [85]. In another study where pregnant rats were dosed daily by gavage with chlorpyrifos (2.5 mg/Kg) from gestational day seven until post-natal day 21 (PND21) (end of lactation), an abnormal weight gain in the offspring was observed. Similarly, Vitalone and collaborators [86] found that pregnant rats, after being exposed from gestational day 7 to PND21 to low doses of methylmercury (0.5 mg/Kg/day in drinking water) and PCB126 (100 ng/Kg/day in food), exhibited increased weight gain in the males of the offspring. This not only illustrates the obesogenicity of such contaminants, but also elicits a gender-specific development of obesity. The obesogenic action of BPA in male rats perinatally exposed has also been described, which exhibited overweight and significant fat accumulation.
All this crucial evidence demonstrates how obesogens act in a specific model, whether cellular or animal; however, it must be taken into account that in some instances the mechanisms may be merely indicatively associated with specific endpoints (weight, fat accumulation, hormones levels, and others). The association between EDCs and health outcomes in humans is nevertheless even more difficult to prove. Most of the unequivocal evidences available so far are derived from acute high dose exposures that occurred throughout history due to regrettable accidents [105,106]. The establishment of cause-and-effect links between exposure and diseases comes from examples such as industrial disasters with dioxins in Seveso, Italy and in Yusho, Japan, which resulted in severe births defects and neurocognitive impairments in children. Those were moments of acute exposure to high levels of EDCs, but demonstrates the adverse health outcomes [105,106]. However, the exposure to EDCs occurs at low doses in a chronic way, throughout our lives, from pre-conception until late age. Thus, such continuous exposure to a myriad of chemicals renders the establishment of a causation almost impossible, nevertheless several epidemiological evidences strongly suggest associations between exposure to EDCs and obesity. Carwille and Michels, for example, found an association between urinary BPA levels and obesity when analyzing data from the 2003–2006 National Health and Nutrition Examination Survey (NHANES) [107]. Similarly, Shankar and collaborators [108] found a positive association among urinary BPA levels, body mass index and waist circumference based on data from 2003–2008 NHANES. Indeed, these studies have suggested that obesogens are contributing to obesity epidemics, regardless of poor nutritional diet content or physical activity. In a longitudinal observational study, exposure to POPs was associated with rapid growth and with being overweight [109]. This is consistent with what was found by Mendez and collaborators [110], who explored the effects of prenatal exposure to organochlorine compounds and found an association with rapid growth in the first months of life and increased BMI later in the infancy. In a Swedish cohort of individuals aged > 70 years, it was demonstrated that serum levels of mono-isobutyl phthalate were associated with BMI, waist circumference and total fat mass [111]. Recently, Tang-Peronard and collaborators [112] observed that prenatal exposure to PCBs and p,p’-dichlorodiphenyldichloroethylene (DDE) might play a role for subsequent obesity development in infancy, with the most pronounced effects being associated with the exposure to low doses. The pharmacological route can also be pointed out regarding the exposure to obesogens, for example, through the use of antidiabetic drugs. Thiazolidinediones belong to the class of oral antidiabetic drugs that improve insulin sensitivity. These drugs exert their antidiabetic effects through a mechanism involving the activation of PPARγ and had been already associated with persistent weight gain [113,114]. Although the epidemiological evidence demonstrates an association between exposure and health outcomes, it is essential to bear in mind that most of these works lack mechanistic explanation due to the limitations of these studies. Most of them fail to consider the exposure levels relative to critical exposure windows. For some studies, there is no consideration regarding confounding factors, such as diet, stress, and others, as well as the latency of obesogens effects. These are vital points that must be taken into account in the design of future studies. Besides, it is also necessary to consider that some obesogens present short-half lives and that they may be converted into their metabolites, so researchers conducting studies on the effects of certain obesogens may not evaluate the real effects of the former. Furthermore, obesogens exert their action through several pathways [115], and most of the human studies were not designed for such an elaborated framework.
4. Obesogens as a Threat to Male Fertility
The obesogen hypothesis proposes that exposure to obesogens affects the biochemical pathways that control the whole metabolic homeostasis. Obesogens are present everywhere, and one of the first studies demonstrating that obesogens could affect the reproductive capacity through “modulation” of the endocrine system dates from the late 1960s [116]. Since then, several reports evidenced that the primary concern regarding obesogens is based on their capacity to induce endocrine disruption [1]. Taking into account that today the time period in which men are exposed to obesogens is large, this issue deserves special attention from all professionals of the reproductive area to understand how these toxics impact male fertility. Compelling evidences have linked the decline of male reproductive health with permanent exposure to obesogens in both developed and developing countries [117,118]. Obesogens alter the male reproductive function primarily by exerting adverse effects on the central nervous system. Below, a brief description of the effects of obesogens in the neuroendocrine control of male fertility, namely in the hypothalamus, is provided. Male reproductive organs may be a free spot where obesogens can be stored, albeit this is a subject of conjecture [119]. It is known that obesogens effectively cross the biological barriers [120], but the extent to which environmental chemicals pass the blood–testis barrier (BTB) remains mostly unknown. There is a correlation between obesogen exposure and decreased testosterone (T) levels, as they modulate the steroidogenic function of LCs (Table 2) and all related events with the synthesis of sex steroids [121]. Furthermore, studies also proposed that obesogens affect testicular metabolism, which is highly dependent on glucose [122,123,124]. Here we will explore how the metabolic cooperation between SCs and germ cells is affected by obesogens (Table 2) and possible consequences to spermatogenesis, and therefore male fertility. The effects of obesogens in the germ cells and sperm quality will also be examined.
Table 2.
Summary of the main obesogens and their proposed effects in both glucose and lipid metabolism, and their toxic effects in reproductive cells/tissues of mammals.
5. Reproductive Axis at the Interface with Environmental Obesogens
The central neuroendocrine system controls all processes in the body, reproduction included. All molecular events are initiated at the base of the brain, where the hypothalamus is located, which serves as the primary interface between the central nervous system and the testicles, in this case. The hypothalamic neural cells synthesize and release the gonadotropin-releasing hormone (GnRH) into the capillary portal system that vascularizes the anterior pituitary. The gonadotrophs in the pituitary response to this stimulus is the release of the corresponding hormones, luteinizing hormone (LH) and follicle-stimulating hormone (FSH) into the bloodstream. FSH and LH act as a functional link between the brain and testes by working on testicular cells to regulate the spermatogenic event [134]. LH binds to membrane receptors of LCs and stimulates T synthesis, influencing the development of peritubular cells, SCs and, consequently, germ cells [135]. LCs irreversibly convert T into E2 by aromatase P450 [136]. On the other hand, FSH binds to membrane receptors on SCs, stimulating the production of E2, activin and inhibin B. E2 inhibits T production by LCs, while activin and inhibin B produce a positive/negative feedback on the pituitary, respectively. There is evidence that endocrine disruptors affect the neuroendocrine systems, and most of the studies have focused on the hypothalamus–pituitary–testicular (HPT) axis, termed the reproductive axis. Endocrine disruptors can exert diverse actions over the target cells according to their chemical structure and activities. In vitro evidences demonstrate that GnRH GT1–7 cell lines exposed to low doses of PCB mixtures, Aroclor 1221 or Aroclor 1254, increased the expression of the GnRH gene [137]. In this case, the endocrine disruption occurs at the level of the hypothalamus [137]. Similar studies using GT1–7 cell lines exposed to small doses of organochlorine pesticides demonstrated an increased expression of GnRH, whereas when exposed to high doses, the opposite effect was observed [138]. Although these works showed a positive response of GnRH cell lines to environmental obesogens, other studies demonstrated different outcomes. In fact, explanted hypothalamus from male rats exposed in utero to dioxins displayed an increased content of GnRH peptide, which was, however, accompanied by an impaired release [139]. Obesogens can favor or reduce the release of neuroendocrine hormones, but this will depend on the type of obesogens, the time of exposure and perhaps the levels to which disruption in the reproductive axis occurs. Different environmental compounds may target the HPT axis at various sites with different intensities, disrupting its regulation. BPA is an example of an obesogen acting through a variety of physiological receptors. BPA molecules are linked by ester bonds that are subject to hydrolysis when exposed to high temperatures or too acidic or basic substances [140]. BPA was thought to exhibit a weak estrogenic activity, based on the relative binding affinity of BPA for the nuclear estrogen receptors (ERs) α and β, which were estimated to be over 1000–10,000-fold lower when compared to E2 [141]. However, BPA binds to both estrogen receptors α and β [142,143] and plasma membrane-bound ERs [144], as well as to the G-protein-coupled receptor 30, showing that beyond the genomic route, BPA also act by non-genomic pathways. Rodents exposed to BPA during the perinatal and postnatal periods exhibited an upregulation of kisspeptin (KiSS-1) expression levels, GnRH and mRNA of FSH [145]. KiSS-1 acts as a guardian to the onset of puberty and for the regulation of gene expression in the HPT axis. The upregulation of KiSS-1 expression stimulated the synthesis and release of GnRH and gonadotropins in the hypothalamus and pituitary, respectively [145]. However, it was later shown that the hypothalamus is not involved in the disruption caused on the HPT axis by an adult exposure to BPA [146]. In this study, the relative transcript levels of the GnRH receptor, the gene which encodes the receptor for type 1 GnRH, was increased by five-fold in the pituitary of BPA-exposed groups [146]. Since GnRH is responsible for the release of FSH and LH from the anterior pituitary, as expected, both relative levels of LHb mRNA and FSHb mRNA, the corresponding genes, were also increased. Although the increased GnRH receptor expression may correspond to augmented T levels, this increase in androgen and E2 receptors suggests the stimulation of negative feedback mechanisms [147]. The same compound was also related to an imbalance in the ratio T/E2 due to an enhanced aromatase activity that increases E2 levels [148]. As a result, the release of gonadotropins at the hypothalamic-pituitary axis is inhibited, compromising all downstream events of the HPT axis, namely spermatogenesis. TBT is also involved in the dysregulation of the HPT axis by inducing leptin resistance and increasing plasma insulin levels, accounting for reduced gonadotropin secretion at the hypothalamic-pituitary level [82]. Phthalates may promote insulin resistance, considering reduced gonadotropin secretion at the hypothalamic-pituitary level [149]. Insulin may directly act through its receptor located in the hypothalamus and pituitary [150], being pivotal for a normal function of the HPT axis [151]. From the perspective of potential reproductive onset, puberty can be defined as activation of the HPT axis, leading to steroidogenesis, gametogenesis and the development of secondary sexual characteristics. Thus, many aspects resulting from the functioning of this complex system are also susceptible to the exposure of environmental contaminants and modulation of the HPT axis.
6. Leydig Cells Are a Sensitive Target of Obesogens
The synthesis of sex steroid hormones occurs in the Leydig cells (LCs) which are extremely sensitive to the toxic effects of environmental contaminants. The deleterious effects of EDCs often conduct the impairment of hormone synthesis since they are associated with the inhibition of the activities of enzymes involved in steroidogenesis. Lipids are essential for this process and cholesterol is one of the primary precursors, so it would be expected that any dysregulation in the homeostasis of these components compromises the synthesis of steroid hormones. Indeed, the pesticide 2,4-dichlorophenoxyacetic acid (2,4-D) decreases the expression of 3-hydroxy-3-methylglutaryl coenzyme A synthase (HMG-CoA synthase) and reductase (HMG-CoA reductase) in LCs of mice [152]. These enzymes are pivotal for cholesterol synthesis, where HMG-CoA synthase is responsible for the condensation of acetyl-CoA with acetoacetyl-CoA to form HMG-CoA, which is reduced to mevalonate for the synthesis of cholesterol. This is one of the most important steps of the cholesterol biosynthesis, where the enzyme responsible for this committing conversion is HMG-CoA reductase. The impairment of cholesterol synthesis after exposure to 2,4-D disturbs testosterone synthesis. In the steroidogenesis, cholesterol is then carried from the outer mitochondrial membrane by steroid acute regulatory protein (StAR), where it is converted to pregnenolone via P450 side chain cleavage enzyme (P450scc). Then, this intermediate proceeds to the smooth endoplasmic reticulum where 3β-hydroxysteroid dehydrogenase (3β-HSD) turns it into progesterone. Progesterone is converted by P450c17 into 17-hydroxyprogesterone and into androstenedione, where the enzyme 17β-hydroxysteroid dehydrogenase converts it into T. It has been demonstrated that exposure to plasticizers such as di(n-butyl)phthalate decreased the expression of cholesterol transport genes such as StAR, the high-density lipoprotein receptor, also known as SRB1 [153]. Furthermore, the expression of genes involved in T biosynthesis, namely P450scc, 3β-HSD, and P450c17, was downregulated, which probably contributes to T deficiency [154]. It seems that the plasticizer DEHP exerts anti-androgenic effects directly onto LCs, inhibiting T synthesis probably through dysfunction of CYP17 [155]. However, the impact of its metabolite, MEHP, is much more pronounced since it inhibited the expression of all steroidogenic enzymes, as well as all T precursors, in both the Δ4 and Δ5 steroidogenic pathways [155]. The effects of both phthalates appeared to be specific for steroidogenesis since they did not alter the expression of the insulin-like 3 gene, a specific marker of LCs. In vivo models have also provided evidence of the cytotoxic effects of similar obesogens in testicular function as observed after daily exposure to 200 mg/kg/day of BPA for six weeks. Such exposure not only revealed the same results, but also disclosed a reduced number of LCs [156]. Similarly, Jin and collaborators [157] reported that after oral administration of 2 μg/kg/day of BPA during 14 days, male rats showed T deficiency due to decreased expression of steroidogenic enzymes. An in utero exposure to linuron, a urea-based herbicide, was revealed to affect fetal testis gene expression and to reduce T production in adult male rat offspring [158]. Curiously, unlike phthalate esters that reduce T production, linuron did not affect the expression of steroidogenic genes [158]. Linuron seems to bind to the androgen receptor but, interestingly, the set of malformations displayed after an in utero exposure to this compound [159] differs from other androgen receptor antagonists [160]. Thus, it is suggested that decreased T production induced by linorun may be due to an alternative mechanism in the absence of cytotoxicity. Linuron is also causally related to changes in sex differentiation of the male reproductive tract [161], which allows hypothesizing that the inhibition of T synthesis along with androgen receptor antagonism may contribute to impact the androgen signaling pathway. Alterations in sex differentiation were also found after exposure to vinclozolin, a fungicide with anti-androgenic properties as well [162]. Indeed, the female-like anogenital distance at birth, retained nipples, undescended testes, and small sex accessory glands, were associated with an in utero vinclozolin exposure in male rat offspring [162]. The anti-androgen effects of vinclozolin were already related to increased levels of LH and T [163], and also inhibited DHT-induced transcriptional activation, through blockage of the androgen receptor-binding to androgen response elements in DNA [164].
7. Mechanisms Mediating Obesogen-Related Sertoli Cell Dysfunction: A Metabolic Standpoint
Typically known as “nurse cells”, Sertoli cells (SCs) are extremely important in male fertility, ensuring all physical and nutritional support of the germ cell line. Spermatogenesis is under the strict control of hormonal and endogenous factors [165,166], so SCs become an easy target of obesogenic EDCs mimicking endogenous hormones [167]. EDCs (obesogens included), not only compromise all structure of SCs, but also induce various cellular and molecular insults [168,169]. Additionally, the interactions between adjacent SCs are disrupted, provoking the premature exfoliation of germ cells [170]. The impact of EDCs on the integrity of cytoskeleton disruption of SCs and BTB has been extensively explored and well documented by Cheng and his collaborators [22,120,169,171,172,173,174,175,176]. Here, we will focus on the significant findings concerning the effects of EDCs, particularly obesogens, in the modulation of SC’s metabolism, since it is vital for the control of spermatogenesis [165,177,178] and because several EDCs have already been reported to alter factors that control the metabolism of these cells (Table 2). During the 1980s, researchers focused in demonstrating how environmental contaminants modulate the metabolism of SCs. Batarseh and collaborators exposed SCs under ex vivo conditions to 10 µM, 50 µM, and 100 µM lead acetate and observed a significant production of lactate in all concentrations tested [127]. SCs increased lactate production in a dose- and time-dependent manner after being exposed to lead acetate [127]. Similarly, a dose-dependent increase in lactate secretion was also reported in primary cultures of SCs exposed to other contaminants with recognized obesogenic activity, such as phthalate esters [124] and PCBs [128]. Increased lactate levels induced by these obesogens are often viewed as evidence of enhanced lactic fermentation. Those reports demonstrated that the quantification of the lactate released from cultured SCs can be used as a measure of the increased glycolytic pathway. Although these works were based on a basic approach, they provided unequivocal proof that EDCs may reprogram the metabolism of SCs with possible adverse consequences on male reproduction. However, the information that existed to date did not allow mechanistic perception of the toxicants’ performance. A few years ago, Premendu P. Mathur and his team shed light on how some obesogens compromise glucose metabolism. Their work showed that BPA disrupts the glycolytic metabolism of SCs by blocking the glucose movement through binding to the glucose transporters in the pore region [123]. BPA also disrupts testicular insulin signaling, by decreasing the expression of insulin receptor substrate 1, insulin receptor substrate 2, and PI3K [123]. PI3K is involved in the control of glucose uptake, namely in the translocation of glucose transporter isoform 1 (GLUT1) to the cell surface [179], so this leads to significant alterations on the glycolytic pathway. Under normal conditions, glucose is taken up by SCs through GLUT1, glucose transporter isoform 2, or glucose transporter isoform 3 (GLUT3), and readily converted to pyruvate through the glycolytic pathway, where phosphofructokinase 1 (PFK1) is the first rate-limiting step [180]. The resulting pyruvate is mostly converted to lactate by the action of lactate dehydrogenase (LDH), with a part being also converted to alanine. Previous reports showed that plasmatic concentrations detected in men exposed to 2,4-D are enough to decrease not only the intracellular levels of glucose, but also the transcript levels of glucose transporter 3 (GLUT3), PFK1 and LDH [122]. Similarly, lactate, alanine, and monocarboxylate transporter isoform 4 (MCT4), the carrier through which lactate is exported to the luminal fluid to be used as a metabolic fuel by the developing germ cells, were decreased as well. Besides, to compromise the intracellular levels of lactate, the exposure to these doses of 2,4-D also reduced the lactate/alanine ratio, which is an indicator of a reduced cytosolic state, and thus hampers the glycolytic metabolism of rat SCs [122,181]. The decrease on PI3K also downregulates the stimulation of lactate production and LDH activity, illustrating the impact of the PI3K/AKT signaling pathway in SCs metabolism [179]. Besides, these changes are also closely related to the apoptotic process [182], suggesting that these metabolic alterations may lead to abnormal apoptosis in SCs and thus induce problems on spermatogenesis and male fertility. It is known that model obesogen TBT alters glucose consumption mainly through disruption of the translocation mechanism mediated by GLUT1 [183]. Cardoso et al. [132] showed that SCs exposed to nanomolar concentrations of TBT exhibit alterations in the glycolytic profile. Ex vivo cultured rat SCs were exposed to 10 nM of TBT for 6 h and exhibited a significant decrease in glucose consumption even though the expression of GLUTs was unaltered. Moreover, these cells also decreased the consumption of pyruvate. Indeed, TBT at nanomolar concentrations reduced the consumption of the two significant substrates used to ensure lactate production. Hence, the striking decrease in the consumption of these two substrates was reflected in a substantial reduction in the production of lactate in SCs exposed to 10 nM of TBT. However, the most remarkable effects were observed when SCs were exposed to subnanomolar concentrations TBT (10−2 nM). In this group, SCs exhibited a different metabolic response since the decreased glucose consumption was concurrent with a down-regulation of the expression of GLUT1, and the production of lactate was lower than SCs exposed to 10 nM TBT groups [132]. TBT inhibits glucose uptake by primarily affecting the translocation of GLUT1 to the plasma membrane. The reduced levels of GLUT1 may be associated with the decreased activity of 5′ adenosine monophosphate-activated protein kinase (AMPK), which is involved in the translocation of GLUT1 for membrane surface, thus enhancing glucose uptake [184]. Pioglitazone is an antidiabetic drug, and similar to the TBT, is a potent PPARγ agonist [54]. Pioglitazone exerts positive effects in male reproductive health, and it has been demonstrated that pioglitazone modulates the glycolytic metabolism of human SCs [104]. Human SCs exposed to 1 and 10 µM of pioglitazone did not alter glucose uptake, but when exposed to pharmacological concentrations (100 µM) significantly increased glucose uptake via GLUTs without changing their protein levels [104]. SCs exposed to pioglitazone 100 µM increased the efficiency of the glycolytic flux and lactate production since the lactate production doubled and was positively correlated with key intervenients of glycolysis, such as MCT4 protein levels [104].
8. How Can Germ Cells Be Affected by Obesogens?
Compelling evidence shows that environmental contaminants induce several dysfunctions associated with the testicular dysgenesis syndrome, including seminiferous tubules atrophy and germ cell degeneration [185]. Furthermore, lipids are essential components of the membranes of germ cells. Deficient lipid incorporation into these cells leads to a defective germ cell structure and contributes to the impairment of sperm parameters, and consequently to alterations of sperm functionality. The oral administration of TBT has also been associated with induced apoptosis in testicular germ cells in prepubertal mice [119]. Those authors did not describe the molecular mechanisms by which germ cell apoptosis was induced, but according to recent evidence, it seems that environmental contaminants activate both intrinsic and extrinsic pathways of apoptosis. Mitra and collaborators [133] observed a caspase-3 activation and increased levels of pathways of phosphorylated c-Jun N-terminal kinase (JNK) and mitogen-activated protein kinase p-38 (p38-MAPK) in SC–germ cell co-cultures after being exposed to 600 nM of TBT. JNK activation serves as a pro-apoptotic signal and regulates the mitochondrial apoptotic pathway through a balancing act between the activation of pro-apoptotic members and inhibition of anti-apoptotic members of the Bcl2-related protein family [186]. On the other hand, the p-38 caspase-independent pathway seems to be also activated by TBT and thus plays a role in the germ cells’ death [187]. Other studies have also associated TBT exposure to the induction of apoptosis in testicular germ cells, since more apoptotic cells were found in the seminiferous tubules of 21-day mice after oral administration of 25, 50 or 100 mg of TBT/kg/day, for three days [188]. Germ cells are also targeted by DES since apoptosis of spermatogenic cells was observed in prenatal and neonatal mice exposed to this compound from gestational day 12 to PND20 [189]. In healthy individuals, germ cells present high levels of polyunsaturated fatty acids (PUFAs), which are crucial to their membrane fluidity and flexibility, and thus for fertilization. Since these cells are, however, unable to synthesize PUFAs, they take these fatty acids from SCs and incorporate them into phospholipids, via lysophosphatidic acid acyltransferase 3 [190]. Several studies already suggested that the current dietary habits, particularly the excessive consumption of high-energy diets, with high content in saturated fats and trans fatty acids, decreases the activity of Δ5 and Δ6 desaturases [191]. It was proposed that the limitation of PUFAs incorporation in membranes of germ cells adversely affects testicular lipid metabolism and thus contributes to the impaired production of sperm [192].
9. Epigenetic Changes in Germ Cells
The permanent exposure to environmental chemicals may induce epigenetic modifications. These modifications do not alter the DNA sequence and mostly occur through DNA methylation, histone, and long non-coding RNAs modifications, remodeling of nucleosomes and higher order chromatin reorganization, thus defining gene expression [193]. Germ cells are the “vehicles” in the transmission of genetic information to the next generation. Epigenetics also ensure that daughter cells have the same phenotype as the parental cell, controlling gene function from one generation to the next [193]. Indeed, the role of epigenetics varies between somatic and germ cells, as the latter undergo meiosis, which is of particular importance to maintain genomic integrity [194]. The epigenetic programming of the germline begins during the migration of primordial germ cells in the embryo [195]. Upon initiation of determination of gonadal sex, the primordial germ cells remethylate DNA and develop the germ cell lineage in a gender-specific manner [195]. Between the 12th and 15th days of embryonic development [196], the fetal testes start expressing steroid receptors, thus becoming more sensitive to the adverse effects of EDCs [197]. So, EDCs acting up during gonadal sex determination could reprogram the germ line through DNA methylation [5]. Because of the elapsed time between exposure and the detectable impacts in offspring (mainly at their adulthood), animal models have been beneficial to study those mechanisms. Data from animal studies supported that such exposure may affect the male reproductive function of offspring, determining the success of fertility. It has been shown that exposure to vinclozolin or methoxychlor during the remethylation programming of the germ line in rats is associated with induced aberrant sexual differentiation, gonad formation, and reproductive functions of the following generation [198,199]. Similar findings were found until the fourth generation, with an increased spermatogenic cell apoptosis and reduced number and motility of sperm being observed [196]. This transmission of the exposure-induced phenotype in a transgenerational manner suggests an epigenetic alteration in the DNA methylation pattern of the male germ line, namely an epigenetic mechanism involving reprogramming of the germ line. Possibly through abnormal regulation of DNA (cytosine-5)-methyltransferase 3 alpha and DNA (cytosine-5)-methyltransferase 3-like, methyltransferases are required for methylation of most imprinted loci in germ cells [200]. It seems that these changes are implicated in the transgenerational effects aforementioned. Thus, male fertility problems were suggested to be caused by a change in genome-wide methylation and, consequently, in gene expression. Dolinoy and collaborators also confirmed the adverse effects in males that were not directly exposed to BPA. The authors showed that early developmental exposure to BPA can change offspring phenotype by stably altering the epigenome, an effect that can be counteracted by maternal dietary supplements [201]. Interestingly, DNA methylation can be counteracted by maternal dietary supplementation with a source of methyl groups such as the phytoestrogen genistein, which has been considered an endocrine disruptor [201]. Regarding phthalates, namely DEHP, it was reported that when given to outbred mice between days 7–14 of embryonic development, it reduced both counts and motility of sperm in male offspring beyond the F3 generation [202]. Accordingly, it was suggested that DNA methylation was the potential epigenetic mechanism underlying these alterations since 16 differentially methylated and expressed genes were identified in newborn pups. Thus, this group of genes is suggested to hold clues on how DEHP acts transgenerationally. Studies in humans were already performed in order to understand the environmental influence in epigenetics. DES, which has been associated decades ago to the presence of hypospadias in human males exposed in utero [203] or whose mothers had been exposed in utero [204], was also reported among unexposed grandsons of women who had been exposed in utero [205]. The exposure occurred during the development of the reproductive system in primordial germ cells of the fetuses, being subsequently transmitted across generations to affected sons and grandsons [205]; although the underlying mechanism is not entirely explained, it may rely on epigenetic changes in the androgen receptor. Modifications in the histones also play a crucial role in epigenetics, since the placement of histone modifications in sperm contributes to the establishment of embryo constitutive heterochromatin [206]. Histone modifications have been shown to be pivotal in changing chromatin structure and, therefore, DNA accessibility [207], being able to regulate gene expression, chromatin remodeling, cell survival and cell death [207]. Histone modifications comprise methylation, acetylation, phosphorylation, ubiquitination, ribosylation, and sumoylation, which can be easily induced and removed by a wide range of enzymes [207]. The functional effects of these modifications depend both on the modified amino acid and the specific covalently attached group [208]. Acetylation is involved in direct histone assembly and helps regulate the unfolding and activation of gene expression, whereas methylation restricts the access to the transcription machinery to DNA [208]. It seems that valproic acid, a commonly prescribed anticonvulsant and mood stabilizer [209], and methoxyacetic acid, a solvent usually used in paints, dyes and fuel additives, can inhibit histone deacetylases exerting broad effects on DNA [210]. Additionally, paternal dietary folate seems also to improve offspring’s susceptibility to congenital disabilities through changes in histone methylation [211]. Interestingly, it has been observed that fathers fed with a high-fat diet evidenced changes in sperm histone composition, although DNA methylation of several imprinted loci appears to remain static [212]. These findings illustrate that men exposed to these contaminants are more predisposed to experience histone modifications. Long non-coding RNAs (lncRNAs) modifications are another epigenetic-related mechanism that can be pointed out as an effect of exposure to environmental contaminants. The role of lncRNAs in epigenetic processes has been highlighted, and several data have suggested that lncRNAs are involved in the cellular stress response [213]. LncRNAs are defined as transcribed RNA molecules greater than 200 nucleotides in length with little or no protein-coding competence. It is known that lncRNAs regulate gene expression, although the mechanisms need to be clarified in-depth [214,215]. It has been described how lncRNAs control gene regulation at every level including transcriptional gene silencing via DNA methylation and regulation of the chromatin structure [215,216]. It is likely that lncRNAs drive significant exposure–disease associations and may also function as biomarkers of environmental exposure. Studies have reported an increased transcription of telomeric repeat-containing RNA and satellite sequences (Sat III) in humans, following physiological stress induced by heat or ethanol [217,218]. This may indicate that lncRNAs have a transcriptional regulatory role, as suggested for other similar RNAs such as telomeric repeat-containing RNA and Sat III [217,218]. Likewise, satellite DNA transcription also appears to be regulated by environmental cues such as temperature [219]. Similarly to lncRNAs, small non-coding RNAs (sncRNAs) seem to be involved in mechanisms of histone modifications, specifically piwi-interacting RNAs (piRNAs). In fact, it has been observed that lncRNAs and piRNAs in the landscape of epigenetic modifications of chromatin state and histone codes are the productions of piRNAs by the lncRNAs that induce up-regulation of the tumor necrosis factor related to apoptosis-inducing ligand protein via H3K4/H3K27 methylation/demethylation [220]. It seems that these RNAs may be important mediators in the response to a broader spectrum of contaminants, and reproductive biologists are searching for further understanding of their biological functions. This is pivotal to identify the functional role of these RNAs in the toxic response of mammalian cells. There is consistent data which links epigenetic changes both in experimental and epidemiological studies to environmental contamination and subsequent effects in sperm quality, but there is a lot of work to do in this topic. Because these epigenetic changes are small, potentially cumulative, and they may develop over time, it may be difficult to establish real cause–effect relationships among environmental contaminants, epigenetic changes, and diseases etiology. However, knowing the relevance of the transgenerational epigenetic inheritance to many human disorders will undoubtedly become an exciting area, for which research on germ cells is particularly important.
10. Obesogens Compromise Sperm Quality and Sperm Function-Related Events
Spermatogenesis is extremely sensitive to the effects of EDCs. Any damage in this event will compromise germ cell differentiation into fully developed sperm. In this section, the adverse effects of EDCs on sperm and the putative impact ECDs on the molecular events associated with their function since their release from seminiferous tubules will be addressed. EDCs interfere with several aspects of sperm function and induce effects that, although are not so evident as the conventional parameters used by clinicians, should be considered as specific biomarkers of exposure. These biomarkers include, for example, oxidative stress induction, DNA damage, fluctuations in the intracellular calcium concentration [Ca2+]i and mitochondrial dysfunction. There are several studies on this topic, however with inconsistent results (for more details see Table 3 and references therein). The lack of consistency between the results in the literature may be explained by the fact that the majority of the studies do not consider relevant biochemical and molecular aspects occurring in sperm or in the surrounding environment. However, we must also recognize that in these studies only the effect of a single EDC is generally evaluated. These single exposure studies are still the norm because it is still difficult to decipher the real impact of a particular contaminant if it is not tested alone. Such individual exposure assessment contrasts with the real word scenario in which we are permanently exposed to a myriad of EDCs, and thus such studies will misrepresent the real scenario. The accumulation of toxic substances in the human body has some clinical implications, especially in male fertility. Annually nearly 400,000 tonnes of pesticides are applied in the Europe [221], while in the United States the application of pesticides surpasses 450,000 tonnes, with the vast majority used in the agricultural sector [222]. Some studies have measured the levels of environmental contaminants in male reproductive fluids, with most of them dealing with metals (see systematic review by Sun and collaborators [223]). Other EDCs including pesticides [224], BPA [225], phtalates [226], perfluorooctane sulfonate (PFOS) and perfluorooctanoic acid (PFOA) [227] have also been detected in seminal male reproductive fluids including seminal plasma. After leaving seminiferous tubules, sperm undergoes several biochemical and morphological changes as they pass through the epididymis to become functionally competent, an intricate process that only ends after a series of several molecular events occurring within the female reproductive tract, called “sperm capacitation”. Sperm is transported against an increased hydrostatic pressure gradient from the seminiferous tubules to the cauda of epididymis [228]. Sex steroids mainly control this process, so it is expected that obesogens mimicking these hormones may disrupt it. Klinefelter and collaborators [229] reported alterations in sperm transit induced by environmental contaminants. These authors found that chloroethylmethanesulphonate, through the proximal segment of the epididymis, reduced T levels, accelerating sperm transit [229], eliciting androgen-dependent mechanisms. Evidence from castrated animals showed that T plays a crucial role in the sperm transport since a significant decrease of epididymal sperm storage [228,230] and an increased intraluminal pressure and contractility of the epididymis was found in these animals [231]. On the other hand, analogs of estrogens also modulate sperm transit, since Wisniewski and collaborators found that BPA administration decreased the sperm transit time through the caput and cauda of the epididymis. Yan and collaborators [232] also showed that TBT alters the secretory and absorptive activities of epididymal epithelium, so it may be expected that sperm transport is affected by organotins. As they pass through the epididymal duct, sperm motility gradually increases, as well as its fertilizing ability. This is attained due to rigorous crosstalk between sperm and epididymal epithelium resulting in several changes in the sperm plasma membrane [233]. The proteins secreted by the epithelium reaches the sperm membrane through apical blebs or small vesicles called epididymosomes, becoming a coating protein or an integral membrane protein. Alterations in the activity of epididymal proteins can also provide information concerning the maturation of sperm. This process is targeted by obesogens [234] as observed by Yan and collaborators [232], who found an abnormal morphology in sperm from TBT-treated rats which might have resulted in part from the decreased activity of epididymal acid phosphatase. TBT inhibits the activity of acid phosphatase [232], a lysosomal enzyme in the epididymal cells, involved in the lysosomal digestion and the removal of old and dead cells by phagocytosis [235]. Furthermore, acid phosphatase is also recruited for phosphorylation/dephosphorylation events associated with sperm maturation [236]. The acquisition of motility is one of the significant changes mediated by protein phosphorylation. Sperm motility is regulated by the phosphorylation profile in several parts of the flagellum and axoneme. Protein kinases and phosphatases coordinate this process, so obesogens that compromise the activity of these proteins affect motility. For instance, genistein is considered a natural obesogen and has been associated with male reproductive problems [237]. This isoflavonoid derived from soy is a competitive inhibitor of protein Tyrosine (Tyr) kinase, so it is expected that the exposure to this obesogen may hamper sperm maturation-related events, though the mechanism of action for genistein needs to be completely elucidated. Indeed, rat sperm incubated with genistein exhibited a decrease in both motility and morphology, with the most remarkable effects being observed for doses of genistein higher than 50 μg/mL and long-time exposure, illustrating the adverse effects of genistein on male fertility [237]. For lower doses (<50 μg/mL), no damages in the epididymal tissue and no significant alterations were observed in sperm parameters [238]. It is possible that high concentrations of genistein inhibit Tyr phosphorylation signaling. Recently, an association between the consumption of fruits and vegetables rich in pesticide residues and decreased sperm parameters was found [239]. The authors found that sperm count and sperm with normal morphology were reduced by 49% and 32%, respectively, when compared with men who had not been exposed to the same substances [239]. Another study also demonstrated that the conventional sperm parameters, specifically semen volume, concentration, morphology and viability of sperm, were altered in men chronically exposed to lead [240]. The majority of studies have focused on the adverse effects of EDCs on sperm quality (see Table 3), since this is the ultimate end-point for reproductive professionals and the one that defines male fertility. A meta-analysis by Wang and collaborators [241] has shown how classic EDCs compromise sperm quality, and although some of them were already banned from circulation, humans continue to be exposed and thus they continue to exert adverse effects. For instance, BPA has been consistently associated with changes in sperm parameters, especially in motility [242] and morphology [243]. Sperm from mice decreased in motility and all kinematic motion parameters after being exposed to 100 μM of BPA for six hours. The decreased sperm motility may be related to mitochondrial dysfunction since a concomitant decrease of ATP production was observed [244]. The majority of ATP required for sperm motility comes from mitochondrial respiration [245], therefore any condition/exposure leading to dysfunction of mitochondrial respiration will certainly compromise sperm motility [244]. Mitochondria are essential organelles for male fertility (see review [246]) and are localized in the sperm midpiece, forming a helicoidal sheath with the number of mitochondria and midpiece length varying with species [247]. Mitochondria in mature sperm have a condensed morphology with a compacted matrix, allowing for greater efficiency of energy production, but there are some species differences [248]. The relevance of mitochondria in sperm function was initially studied strictly concerning ATP production for motility. For instance, the activity of several complexes in the electron transfer chain of the inner mitochondrial membrane, as well as mitochondrial membrane potential (MMP), is correlated with sperm motility [249,250,251,252]. However, sperm mitochondria have unique features different than those of somatic cells, allowing them to efficiently drive important functions beyond sperm motility such as hyperactivation, capacitation, acrosome reaction, and fertilization [253,254]. Mitochondrial (dys)function has clinical relevance since it has been associated with male infertility [255,256]. Even changes in the mitochondrial genome have also been associated with male infertility due to compromised sperm quality, motility, and function [246]. Mitochondria are particularly sensitive to environmental exposures. It has been conjectured that the interaction between mitochondria and environmental exposure may be at the basis of several age-related diseases, such as Parkinson’s and Alzheimer’s diseases [257]. However, a detailed characterization of the “mitochondrial exposome” that allows us to understand the specific contribution of environmental chemicals in sperm mitochondrial dysfunction is necessary. Skibinska and collaborators exposed human sperm to different concentrations (10−8 and 10−6 M) of BPA and found alterations in the MMP [258]. In addition, these authors also found that human sperm incubated with E2, genistein, and BPA simultaneously decreased MMP and increased the production of superoxide anion. PCBs also induce sperm dysfunction since, under in vitro exposure to the mixture Aroclor 1254, a decreased motility, viability and acrosome reaction of rat sperm at all concentrations tested (10−7 M, 10−8 M, 10−9 M) was observed [259]. It was also found that Aroclor 1254 decreased the synthesis of ATP in a concentration-dependent manner, which elicits a decrease in the MMP. Additionally, in vitro exposure to Aroclor 1254 induced oxidative stress (OS), increased lipid peroxidation and induced mitochondrial cytochrome c release. The translocation of cytochrome c from mitochondria to the cytosol is the primary event in the mitochondrial pathway leading to the formation of apoptosomes and activation of the caspase cascade, which was confirmed by caspase-3 activation [259]. Although this has been observed in rat sperm, similar results were also reported in human sperm exposed to this pesticide [260]. Evidence from in vivo models has also corroborated in vitro findings (Table 3). Pant and collaborators [261] measured the levels of DEHP in the seminal fluid of men from rural (mean: 0.13–0.77 μg/mL) and urban areas and found negative associations with both sperm motility and mitochondrial integrity; however, the levels of DEHP were positively associated with sperm reactive oxygen species (ROS) production, lipid peroxidation, and nuclear DNA damages [261]. Obesogens have also been linked to developmental toxicity via DNA damage [262] and OS [263] (see Table 3). This is one of the most well-documented effects of environmental chemicals in male reproductive health [264,265]. OS may be part of the molecular basis of male infertility associated with environmental toxicants exposure. Chitra and collaborators [266] showed that administration of increasing doses of BPA is associated with a decreased activity of enzymes involved in the antioxidant system. Specifically, BPA decreased the activities of superoxide dismutase, catalase, glutathione reductase and glutathione peroxidase in the epididymal sperm of rats. This is of great relevance since an inefficient antioxidant system favors the production of ROS, as observed by the increased levels of hydrogen peroxide and consequent lipid peroxidation [266]. Recent data suggests that the anti-androgenic effects of DEHP may cause ROS production, lipid peroxidation and apoptosis of spermatocytes [267]. The production of these ROS seems to be related to Ca2+ mediated activation of the nicotinamide adenine dinucleotide phosphate complex, which may correlate with DEHP-induced Ca2+ entry [267]. In addition to the previous effects aforementioned, it should be further noted that obesogens also compromise the fertilizing ability of sperm. Even if the spermatozoa were not exposed to the effects of obesogens (both direct or indirect), they could always find a “hostile environment” in the female reproductive tract, since significant concentrations of PCBs in secretions and follicular fluid have been reported [268,269]. Thus, it is plausible that obesogens may, to some extent, compromise molecular mechanisms related to fertilization. For instance, it was reported that TBT decreased the activity of acrosin [232]. Acrosin is part of the mammalian sperm acrosome and is primarily found in an inactive form called proacrosin, which is only converted into the active form upon acrosome reaction. Acrosin activity is positively correlated with sperm morphology (regarding percentages of normal morphology) and negatively correlated with percentages of sperm with acrosome deficiency [270,271,272], so sperm with impaired acrosin activity will not be able to penetrate zona pellucida and fuse with the oocyte, thus contributing to male subfertility/infertility. Curiously, it has been shown that the administration of BPA to sperm leads to a dose-dependent induction of acrosome reaction, suggesting that BPA, especially at high concentrations, modulates Tyr phosphorylation by regulating PKA activity. A significant inhibitory effect on fertilization and early embryonic development was also found, which might be due to decreased sperm motility and early acrosome reaction [244]. Rahman and collaborators [244] proposed that decreased sperm motility and early acrosome reaction might result in part of the downregulation of sperm’s actin. Actin is a cytoskeletal protein localized in the acrosome, equatorial region, and tail [273]. The localization of actin in the tail illustrates its function in sperm motility, while actin in the sperm head may be involved in acrosome reaction [273]. Sperm functionality is reflected by the ability to fertilize oocytes, where sperm motility, membrane integrity, and even mitochondrial activity are crucial to allow the interaction between sperm and the female gamete [274,275,276]. Overall, there is a need for further evaluation concerning the properties of these contaminants and their reproductive toxicity to establish safety thresholds. This might be important to confirm which of environmental pollutants are contributing to the current decline of male fertility.
Table 3.
Summary of the main studies focusing on the effects of obesogens on sperm quality and sperm parameters.
11. Conclusions
There is an intense debate concerning the contribution of environmental cues to male fertility. Several studies concerning this topic reveal a negative impact of external compounds in the reproductive function of individuals exposed from very early periods (even in prenatal), not only through indirect effects mediated by changes in hormonal levels and the HPT axis, but also by direct changes in the reproductive tissues. The toxic effects may compromise spermatogenesis and end up in the impairment of the molecular composition of sperm quality and function. Definitive conclusions on how obesogens affect the cellular and molecular mechanisms involved in the production and function of sperm are, however, hard to establish. First, when exposure occurs, it is often through a combined mixture of environmental chemicals and not due to a single compound. Besides, sometimes there is also a long latency period between the exposure and the noticed effects, which further hampers the association to a specific agent. However, from a clinical point of view, it is imperative to identify mechanisms by which testicular metabolic pathways are compromised after exposure to obesogens. SCs have arisen as one of the best in vitro and/or ex vivo models that have allowed researchers from this area to understand the molecular basis by which obesogens impact male fertility. In addition, the use of experimental animal models must be applied to permit detailed knowledge on the effects of obesogens on testicular cell-specific functions. Apart from the elucidation of all those points, it is needed to unveil why and how the metabolic alterations caused by what male individuals are exposed to may result in molecular changes that are carried out by male gametes and thus may be passed to the next generations. In fact, in this work we gathered several studies showing the relationship between the exposure to environmental contaminants and epigenetics, and identified several toxicants that modify epigenetic marks, particularly regarding DNA methylation. This field reveals a considerable potential for precocious disease diagnosis, since future research may allow prediction of which contaminants would put exposed individuals at risk, which individuals will be more susceptible to develop a disease, and whether such epigenetic alterations increase the risk of illness. Since we are permanently exposed to environmental contaminants that may induce a myriad of effects that persist through the generations, this topic will undoubtedly become a matter of debate for professionals of the environmental toxicology and reproduction fields.
Author Contributions
Conceptualization, L.R. and A.C.A.S.; methodology, L.R. and A.C.A.S.; validation, L.R. and A.C.A.S.; formal analysis, L.R. and A.C.A.S.; investigation, L.R. and A.C.A.S.; writing—original draft preparation, L.R. and A.C.A.S.; writing—review and editing, L.R. and A.C.A.S.; supervision, L.R. and A.C.A.S.; project administration, L.R. and A.C.A.S.; funding acquisition, A.C.A.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Portuguese Foundation for Science and Technology through the Comprehensive Health Research Centre (CHRC) project UIDP/04923/2020 and the La Mer Project funded by the Japanese Ministry of Education, Culture, Sports, Science, and Technology (MEXT).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gore, A.C.; Chappell, V.A.; Fenton, S.E.; Flaws, J.A.; Nadal, A.; Prins, G.S.; Toppari, J.; Zoeller, R.T. EDC-2: The Endocrine Society’s Second Scientific Statement on Endocrine-Disrupting Chemicals. Endocr. Rev. 2015, 36, E1–E150. [Google Scholar] [CrossRef] [PubMed]
- Zoeller, R.T.; Brown, T.R.; Doan, L.L.; Gore, A.C.; Skakkebaek, N.; Soto, A.; Woodruff, T.; Vom Saal, F. Endocrine-disrupting chemicals and public health protection: A statement of principles from The Endocrine Society. Endocrinology 2012, 153, 4097–4110. [Google Scholar] [CrossRef]
- Kahn, L.G.; Philippat, C.; Nakayama, S.F.; Slama, R.; Trasande, L. Endocrine-disrupting chemicals: Implications for human health. Lancet Diabetes Endocrinol. 2020, 8, 703–718. [Google Scholar] [CrossRef]
- Bergman, A.; Heindel, J.J.; Kasten, T.; Kidd, K.A.; Jobling, S.; Neira, M.; Zoeller, R.T.; Becher, G.; Bjerregaard, P.; Bornman, R.; et al. The impact of endocrine disruption: A consensus statement on the state of the science. Environ. Health Perspect. 2013, 121, A104–A106. [Google Scholar] [CrossRef]
- Schug, T.T.; Janesick, A.; Blumberg, B.; Heindel, J.J. Endocrine disrupting chemicals and disease susceptibility. J. Steroid Biochem. Mol. Biol. 2011, 127, 204–215. [Google Scholar] [CrossRef]
- Grun, F.; Watanabe, H.; Zamanian, Z.; Maeda, L.; Arima, K.; Cubacha, R.; Gardiner, D.M.; Kanno, J.; Iguchi, T.; Blumberg, B. Endocrine-disrupting organotin compounds are potent inducers of adipogenesis in vertebrates. Mol. Endocrinol. 2006, 20, 2141–2155. [Google Scholar] [CrossRef]
- Grün, F.; Blumberg, B. Environmental obesogens: Organotins and endocrine disruption via nuclear receptor signaling. Endocrinology 2006, 147, s50–s55. [Google Scholar] [CrossRef]
- Trasande, L.; Zoeller, R.T.; Hass, U.; Kortenkamp, A.; Grandjean, P.; Myers, J.P.; DiGangi, J.; Bellanger, M.; Hauser, R.; Legler, J.; et al. Estimating burden and disease costs of exposure to endocrine-disrupting chemicals in the European union. J. Clin. Endocrinol. Metab. 2015, 100, 1245–1255. [Google Scholar] [CrossRef]
- Trasande, L. Sicker, Fatter, Poorer: The Urgent Threat of Hormone-Disrupting Chemicals on Our Health and Future... and What We Can Do about It; Houghton Mifflin: Boston, MA, USA, 2019. [Google Scholar]
- Bruce, B.; Kristin, L. The Obesogen Effect, 1st ed.; Grand Central Life & Style: New York, NY, USA, 2018; p. 307. [Google Scholar]
- Darbre, P.D. Endocrine Disruptors and Obesity. Curr. Obes. Rep. 2017, 6, 18–27. [Google Scholar] [CrossRef]
- Veiga-Lopez, A.; Pu, Y.; Gingrich, J.; Padmanabhan, V. Obesogenic Endocrine Disrupting Chemicals: Identifying Knowledge Gaps. Trends Endocrinol. Metab. 2018, 29, 607–625. [Google Scholar] [CrossRef]
- Heindel, J.J.; Blumberg, B. Environmental Obesogens: Mechanisms and Controversies. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 89–106. [Google Scholar] [CrossRef] [PubMed]
- Chamorro-Garcia, R.; Blumberg, B. Current Research Approaches and Challenges in the Obesogen Field. Front. Endocrinol. 2019, 10, 167. [Google Scholar] [CrossRef] [PubMed]
- Heindel, J.J. History of the Obesogen Field: Looking Back to Look Forward. Front. Endocrinol. 2019, 10, 14. [Google Scholar] [CrossRef] [PubMed]
- Legler, J.; Fletcher, T.; Govarts, E.; Porta, M.; Blumberg, B.; Heindel, J.J.; Trasande, L. Obesity, diabetes, and associated costs of exposure to endocrine-disrupting chemicals in the European Union. J. Clin. Endocrinol. Metab. 2015, 100, 1278–1288. [Google Scholar] [CrossRef]
- Alonso-Magdalena, P.; Quesada, I.; Nadal, A. Endocrine disruptors in the etiology of type 2 diabetes mellitus. Nat. Rev. Endocrinol 2011, 7, 346–353. [Google Scholar] [CrossRef]
- Kuo, C.C.; Moon, K.; Thayer, K.A.; Navas-Acien, A. Environmental chemicals and type 2 diabetes: An updated systematic review of the epidemiologic evidence. Curr. Diab. Rep. 2013, 13, 831–849. [Google Scholar] [CrossRef]
- Braun, J.M. Early-life exposure to EDCs: Role in childhood obesity and neurodevelopment. Nat. Rev. Endocrinol. 2017, 13, 161–173. [Google Scholar] [CrossRef]
- Fallara, G.; Cazzaniga, W.; Boeri, L.; Capogrosso, P.; Candela, L.; Pozzi, E.; Belladelli, F.; Schifano, N.; Ventimiglia, E.; Abbate, C. Male factor infertility trends throughout the last 10 years: Report from a tertiary-referral academic andrology centre. Andrology 2021, 9, 610–617. [Google Scholar] [CrossRef]
- Rodprasert, W.; Main, K.M.; Toppari, J.; Virtanen, H.E. Associations between male reproductive health and exposure to endocrine-disrupting chemicals. Curr. Opin. Endocr. Metab. Res. 2019, 7, 49–61. [Google Scholar] [CrossRef]
- Wong, E.W.; Cheng, C.Y. Impacts of environmental toxicants on male reproductive dysfunction. Trends Pharmacol. Sci. 2011, 32, 290–299. [Google Scholar] [CrossRef]
- Mathur, P.P.; D’Cruz, S.C. The effect of environmental contaminants on testicular function. Asian J. Androl. 2011, 13, 585–591. [Google Scholar] [CrossRef]
- De Coster, S.; van Larebeke, N. Endocrine-disrupting chemicals: Associated disorders and mechanisms of action. J. Environ. Public Health 2012, 2012, 713696. [Google Scholar] [CrossRef] [PubMed]
- Kelce, W.R.; Wilson, E.M. Environmental antiandrogens: Developmental effects, molecular mechanisms, and clinical implications. J. Mol. Med. 1997, 75, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Sohoni, P.; Sumpter, J. Several environmental oestrogens are also anti-androgens. J. Endocrinol. 1998, 158, 327–339. [Google Scholar] [CrossRef]
- Phillips, K.P.; Foster, W.G. Key developments in endocrine disrupter research and human health. J. Toxicol. Environ. Health B 2008, 11, 322–344. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, S.A.; Rice, S. Endocrine-disrupting chemicals as modulators of sex steroid synthesis. Best Pract. Res. Clin. Endocrinol. Metab. 2006, 20, 45–61. [Google Scholar] [CrossRef] [PubMed]
- Farag, A.T.; Radwan, A.H.; Sorour, F.; El Okazy, A.; El-Agamy, E.-S.; El-Sebae, A.E.-K. Chlorpyrifos induced reproductive toxicity in male mice. Reprod. Toxicol. 2010, 29, 80–85. [Google Scholar] [CrossRef]
- Han, X.D.; Tu, Z.G.; Gong, Y.; Shen, S.N.; Wang, X.Y.; Kang, L.N.; Hou, Y.Y.; Chen, J.X. The toxic effects of nonylphenol on the reproductive system of male rats. Reprod. Toxicol. 2004, 19, 215–221. [Google Scholar] [CrossRef]
- Lee, B.-J.; Jung, E.-Y.; Yun, Y.-W.; Kang, J.-K.; Baek, I.-J.; Yon, J.-M.; Lee, Y.-B.; Sohn, H.-S.; Lee, J.-Y.; Kim, K.-S. Effects of exposure to genistein during pubertal development on the reproductive system of male mice. J. Reprod. Dev. 2004, 50, 399–409. [Google Scholar] [CrossRef][Green Version]
- McNutt, S.H.; Purwin, P.; Murray, C. Vulvo-vaginitis in swine: Preliminary report. J. Am. Vet. Med. Assoc. 1928, 73, 484. [Google Scholar]
- Bennetts, H.W.; Underwood, E.J.; Shier, F.L. A specific breeding problem of sheep on subterranean clover pastures in Western Australia. Aust. Vet. J. 1946, 22, 2–12. [Google Scholar] [CrossRef]
- Gassner, F.X.; Reifenstein, E.C., Jr.; Algeo, J.W.; Mattox, W.E. Effects of hormones on growth, fattening, and meat production potential of livestock. Recent Prog. Horm. Res. 1958, 14, 183–210; discussion 210–217. [Google Scholar] [PubMed]
- Department of Health, Environmental Assessment Worksheet. The Advancement of Medical Research and Education through the Department of Health, Education, and Welfare. Final Report of the Secretary’s Consultants of Medical Research Education; Department of Health: Washington, DC, USA, 1958. [Google Scholar]
- Aulerich, R.J.; Ringer, R.K.; Iwamoto, S. Reproductive failure and mortality in mink fed on Great Lakes fish. J. Reprod. Fertil. Suppl. 1973, 19, 365–376. [Google Scholar] [PubMed]
- Aulerich, R.J.; Ringer, R.K. Current status of PCB toxicity to mink, and effect on their reproduction. Arch. Environ. Contam. Toxicol. 1977, 6, 279–292. [Google Scholar] [CrossRef]
- Gilbertson, M.; Reynolds, L.M. Hexachlorobenzene (HCB) in the eggs of common terns in Hamilton Harbour, Ontario. Bull. Environ. Contam. Toxicol. 1972, 7, 371–373. [Google Scholar] [CrossRef] [PubMed]
- Kioumourtzoglou, M.A.; Coull, B.A.; O’Reilly, E.J.; Ascherio, A.; Weisskopf, M.G. Association of Exposure to Diethylstilbestrol During Pregnancy with Multigenerational Neurodevelopmental Deficits. JAMA Pediatrics 2018, 172, 670–677. [Google Scholar] [CrossRef]
- Huo, D.; Anderson, D.; Herbst, A.L. Follow-up of Patients with Clear-Cell Adenocarcinoma of the Vagina and Cervix. N. Engl. J. Med. 2018, 378, 1746–1748. [Google Scholar] [CrossRef]
- McLachlan, J. Estrogens in the Environment; Elsevier Inc.: New York, NY, USA, 1980. [Google Scholar]
- Colborn, T.; Clement, C. Chemically-Induced Alterations in Sexual and Functional Development: The Wildlife/Human Connection; Princeton Scientific Pub. Co.: Princeton, NJ, USA, 1992. [Google Scholar]
- Carlsen, E.; Giwercman, A.; Keiding, N.; Skakkebaek, N.E. Evidence for decreasing quality of semen during past 50 years. BMJ Br. Med. J. 1992, 305, 609–613. [Google Scholar] [CrossRef] [PubMed]
- European Environment Agency. The Impacts of Endocrine Disrupters on Wildlife, People and Their Environments. The Weybridge + 15 (1996–2011) Report; Publications Office of the European Union: Copenhagen, Denmark, 2012. [Google Scholar]
- Hotchkiss, A.K.; Rider, C.V.; Blystone, C.R.; Wilson, V.S.; Hartig, P.C.; Ankley, G.T.; Foster, P.M.; Gray, C.L.; Gray, L.E. Fifteen years after “Wingspread”—environmental endocrine disrupters and human and wildlife health: Where we are today and where we need to go. Toxicol. Sci. 2008, 105, 235–259. [Google Scholar] [CrossRef]
- Damstra, T.; Barlow, S.; Bergman, A.; Kavlock, R.; Van Der Kraak, G. Global Assessment of the State-of-the-Science of Endocrine Disruptors; World Health Organization: Geneva, Switzerland, 2002; pp. 11–32. [Google Scholar]
- Diamanti-Kandarakis, E.; Bourguignon, J.-P.; Giudice, L.C.; Hauser, R.; Prins, G.S.; Soto, A.M.; Zoeller, R.T.; Gore, A.C. Endocrine-disrupting chemicals: An Endocrine Society scientific statement. Endocr. Rev. 2009, 30, 293–342. [Google Scholar] [CrossRef]
- Bergman, Å.; Heindel, J.J.; Jobling, S.; Kidd, K.; Zoeller, T.R. State of the Science of Endocrine Disrupting Chemicals 2012: Summary for Decision-Makers; W.H.O.: Geneva, Switzerland, 2013. [Google Scholar]
- Heindel, J.J.; Vom Saal, F.S.; Blumberg, B.; Bovolin, P.; Calamandrei, G.; Ceresini, G.; Cohn, B.A.; Fabbri, E.; Gioiosa, L.; Kassotis, C.; et al. Parma consensus statement on metabolic disruptors. Environ. Health 2015, 14, 54. [Google Scholar] [CrossRef] [PubMed]
- Janesick, A.S.; Schug, T.T.; Heindel, J.J.; Blumberg, B. Environmental Chemicals and Obesity. In Handbook of Obesity-Epidemiology, Etiology, and Physiopathology; Bray, G.A., Bouchard, C., Eds.; CRC Press: Boca Raton, FL, USA, 2014; Volume 1, pp. 471–488. [Google Scholar]
- Lee, D.H.; Porta, M.; Jacobs, D.R., Jr.; Vandenberg, L.N. Chlorinated persistent organic pollutants, obesity, and type 2 diabetes. Endocr. Rev. 2014, 35, 557–601. [Google Scholar] [CrossRef] [PubMed]
- Janesick, A.; Blumberg, B. Minireview: PPARγ as the target of obesogens. J. Steroid Biochem. Mol. Biol. 2011, 127, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Janesick, A.S.; Dimastrogiovanni, G.; Vanek, L.; Boulos, C.; Chamorro-García, R.; Tang, W.; Blumberg, B. On the utility of ToxCast™ and ToxPi as methods for identifying new obesogens. Environ. Health Perspect. 2016, 124, 1214. [Google Scholar] [CrossRef] [PubMed]
- Grun, F.; Blumberg, B. Minireview: The case for obesogens. Mol. Endocrinol. 2009, 23, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Le Maire, A.; Grimaldi, M.; Roecklin, D.; Dagnino, S.; Vivat-Hannah, V.; Balaguer, P.; Bourguet, W. Activation of RXR-PPAR heterodimers by organotin environmental endocrine disruptors. EMBO Rep. 2009, 10, 367–373. [Google Scholar] [CrossRef]
- Kanayama, T.; Kobayashi, N.; Mamiya, S.; Nakanishi, T.; Nishikawa, J. Organotin compounds promote adipocyte differentiation as agonists of the peroxisome proliferator-activated receptor gamma/retinoid X receptor pathway. Mol. Pharmacol. 2005, 67, 766–774. [Google Scholar] [CrossRef]
- Fang, L.; Xu, C.; Li, J.; Borggaard, O.K.; Wang, D. The importance of environmental factors and matrices in the adsorption, desorption, and toxicity of butyltins: A review. Environ. Sci. Pollut. Res. Int. 2017, 24, 9159–9173. [Google Scholar] [CrossRef]
- Brtko, J.; Dvorak, Z. Triorganotin compounds--ligands for rexinoid inducible transcription factors: Biological effects. Toxicol. Lett. 2015, 234, 50–58. [Google Scholar] [CrossRef]
- Sousa, A.C.A.; Tanabe, S.; Pastorinho, M.R. Organotins: Sources and Impacts on Health and Environment A2. In Encyclopedia on Anthropocene; DellaSala, D.A., Goldstein, M.I., Eds.; Elsevier: Oxford, UK, 2018; Volume 1, pp. 133–139. [Google Scholar]
- Nielsen, J.B.; Strand, J. Butyltin compounds in human liver. Environ. Res. 2002, 88, 129–133. [Google Scholar] [CrossRef]
- Rantakokko, P.; Main, K.M.; Wohlfart-Veje, C.; Kiviranta, H.; Airaksinen, R.; Vartiainen, T.; Skakkebaek, N.E.; Toppari, J.; Virtanen, H.E. Association of placenta organotin concentrations with congenital cryptorchidism and reproductive hormone levels in 280 newborn boys from Denmark and Finland. Hum. Reprod. 2013, 28, 1647–1660. [Google Scholar] [CrossRef] [PubMed]
- Rantakokko, P.; Main, K.M.; Wohlfart-Veje, C.; Kiviranta, H.; Airaksinen, R.; Vartiainen, T.; Skakkebaek, N.E.; Toppari, J.; Virtanen, H.E. Association of placenta organotin concentrations with growth and ponderal index in 110 newborn boys from Finland during the first 18 months of life: A cohort study. Environ. Health 2014, 13, 45. [Google Scholar] [CrossRef] [PubMed]
- Rantakokko, P.; Turunen, A.; Verkasalo, P.K.; Kiviranta, H.; Mannisto, S.; Vartiainen, T. Blood levels of organotin compounds and their relation to fish consumption in Finland. Sci. Total Environ. 2008, 399, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Mukai, H.; Tanabe, S.; Sakayama, K.; Miyazaki, T.; Masuno, H. Butyltin residues in livers of humans and wild terrestrial mammals and in plastic products. Environ. Pollut. 1999, 106, 213–218. [Google Scholar] [CrossRef]
- Sousa, A.C.; Pastorinho, M.R.; Takahashi, S.; Tanabe, S. History on organotin compounds, from snails to humans. Environ. Chem. Lett. 2014, 12, 117–137. [Google Scholar] [CrossRef]
- Shoucri, B.M.; Martinez, E.S.; Abreo, T.J.; Hung, V.T.; Moosova, Z.; Shioda, T.; Blumberg, B. Retinoid X Receptor Activation Alters the Chromatin Landscape to Commit Mesenchymal Stem Cells to the Adipose Lineage. Endocrinology 2017, 158, 3109–3125. [Google Scholar] [CrossRef]
- Asakawa, H.; Tsunoda, M.; Kaido, T.; Hosokawa, M.; Sugaya, C.; Inoue, Y.; Kudo, Y.; Satoh, T.; Katagiri, H.; Akita, H.; et al. Enhanced inhibitory effects of TBT chloride on the development of F1 rats. Arch. Environ. Contam. Toxicol. 2010, 58, 1065–1073. [Google Scholar] [CrossRef]
- Hao, C.; Cheng, X.; Xia, H.; Ma, X. The endocrine disruptor mono-(2-ethylhexyl) phthalate promotes adipocyte differentiation and induces obesity in mice. Biosci. Rep. 2012, 32, 619–629. [Google Scholar] [CrossRef]
- Biemann, R.; Navarrete Santos, A.; Navarrete Santos, A.; Riemann, D.; Knelangen, J.; Bluher, M.; Koch, H.; Fischer, B. Endocrine disrupting chemicals affect the adipogenic differentiation of mesenchymal stem cells in distinct ontogenetic windows. Biochem. Biophys. Res. Commun. 2012, 417, 747–752. [Google Scholar] [CrossRef]
- Thomas, P.; Dong, J. Binding and activation of the seven-transmembrane estrogen receptor GPR30 by environmental estrogens: A potential novel mechanism of endocrine disruption. J. Steroid Biochem. Mol. Biol. 2006, 102, 175–179. [Google Scholar] [CrossRef]
- Watson, C.S.; Bulayeva, N.N.; Wozniak, A.L.; Alyea, R.A. Xenoestrogens are potent activators of nongenomic estrogenic responses. Steroids 2007, 72, 124–134. [Google Scholar] [CrossRef]
- Vandenberg, L.N.; Maffini, M.V.; Sonnenschein, C.; Rubin, B.S.; Soto, A.M. Bisphenol-A and the great divide: A review of controversies in the field of endocrine disruption. Endocr. Rev. 2009, 30, 75–95. [Google Scholar] [CrossRef] [PubMed]
- Masuno, H.; Iwanami, J.; Kidani, T.; Sakayama, K.; Honda, K. Bisphenol a accelerates terminal differentiation of 3T3-L1 cells into adipocytes through the phosphatidylinositol 3-kinase pathway. Toxicol. Sci. 2005, 84, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Wu, Q.; Sakthivel, S.; Pavithran, P.V.; Vasukutty, J.R.; Kannan, K. Urinary levels of endocrine-disrupting chemicals, including bisphenols, bisphenol A diglycidyl ethers, benzophenones, parabens, and triclosan in obese and non-obese Indian children. Environ. Res. 2015, 137, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Overby, H.; Heal, E.; Wang, S.; Chen, J.; Shen, C.L.; Zhao, L. Methylparaben and butylparaben alter multipotent mesenchymal stem cell fates towards adipocyte lineage. Toxicol. Appl. Pharmacol. 2017, 329, 48–57. [Google Scholar] [CrossRef]
- Kodani, S.D.; Overby, H.B.; Morisseau, C.; Chen, J.; Zhao, L.; Hammock, B.D. Parabens inhibit fatty acid amide hydrolase: A potential role in paraben-enhanced 3T3-L1 adipocyte differentiation. Toxicol. Lett. 2016, 262, 92–99. [Google Scholar] [CrossRef]
- Hu, P.; Chen, X.; Whitener, R.J.; Boder, E.T.; Jones, J.O.; Porollo, A.; Chen, J.; Zhao, L. Effects of parabens on adipocyte differentiation. Toxicol. Sci. 2013, 131, 56–70. [Google Scholar] [CrossRef]
- Ferrante, M.C.; Amero, P.; Santoro, A.; Monnolo, A.; Simeoli, R.; Di Guida, F.; Mattace Raso, G.; Meli, R. Polychlorinated biphenyls (PCB 101, PCB 153 and PCB 180) alter leptin signaling and lipid metabolism in differentiated 3T3-L1 adipocytes. Toxicol. Appl. Pharmacol. 2014, 279, 401–408. [Google Scholar] [CrossRef]
- Howell, G., 3rd; Mangum, L. Exposure to bioaccumulative organochlorine compounds alters adipogenesis, fatty acid uptake, and adipokine production in NIH3T3-L1 cells. Toxicol. Vitr. 2011, 25, 394–402. [Google Scholar] [CrossRef]
- Penza, M.; Montani, C.; Romani, A.; Vignolini, P.; Pampaloni, B.; Tanini, A.; Brandi, M.; Alonso-Magdalena, P.; Nadal, A.; Ottobrini, L. Genistein affects adipose tissue deposition in a dose-dependent and gender-specific manner. Endocrinology 2006, 147, 5740–5751. [Google Scholar] [CrossRef]
- Ruhlen, R.L.; Howdeshell, K.L.; Mao, J.; Taylor, J.A.; Bronson, F.H.; Newbold, R.R.; Welshons, W.V.; vom Saal, F.S. Low phytoestrogen levels in feed increase fetal serum estradiol resulting in the fetal estrogenization syndrome and obesity in CD-1 mice. Environ. Health Perspect. 2008, 116, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Z.; Chen, S.; Wu, T.; Zhang, J.; Su, Y.; Chen, Y.; Wang, C. Tributyltin causes obesity and hepatic steatosis in male mice. Environ. Toxicol. 2011, 26, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Silha, J.V.; Krsek, M.; Skrha, J.V.; Sucharda, P.; Nyomba, B.L.; Murphy, L.J. Plasma resistin, adiponectin and leptin levels in lean and obese subjects: Correlations with insulin resistance. Eur. J. Endocrinol. 2003, 149, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Degawa-Yamauchi, M.; Bovenkerk, J.E.; Juliar, B.E.; Watson, W.; Kerr, K.; Jones, R.; Zhu, Q.; Considine, R.V. Serum resistin (FIZZ3) protein is increased in obese humans. J. Clin. Endocrinol. Metab. 2003, 88, 5452–5455. [Google Scholar] [CrossRef]
- Penza, M.; Jeremic, M.; Marrazzo, E.; Maggi, A.; Ciana, P.; Rando, G.; Grigolato, P.G.; Di Lorenzo, D. The environmental chemical tributyltin chloride (TBT) shows both estrogenic and adipogenic activities in mice which might depend on the exposure dose. Toxicol. Appl. Pharmacol. 2011, 255, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Vitalone, A.; Catalani, A.; Cinque, C.; Fattori, V.; Matteucci, P.; Zuena, A.R.; Costa, L.G. Long-term effects of developmental exposure to low doses of PCB 126 and methylmercury. Toxicol. Lett. 2010, 197, 38–45. [Google Scholar] [CrossRef]
- Marth, P.C.; Mitchell, J.W. 2, 4-Dichlorophenoxyacetic acid as a differential herbicide. Botanical. Gazette. 1944, 106, 224–232. [Google Scholar] [CrossRef]
- Cook, J.; Hewett, C.; Hieger, I. The isolation ofa cancer-producing hydrocarbon from coal tar. Parts, I, II and III. J. Chem. Soc. 1933, 1, 395–405. [Google Scholar] [CrossRef]
- Conix, A. Thermoplastic polyesters from bisphenols. Ind. Eng. Chem. 1959, 51, 147–150. [Google Scholar] [CrossRef]
- Gray, H. DURSBAN—A new organo-phosphorus insecticide. Down Earth 1965, 21, 2. [Google Scholar]
- Hansens, E.J.; Bartley, C. Three new Insecticides for Housefly Control in Barns. J. Econ. Entomol. 1953, 46, 372–374. [Google Scholar] [CrossRef]
- Burroughs, W.; Culbertson, C.; Cheng, E.; Hale, W.; Homeyer, P. The influence of oral administration of diethylstilbestrol to beef cattle. J. Anim. Sci. 1955, 14, 1015–1024. [Google Scholar] [CrossRef]
- Dubrunfaut, A.P. Sur une propriété analytique des fermentations alcoolique et lactique, et sur leur application à l’étude des sucres. Ann. Chim. Phys. 1847, 21, 169–178. [Google Scholar]
- Akiyama, T.; Ishida, J.; Nakagawa, S.; Ogawara, H.; Watanabe, S.-I.; Itoh, N.; Shibuya, M.; Fukami, Y. Genistein, a specific inhibitor of tyrosine-specific protein kinases. J. Biol. Chem. 1987, 262, 5592–5595. [Google Scholar] [CrossRef]
- Bacon, A.; Froome, K.; Gent, A.; Cooke, T.; Sowerby, P. Lead poisoning from drinking soft water. Lancet 1967, 289, 264–266. [Google Scholar] [CrossRef]
- Ikeda, K. On the taste of the salt of glutamic acid. Proc. 8th Int. Congr. Appl. Chem. 1912, 38, 147. [Google Scholar]
- Posselt, W.; Reimann, L. Chemische Untersuchungen des Tabaks und Darstellung des eigenhumlichen wirksamen Principes dieser Pflanze. Geigers Magazin der Pharmazie 1828, 24, 138–161. [Google Scholar]
- Leppert, B.; Strunz, S.; Seiwert, B.; Schlittenbauer, L.; Schlichting, R.; Pfeiffer, C.; Röder, S.; Bauer, M.; Borte, M.; Stangl, G.I.; et al. Maternal paraben exposure triggers childhood overweight development. Nat. Commun. 2020, 11, 561. [Google Scholar] [CrossRef]
- Creighton, J.T.; Gresham, W.B. Parathion for control of green peach aphid on shade-grown tobacco. J. Econ. Entomol. 1947, 40, 915–917. [Google Scholar] [CrossRef]
- De Kok, J.; De Kok, A.; Brinkman, U.T. Analysis of polybrominated aromatic ethers. J. Chromatogr. A 1979, 171, 269–278. [Google Scholar] [CrossRef]
- Gauger, K.J.; Kato, Y.; Haraguchi, K.; Lehmler, H.-J.; Robertson, L.W.; Bansal, R.; Zoeller, R.T. Polychlorinated biphenyls (PCBs) exert thyroid hormone-like effects in the fetal rat brain but do not bind to thyroid hormone receptors. Environ. Health Perspect. 2004, 112, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Gilliland, F.D.; Mandel, J.S. Serum perfluorooctanoic acid and hepatic enzymes, lipoproteins, and cholesterol: A study of occupationally exposed men. Am. J. Ind. Med. 1996, 29, 560–568. [Google Scholar] [CrossRef]
- Hatch, E.E.; Nelson, J.W.; Qureshi, M.M.; Weinberg, J.; Moore, L.L.; Singer, M.; Webster, T.F. Association of urinary phthalate metabolite concentrations with body mass index and waist circumference: A cross-sectional study of NHANES data, 1999–2002. Environ. Health 2008, 7, 1–15. [Google Scholar] [CrossRef]
- Meneses, M.J.; Bernardino, R.L.; Sa, R.; Silva, J.; Barros, A.; Sousa, M.; Silva, B.M.; Oliveira, P.F.; Alves, M.G. Pioglitazone increases the glycolytic efficiency of human Sertoli cells with possible implications for spermatogenesis. Int. J. Biochem. Cell Biol. 2016, 79, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, T.; Kaneko, S.; Hayabuchi, H. Sex ratio in offspring of those affected by dioxin and dioxin-like compounds: The Yusho, Seveso, and Yucheng incidents. Occup. Environ. Med. 2001, 58, 540–541. [Google Scholar] [CrossRef]
- Caputo, R.; Monti, M.; Ermacora, E.; Carminati, G.; Gelmetti, C.; Gianotti, R.; Gianni, E.; Puccinelli, V. Cutaneous manifestations of tetrachlorodibenzo-p-dioxin in children and adolescents: Follow-up 10 years after the Seveso, Italy, accident. J. Am. Acad. Dermatol. 1988, 19, 812–819. [Google Scholar] [CrossRef]
- Carwile, J.L.; Michels, K.B. Urinary bisphenol A and obesity: NHANES 2003–2006. Environ. Res. 2011, 111, 825–830. [Google Scholar] [CrossRef]
- Shankar, A.; Teppala, S.; Sabanayagam, C. Urinary bisphenol a levels and measures of obesity: Results from the national health and nutrition examination survey 2003–2008. ISRN Endocrinol. 2012, 2012, 965243. [Google Scholar] [CrossRef]
- Valvi, D.; Mendez, M.A.; Garcia-Esteban, R.; Ballester, F.; Ibarluzea, J.; Goni, F.; Grimalt, J.O.; Llop, S.; Marina, L.S.; Vizcaino, E.; et al. Prenatal exposure to persistent organic pollutants and rapid weight gain and overweight in infancy. Obesity Silver Spring 2014, 22, 488–496. [Google Scholar] [CrossRef]
- Mendez, M.A.; Garcia-Esteban, R.; Guxens, M.; Vrijheid, M.; Kogevinas, M.; Goni, F.; Fochs, S.; Sunyer, J. Prenatal organochlorine compound exposure, rapid weight gain, and overweight in infancy. Environ. Health Perspect. 2011, 119, 272–278. [Google Scholar] [CrossRef]
- Lind, P.M.; Roos, V.; Ronn, M.; Johansson, L.; Ahlstrom, H.; Kullberg, J.; Lind, L. Serum concentrations of phthalate metabolites are related to abdominal fat distribution two years later in elderly women. Environ. Health 2012, 11, 21. [Google Scholar] [CrossRef]
- Tang-Péronard, J.L.; Heitmann, B.L.; Andersen, H.R.; Steuerwald, U.; Grandjean, P.; Weihe, P.; Jensen, T.K. Association between prenatal polychlorinated biphenyl exposure and obesity development at ages 5 and 7 y: A prospective cohort study of 656 children from the Faroe Islands. Am. Clin. Nutr. 2013, 99, 5–13. [Google Scholar] [CrossRef]
- Larsen, T.; Toubro, S.; Astrup, A. PPARgamma agonists in the treatment of type II diabetes: Is increased fatness commensurate with long-term efficacy? Int. J. Obes. 2003, 27, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Rubenstrunk, A.; Hanf, R.; Hum, D.W.; Fruchart, J.-C.; Staels, B. Safety issues and prospects for future generations of PPAR modulators. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2007, 1771, 1065–1081. [Google Scholar] [CrossRef]
- Legler, J. An integrated approach to assess the role of chemical exposure in obesity. Obesity Silver Spring 2013, 21, 1084–1085. [Google Scholar] [CrossRef] [PubMed]
- Ratcliffe, D.A. Decrease in eggshell weight in certain birds of prey. Nature 1967, 215, 208–210. [Google Scholar] [CrossRef] [PubMed]
- Trasande, L.; Zoeller, R.T.; Hass, U.; Kortenkamp, A.; Grandjean, P.; Myers, J.P.; DiGangi, J.; Hunt, P.M.; Rudel, R.; Sathyanarayana, S.; et al. Burden of disease and costs of exposure to endocrine disrupting chemicals in the European Union: An updated analysis. Andrology 2016, 4, 565–572. [Google Scholar] [CrossRef]
- Attina, T.M.; Hauser, R.; Sathyanarayana, S.; Hunt, P.A.; Bourguignon, J.P.; Myers, J.P.; DiGangi, J.; Zoeller, R.T.; Trasande, L. Exposure to endocrine-disrupting chemicals in the USA: A population-based disease burden and cost analysis. Lancet Diabetes Endocrinol. 2016, 4, 996–1003. [Google Scholar] [CrossRef]
- Reame, V.; Pytlowanciv, E.Z.; Ribeiro, D.L.; Pissolato, T.F.; Taboga, S.R.; Góes, R.M.; Pinto-Fochi, M.E. Obesogenic environment by excess of dietary fats in different phases of development reduces spermatic efficiency of Wistar rats at adulthood: Correlations with metabolic status. Biol. Reprod. 2014, 91, 151–161. [Google Scholar] [CrossRef]
- Wan, H.-T.; Mruk, D.D.; Wong, C.K.; Cheng, C.Y. The apical ES–BTB–BM functional axis is an emerging target for toxicant-induced infertility. Trends Mol. Med. 2013, 19, 396–405. [Google Scholar] [CrossRef][Green Version]
- Akingbemi, B.T.; Youker, R.T.; Sottas, C.M.; Ge, R.; Katz, E.; Klinefelter, G.R.; Zirkin, B.R.; Hardy, M.P. Modulation of rat Leydig cell steroidogenic function by di(2-ethylhexyl)phthalate. Biol. Reprod. 2001, 65, 1252–1259. [Google Scholar] [CrossRef]
- Alves, M.; Neuhaus-Oliveira, A.; Moreira, P.; Socorro, S.; Oliveira, P. Exposure to 2, 4-dichlorophenoxyacetic acid alters glucose metabolism in immature rat Sertoli cells. Reprod. Toxicol. 2013, 38, 81–88. [Google Scholar] [CrossRef]
- D’Cruz, S.C.; Jubendradass, R.; Jayakanthan, M.; Rani, S.J.A.; Mathur, P.P. Bisphenol A impairs insulin signaling and glucose homeostasis and decreases steroidogenesis in rat testis: An in vivo and in silico study. Food Chem. Toxicol. 2012, 50, 1124–1133. [Google Scholar] [CrossRef] [PubMed]
- Moss, E.J.; Cook, M.W.; Thomas, L.V.; Gray, T.J. The effect of mono-(2-ethylhexyl) phthalate and other phthalate esters on lactate production by Sertoli cells in vitro. Toxicol. Lett. 1988, 40, 77–84. [Google Scholar] [CrossRef]
- D’Cruz, S.C.; Jubendradass, R.; Mathur, P.P. Bisphenol A induces oxidative stress and decreases levels of insulin receptor substrate 2 and glucose transporter 8 in rat testis. Reprod. Sci. 2012, 19, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Sai, L.; Li, X.; Liu, Y.; Guo, Q.; Xie, L.; Yu, G.; Bo, C.; Zhang, Z.; Li, L. Effects of chlorpyrifos on reproductive toxicology of male rats. Environ. Toxicol. 2014, 29, 1083–1088. [Google Scholar] [CrossRef] [PubMed]
- Batarseh, L.I.; Welsh, M.J.; Brabec, M.J. Effect of lead acetate on Sertoli cell lactate production and protein synthesis in vitro. Cell Biol. Toxicol. 1986, 2, 283–292. [Google Scholar] [CrossRef]
- Raychoudhury, S.S.; Flowers, A.F.; Millette, C.F.; Finlay, M.F. Toxic effects of polychlorinated biphenyls on cultured rat Sertoli cells. J. Androl. 2000, 21, 964–973. [Google Scholar]
- Itsuki-Yoneda, A.; Kimoto, M.; Tsuji, H.; Hiemori, M.; Yamashita, H. Effect of a hypolipidemic drug, di (2-ethylhexyl) phthalate, on mRNA-expression associated fatty acid and acetate metabolism in rat tissues. Biosci. Biotechnol. Biochem. 2007, 71, 414–420. [Google Scholar] [CrossRef]
- Mitra, S.; Srivastava, A.; Khandelwal, S. Long term impact of the endocrine disruptor tributyltin on male fertility following a single acute exposure. Environ. Toxicol. 2017, 32, 2295–2304. [Google Scholar] [CrossRef]
- Mitra, S.; Srivastava, A.; Khanna, S.; Khandelwal, S. Consequences of tributyltin chloride induced stress in Leydig cells: An ex-vivo approach. Environ. Toxicol. Pharmacol. 2014, 37, 850–860. [Google Scholar] [CrossRef]
- Cardoso, A.M.; Alves, M.G.; Sousa, A.C.; Jarak, I.; Carvalho, R.A.; Oliveira, P.F.; Cavaco, J.E.; Rato, L. The effects of the obesogen tributyltin on the metabolism of Sertoli cells cultured ex vivo. Arch. Toxicol. 2018, 92, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; Srivastava, A.; Khandelwal, S. Tributyltin chloride induced testicular toxicity by JNK and p38 activation, redox imbalance and cell death in sertoli-germ cell co-culture. Toxicology 2013, 314, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.Y.; Mruk, D.D. A local autocrine axis in the testes that regulates spermatogenesis. Nat. Rev. Endocrinol. 2010, 6, 380–395. [Google Scholar] [CrossRef]
- Walker, W.H.; Cheng, J. FSH and testosterone signaling in Sertoli cells. Reproduction 2005, 130, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Rommerts, F.F.; de Jong, F.H.; Brinkmann, A.O.; van der Molen, H.J. Development and cellular localization of rat testicular aromatase activity. J. Reprod. Fertil. 1982, 65, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Gore, A.C. Developmental programming and endocrine disruptor effects on reproductive neuroendocrine systems. Front. Neuroendocrinol. 2008, 29, 358–374. [Google Scholar] [CrossRef]
- Gore, A.C. Organochlorine pesticides directly regulate gonadotropin-releasing hormone gene expression and biosynthesis in the GT1-7 hypothalamic cell line. Mol. Cell. Endocrinol. 2002, 192, 157–170. [Google Scholar] [CrossRef]
- Stoker, T.E.; Parks, L.G.; Gray, L.E.; Cooper, R.L. Endocrine-disrupting chemicals: Prepubertal exposures and effects on sexual maturation and thyroid function in the male rat. A focus on the EDSTAC Recommendations. Crit. Rev. Toxicol. 2000, 30, 197–252. [Google Scholar] [CrossRef]
- Welshons, W.V.; Nagel, S.C.; vom Saal, F.S. Large effects from small exposures. III. Endocrine mechanisms mediating effects of bisphenol A at levels of human exposure. Endocrinology 2006, 147, S56–S69. [Google Scholar] [CrossRef]
- Rubin, B.S. Bisphenol A: An endocrine disruptor with widespread exposure and multiple effects. J. Steroid. Biochem. Mol. Biol. 2011, 127, 27–34. [Google Scholar] [CrossRef]
- Kuiper, G.G.; Lemmen, J.G.; Carlsson, B.; Corton, J.C.; Safe, S.H.; van der Saag, P.T.; van der Burg, B.; Gustafsson, J.A. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology 1998, 139, 4252–4263. [Google Scholar] [CrossRef]
- Gould, J.C.; Leonard, L.S.; Maness, S.C.; Wagner, B.L.; Conner, K.; Zacharewski, T.; Safe, S.; McDonnell, D.P.; Gaido, K.W. Bisphenol A interacts with the estrogen receptor alpha in a distinct manner from estradiol. Mol. Cell Endocrinol. 1998, 142, 203–214. [Google Scholar] [CrossRef]
- Wozniak, A.L.; Bulayeva, N.N.; Watson, C.S. Xenoestrogens at picomolar to nanomolar concentrations trigger membrane estrogen receptor-α-mediated Ca2+ fluxes and prolactin release in GH3/B6 pituitary tumor cells. Environ. Health Perspect. 2005, 113, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Xi, W.; Lee, C.K.; Yeung, W.S.; Giesy, J.P.; Wong, M.H.; Zhang, X.; Hecker, M.; Wong, C.K. Effect of perinatal and postnatal bisphenol A exposure to the regulatory circuits at the hypothalamus-pituitary-gonadal axis of CD-1 mice. Reprod. Toxicol. 2011, 31, 409–417. [Google Scholar] [CrossRef]
- Wisniewski, P.; Romano, R.M.; Kizys, M.M.; Oliveira, K.C.; Kasamatsu, T.; Giannocco, G.; Chiamolera, M.I.; Dias-da-Silva, M.R.; Romano, M.A. Adult exposure to bisphenol A (BPA) in Wistar rats reduces sperm quality with disruption of the hypothalamic–pituitary–testicular axis. Toxicology 2015, 329, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jeong, K.; Kaiser, U. Gonadotropin-releasing hormone regulation of gonadotropin biosynthesis and secretion. In Knobil and Neill’s Physiology of Reproduction; Neill, J.D., Ed.; Elsevier: San Diego, CA, USA, 2006; Volume 1. [Google Scholar]
- Arase, S.; Ishii, K.; Igarashi, K.; Aisaki, K.; Yoshio, Y.; Matsushima, A.; Shimohigashi, Y.; Arima, K.; Kanno, J.; Sugimura, Y. Endocrine disrupter bisphenol A increases in situ estrogen production in the mouse urogenital sinus. Biol. Reprod. 2011, 84, 734–742. [Google Scholar] [CrossRef]
- Stahlhut, R.W.; van Wijngaarden, E.; Dye, T.D.; Cook, S.; Swan, S.H. Concentrations of urinary phthalate metabolites are associated with increased waist circumference and insulin resistance in adult US males. Environ. Health Perspect. 2007, 115, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Havrankova, J.; Schmechel, D.; Roth, J.; Brownstein, M. Identification of insulin in rat brain. Proc. Natl. Acad. Sci. USA 1978, 75, 5737–5741. [Google Scholar] [CrossRef] [PubMed]
- Schoeller, E.L.; Albanna, G.; Frolova, A.I.; Moley, K.H. Insulin rescues impaired spermatogenesis via the hypothalamic-pituitary-gonadal axis in Akita diabetic mice and restores male fertility. Diabetes 2012, 61, 1869–1878. [Google Scholar] [CrossRef]
- Harada, Y.; Tanaka, N.; Ichikawa, M.; Kamijo, Y.; Sugiyama, E.; Gonzalez, F.J.; Aoyama, T. PPARα-dependent cholesterol/testosterone disruption in Leydig cells mediates 2, 4-dichlorophenoxyacetic acid-induced testicular toxicity in mice. Arch. Toxicol. 2016, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Barlow, N.J.; Phillips, S.L.; Wallace, D.G.; Sar, M.; Gaido, K.W.; Foster, P.M. Quantitative changes in gene expression in fetal rat testes following exposure to di (n-butyl) phthalate. Toxicol. Sci. 2003, 73, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Zhang, Z.; Shen, W.-J.; Azhar, S. Cellular cholesterol delivery, intracellular processing and utilization for biosynthesis of steroid hormones. Nutr. Metab. 2010, 7, 1–25. [Google Scholar] [CrossRef]
- Desdoits-Lethimonier, C.; Albert, O.; Le Bizec, B.; Perdu, E.; Zalko, D.; Courant, F.; Lesne, L.; Guille, F.; Dejucq-Rainsford, N.; Jegou, B. Human testis steroidogenesis is inhibited by phthalates. Hum. Reprod. 2012, 27, 1451–1459. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, D.; Yanagiba, Y.; Duan, Z.; Ito, Y.; Okamura, A.; Asaeda, N.; Tagawa, Y.; Li, C.; Taya, K.; Zhang, S.-Y. Bisphenol A may cause testosterone reduction by adversely affecting both testis and pituitary systems similar to estradiol. Toxicol. Lett. 2010, 194, 16–25. [Google Scholar] [CrossRef]
- Jin, P.; Wang, X.; Chang, F.; Bai, Y.; Li, Y.; Zhou, R.; Chen, L. Low dose bisphenol A impairs spermatogenesis by suppressing reproductive hormone production and promoting germ cell apoptosis in adult rats. J. Biomed. Res. 2013, 27, 135–144. [Google Scholar]
- Wilson, V.S.; Lambright, C.R.; Furr, J.R.; Howdeshell, K.L.; Gray, L.E. The herbicide linuron reduces testosterone production from the fetal rat testis during both in utero and in vitro exposures. Toxicol. Lett. 2009, 186, 73–77. [Google Scholar] [CrossRef]
- Lambright, C.; Ostby, J.; Bobseine, K.; Wilson, V.; Hotchkiss, A.; Mann, P.; Gray, L. Cellular and molecular mechanisms of action of linuron: An antiandrogenic herbicide that produces reproductive malformations in male rats. Toxicol. Sci. 2000, 56, 389–399. [Google Scholar] [CrossRef]
- Wolf, C.; Lambright, C.; Mann, P.; Price, M.; Cooper, R.L.; Ostby, J.; Gray, L.E. Administration of potentially antiandrogenic pesticides (procymidone, linuron, iprodione, chlozolinate, p, p′-DDE, and ketoconazole) and toxic substances (dibutyl-and diethylhexyl phthalate, PCB 169, and ethane dimethane sulphonate) during sexual differentiation produces diverse profiles of reproductive malformations in the male rat. Toxicol. Ind. Health 1999, 15, 94–118. [Google Scholar]
- Hotchkiss, A.; Parks-Saldutti, L.; Ostby, J.; Lambright, C.; Furr, J.; Vandenbergh, J.; Gray, L. A mixture of the “antiandrogens” linuron and butyl benzyl phthalate alters sexual differentiation of the male rat in a cumulative fashion. Biol. Reprod. 2004, 71, 1852–1861. [Google Scholar] [CrossRef]
- Ostby, J.; Monosson, E.; Kelce, W.R.; Gray, L.E. Environmental antiandrogens: Low doses of the fungicide vinclozolin alter sexual differentiation of the male rat. Toxicol. Ind. Health 1999, 15, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Monosson, E.; Kelce, W.R.; Lambright, C.; Ostby, J.; Gray, L.E. Peripubertal exposure to the antiandrogenic fungicide, vinclozolin, delays puberty, inhibits the development of androgen-dependent tissues, and alters androgen receptor function in the male rat. Toxicol. Ind. Health 1999, 15, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.-i.; Kelce, W.R.; Sar, M.; Wilson, E.M. Androgen receptor antagonist versus agonist activities of the fungicide vinclozolin relative to hydroxyflutamide. J. Biol. Chem. 1995, 270, 19998–20003. [Google Scholar] [CrossRef]
- Alves, M.G.; Rato, L.; Carvalho, R.A.; Moreira, P.I.; Socorro, S.; Oliveira, P.F. Hormonal control of Sertoli cell metabolism regulates spermatogenesis. Cell. Mol. Life Sci. 2013, 70, 777–793. [Google Scholar] [CrossRef]
- Rato, L.; Meneses, M.J.; Silva, B.M.; Sousa, M.; Alves, M.G.; Oliveira, P.F. New insights on hormones and factors that modulate Sertoli cell metabolism. Histol. Histopathol. 2016, 31, 499–513. [Google Scholar]
- Cardoso, A.M.; Alves, M.G.; Mathur, P.P.; Oliveira, P.F.; Cavaco, J.E.; Rato, L. Obesogens and male fertility. Obes. Rev. 2017, 18, 109–125. [Google Scholar] [CrossRef]
- Foster, P.; Foster, J.R.; Cook, M.W.; Thomas, L.V.; Gangolli, S.D. Changes in ultrastructure and cytochemical localization of zinc in rat testis following the administration of Di-n-pentyl phthalate. Toxicol. Appl. Pharmacol. 1982, 63, 120–132. [Google Scholar] [CrossRef]
- Li, M.W.; Mruk, D.D.; Lee, W.M.; Cheng, C.Y. Disruption of the blood-testis barrier integrity by bisphenol A in vitro: Is this a suitable model for studying blood-testis barrier dynamics? Int. J. Biochem. Cell Biol. 2009, 41, 2302–2314. [Google Scholar] [CrossRef] [PubMed]
- Siu, E.R.; Mruk, D.D.; Porto, C.S.; Cheng, C.Y. Cadmium-induced testicular injury. Toxicol. Appl. Pharmacol. 2009, 238, 240–249. [Google Scholar] [CrossRef]
- Gao, Y.; Mruk, D.D.; Cheng, C.Y. Sertoli cells are the target of environmental toxicants in the testis–a mechanistic and therapeutic insight. Expert Opin. Ther. Targets 2015, 19, 1073–1090. [Google Scholar] [CrossRef]
- Cheng, C.Y. Toxicants target cell junctions in the testis: Insights from the indazole-carboxylic acid model. Spermatogenesis 2014, 4, e981485. [Google Scholar] [CrossRef]
- Su, L.; Mruk, D.D.; Cheng, C.Y. Regulation of drug transporters in the testis by environmental toxicant cadmium, steroids and cytokines. Spermatogenesis 2012, 2, 285–293. [Google Scholar] [CrossRef][Green Version]
- Cheng, C.Y.; Wong, E.W.; Lie, P.P.; Li, M.W.; Su, L.; Siu, E.R.; Yan, H.H.; Mannu, J.; Mathur, P.P.; Bonanomi, M. Environmental toxicants and male reproductive function. Spermatogenesis 2011, 1, 2–13. [Google Scholar] [CrossRef]
- Xiao, X.; Mruk, D.D.; Tang, E.I.; Wong, C.K.; Lee, W.M.; John, C.M.; Turek, P.J.; Silvestrini, B.; Cheng, C.Y. Environmental toxicants perturb human Sertoli cell adhesive function via changes in F-actin organization mediated by actin regulatory proteins. Hum. Reprod. 2014, 29, 1279–1291. [Google Scholar] [CrossRef]
- Mruk, D.D.; Cheng, C.Y. Environmental contaminants: Is male reproductive health at risk? Spermatogenesis 2011, 1, 283–290. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Oliveira, P.F.; Alves, M.G. Sertoli Cell Metabolism and Spermatogenesis; Springer: Cham, Switzerland, 2015; p. 98. [Google Scholar]
- Rato, L.; Alves, M.G.; Socorro, S.; Duarte, A.I.; Cavaco, J.E.; Oliveira, P.F. Metabolic regulation is important for spermatogenesis. Nat. Rev. Urol. 2012, 9, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Meroni, S.; Riera, M.; Pellizzari, E.; Cigorraga, S. Regulation of rat Sertoli cell function by FSH: Possible role of phosphatidylinositol 3-kinase/protein kinase B pathway. J. Endocrinol. 2002, 174, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Chehtane, M.; Khaled, A.R. Interleukin-7 mediates glucose utilization in lymphocytes through transcriptional regulation of the hexokinase II gene. Am. J. Physiol. Cell Physiol. 2010, 298, C1560–C1571. [Google Scholar] [CrossRef] [PubMed]
- Rato, L.; Alves, M.G.; Socorro, S.; Carvalho, R.A.; Cavaco, J.E.; Oliveira, P.F. Metabolic modulation induced by oestradiol and DHT in immature rat Sertoli cells cultured in vitro. Biosci. Rep. 2012, 32, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Majors, B.S.; Betenbaugh, M.J.; Chiang, G.G. Links between metabolism and apoptosis in mammalian cells: Applications for anti-apoptosis engineering. Metab. Eng. 2007, 9, 317–326. [Google Scholar] [CrossRef]
- Yamada, S.; Kotake, Y.; Sekino, Y.; Kanda, Y. AMP-activated protein kinase-mediated glucose transport as a novel target of tributyltin in human embryonic carcinoma cells. Metallomics 2013, 5, 484–491. [Google Scholar] [CrossRef]
- Galardo, M.N.; Riera, M.F.; Pellizzari, E.H.; Cigorraga, S.B.; Meroni, S.B. The AMP-activated protein kinase activator, 5-aminoimidazole-4-carboxamide-1-bD-ribonucleoside, regulates lactate production in rat Sertoli cells. J. Mol. Endocrinol. 2007, 39, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Skakkebaek, N.; Rajpert-De Meyts, E.; Main, K. Testicular dysgenesis syndrome: An increasingly common developmental disorder with environmental aspects: Opinion. Hum. Reprod. 2001, 16, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Weston, C.R.; Davis, R.J. The JNK signal transduction pathway. Curr. Opin. Cell Biol. 2007, 19, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Urushibara, N.; Mitsuhashi, S.; Sasaki, T.; Kasai, H.; Yoshimizu, M.; Fujita, H.; Oda, A. JNK and p38 MAPK are independently involved in tributyltin-mediated cell death in rainbow trout (Oncorhynchus mykiss) RTG-2 cells. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2009, 149, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Kim, J.H.; Han, J.H.; Yoon, Y.D. Inhibitory effect of tributyltin on expression of steroidogenic enzymes in mouse testis. Int. J. Toxicol. 2008, 27, 175–182. [Google Scholar] [CrossRef]
- Fielden, M.R.; Halgren, R.G.; Fong, C.J.; Staub, C.; Johnson, L.; Chou, K.; Zacharewski, T.R. Gestational and lactational exposure of male mice to diethylstilbestrol causes long-term effects on the testis, sperm fertilizing ability in vitro, and testicular gene expression. Endocrinology 2002, 143, 3044–3059. [Google Scholar] [CrossRef]
- Koeberle, A.; Shindou, H.; Harayama, T.; Yuki, K.; Shimizu, T. Polyunsaturated fatty acids are incorporated into maturating male mouse germ cells by lysophosphatidic acid acyltransferase 3. FASEB J. 2012, 26, 169–180. [Google Scholar] [CrossRef]
- Sæther, T.; Tran, T.N.; Rootwelt, H.; Christophersen, B.O.; Haugen, T.B. Expression and regulation of Δ5-desaturase, Δ6-desaturase, stearoyl-coenzyme A (CoA) desaturase 1, and stearoyl-CoA desaturase 2 in rat testis. Biol. Reprod. 2003, 69, 117–124. [Google Scholar] [CrossRef]
- Rato, L.; Alves, M.; Dias, T.; Lopes, G.; Cavaco, J.; Socorro, S.; Oliveira, P. High-energy diets may induce a pre-diabetic state altering testicular glycolytic metabolic profile and male reproductive parameters. Andrology 2013, 1, 495–504. [Google Scholar] [CrossRef]
- Sasaki, H.; Matsui, Y. Epigenetic events in mammalian germ-cell development: Reprogramming and beyond. Nat. Rev. Genet. 2008, 9, 129–140. [Google Scholar] [CrossRef]
- Moore, D.S. The Developing Genome: An Introduction to Behavioral Epigenetics; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Skinner, M.K.; Manikkam, M.; Guerrero-Bosagna, C. Epigenetic transgenerational actions of environmental factors in disease etiology. Trends Endocrinol. Metab. 2010, 21, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Anway, M.D.; Cupp, A.S.; Uzumcu, M.; Skinner, M.K. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 2005, 308, 1466–1469. [Google Scholar] [CrossRef] [PubMed]
- Piprek, R. Genetic mechanisms underlying male sex determination in mammals. J. Appl. Genet. 2009, 50, 347–360. [Google Scholar] [CrossRef]
- Cupp, A.S.; Uzumcu, M.; Suzuki, H.; Dirks, K.; Phillips, B.; Skinner, M.K. Effect of transient embryonic in vivo exposure to the endocrine disruptor methoxychlor on embryonic and postnatal testis development. J. Androl. 2003, 24, 736–745. [Google Scholar] [CrossRef]
- Uzumcu, M.; Suzuki, H.; Skinner, M.K. Effect of the anti-androgenic endocrine disruptor vinclozolin on embryonic testis cord formation and postnatal testis development and function. Reprod. Toxicol. 2004, 18, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, M.; Okano, M.; Hata, K.; Sado, T.; Tsujimoto, N.; Li, E.; Sasaki, H. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 2004, 429, 900–903. [Google Scholar] [CrossRef]
- Dolinoy, D.C.; Huang, D.; Jirtle, R.L. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc. Natl. Acad. Sci. USA 2007, 104, 13056–13061. [Google Scholar] [CrossRef]
- Doyle, T.J.; Bowman, J.L.; Windell, V.L.; McLean, D.J.; Kim, K.H. Transgenerational effects of di-(2-ethylhexyl) phthalate on testicular germ cell associations and spermatogonial stem cells in mice. Biol. Reprod. 2013, 88, 112. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, E.; Leiter, E. Genital abnormalities and abnormal semen analyses in male patients exposed to diethylstilbestrol in utero. J. Urol. 1981, 125, 47–50. [Google Scholar] [CrossRef]
- Brouwers, M.; Feitz, W.; Roelofs, L.; Kiemeney, L.; De Gier, R.; Roeleveld, N. Hypospadias: A transgenerational effect of diethylstilbestrol? Hum. Reprod. 2006, 21, 666–669. [Google Scholar] [CrossRef]
- Kalfa, N.; Paris, F.; Soyer-Gobillard, M.-O.; Daures, J.-P.; Sultan, C. Prevalence of hypospadias in grandsons of women exposed to diethylstilbestrol during pregnancy: A multigenerational national cohort study. Fertil. Steril. 2011, 95, 2574–2577. [Google Scholar] [CrossRef] [PubMed]
- Van de Werken, C.; van der Heijden, G.W.; Eleveld, C.; Teeuwssen, M.; Albert, M.; Baarends, W.M.; Laven, J.S.; Peters, A.H.; Baart, E.B. Paternal heterochromatin formation in human embryos is H3K9/HP1 directed and primed by sperm-derived histone modifications. Nat. Commun. 2014, 18, 1–15. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Collotta, M.; Bertazzi, P.; Bollati, V. Epigenetics and pesticides. Toxicology 2013, 307, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Dansky, L.V.; Finnell, R.H. Parental epilepsy, anticonvulsant drugs, and reproductive outcome: Epidemiologic and experimental findings spanning three decades; 2: Human studies. Reprod. Toxicol. 1991, 5, 301–335. [Google Scholar] [CrossRef]
- Tabb, M.M.; Blumberg, B. New modes of action for endocrine-disrupting chemicals. Mol. Endocrinol. 2006, 20, 475–482. [Google Scholar] [CrossRef]
- Lambrot, R.; Xu, C.; Saint-Phar, S.; Chountalos, G.; Cohen, T.; Paquet, M.; Suderman, M.; Hallett, M.; Kimmins, S. Low paternal dietary folate alters the mouse sperm epigenome and is associated with negative pregnancy outcomes. Nat. Commun. 2013, 4, 1–7. [Google Scholar] [CrossRef]
- Terashima, M.; Barbour, S.; Ren, J.; Yu, W.; Han, Y.; Muegge, K. Effect of high fat diet on paternal sperm histone distribution and male offspring liver gene expression. Epigenetics 2015, 10, 861–871. [Google Scholar] [CrossRef]
- Chen, L.-L.; Carmichael, G.G. Decoding the function of nuclear long non-coding RNAs. Curr. Opin. Cell Biol. 2010, 22, 357–364. [Google Scholar] [CrossRef]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef]
- Bernstein, E.; Allis, C.D. RNA meets chromatin. Genes. Dev. 2005, 19, 1635–1655. [Google Scholar] [CrossRef]
- Whitehead, J.; Pandey, G.K.; Kanduri, C. Regulation of the mammalian epigenome by long noncoding RNAs. Biochim. Biophys. Acta 2009, 1790, 936–947. [Google Scholar] [CrossRef]
- Jolly, C.; Lakhotia, S.C. Human sat III and Drosophila hsrω transcripts: A common paradigm for regulation of nuclear RNA processing in stressed cells. Nucleic Acids Res. 2006, 34, 5508–5514. [Google Scholar] [CrossRef] [PubMed]
- Luke, B.; Lingner, J. TERRA: Telomeric repeat-containing RNA. EMBO J. 2009, 28, 2503–2510. [Google Scholar] [CrossRef] [PubMed]
- Pezer, Ž.; Brajković, J.; Feliciello, I.; Ugarković, Đ. Transcription of satellite DNAs in insects. In Long Non-Coding RNAs; Springer: Berlin/Heidelberg, Germany, 2011; pp. 161–178. [Google Scholar]
- He, X.; Chen, X.; Zhang, X.; Duan, X.; Pan, T.; Hu, Q.; Zhang, Y.; Zhong, F.; Liu, J.; Zhang, H.; et al. An Lnc RNA (GAS5)/SnoRNA-derived piRNA induces activation of TRAIL gene by site-specifically recruiting MLL/COMPASS-like complexes. Nucleic. Acids. Res. 2015, 43, 3712–3725. [Google Scholar] [CrossRef] [PubMed]
- European Environment Agency. Pesticides Sales. Available online: https://www.eea.europa.eu/airs/2017/environment-and-health/pesticides-sales (accessed on 24 October 2021).
- Alavanja, M.C. Introduction: Pesticides use and exposure, extensive worldwide. Rev. Environ. Health 2009, 24, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Yu, G.; Zhang, Y.; Liu, X.; Du, C.; Wang, L.; Li, Z.; Wang, C. Heavy Metal Level in Human Semen with Different Fertility: A Meta-Analysis. Biol. Trace Elem. Res. 2017, 176, 27–36. [Google Scholar] [CrossRef]
- Younglai, E.V.; Foster, W.G.; Hughes, E.G.; Trim, K.; Jarrell, J.F. Levels of environmental contaminants in human follicular fluid, serum, and seminal plasma of couples undergoing in vitro fertilization. Arch. Environ. Contam. Toxicol. 2002, 43, 121–126. [Google Scholar] [CrossRef]
- Vitku, J.; Sosvorova, L.; Chlupacova, T.; Hampl, R.; Hill, M.; Sobotka, V.; Heracek, J.; Bicikova, M.; Starka, L. Differences in bisphenol A and estrogen levels in the plasma and seminal plasma of men with different degrees of infertility. Physiol. Res. 2015, 64, S303–S311. [Google Scholar] [CrossRef]
- Chang, W.-H.; Wu, M.-H.; Pan, H.-A.; Guo, P.-L.; Lee, C.-C. Semen quality and insulin-like factor 3: Associations with urinary and seminal levels of phthalate metabolites in adult males. Chemosphere 2017, 173, 594–602. [Google Scholar] [CrossRef]
- La Rocca, C.; Tait, S.; Guerranti, C.; Busani, L.; Ciardo, F.; Bergamasco, B.; Perra, G.; Mancini, F.R.; Marci, R.; Bordi, G.; et al. Exposure to Endocrine Disruptors and Nuclear Receptors Gene Expression in Infertile and Fertile Men from Italian Areas with Different Environmental Features. Int. J. Environ. Res. Public Health 2015, 12, 12426–12445. [Google Scholar] [CrossRef]
- Foldesy, R.G.; Bedford, J.M. Biology of the scrotum. I. Temperature and androgen as determinants of the sperm storage capacity of the rat cauda epididymidis. Biol. Reprod. 1982, 26, 673–682. [Google Scholar] [CrossRef]
- Klinefelter, G.R.; Suarez, J.D. Toxicant-induced acceleration of epididymal sperm transit: Androgen-dependent proteins may be involved. Reprod. Toxicol. 1997, 11, 511–519. [Google Scholar] [CrossRef]
- Lubicz-Nawrocki, C.M. Effects of castration and testosterone replacement on the number of spermatozoa in the cauda epididymidis of hamsters. J. Reprod. Fertil. 1974, 39, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Din-Udom, A.; Sujarit, S.; Pholpramool, C. Short-term effect of androgen deprivation on intraluminal pressure and contractility of the rat epididymis. J. Reprod. Fertil. 1985, 73, 405–410. [Google Scholar] [CrossRef]
- Yan, F.; Chen, Y.; Zuo, Z.; Chen, Y.; Yang, Z.; Wang, C. Effects of tributyltin on epididymal function and sperm maturation in mice. Environ. Toxicol. Pharmacol. 2009, 28, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.; Ito, C.; Natsume, Y.; Sugitani, Y.; Yamanaka, H.; Kuretake, S.; Yanagida, K.; Sato, A.; Toshimori, K.; Noda, T. Lack of acrosome formation in mice lacking a Golgi protein, GOPC. Proc. Natl. Acad. Sci. USA 2002, 99, 11211–11216. [Google Scholar] [CrossRef] [PubMed]
- Rato, L.; Sousa, A.C.; Oliveira, P.F.; Alves, M.G.; Mathur, P.P. Sperm Maturation as a Possible Target of Obesogens. Immunol. Endocr. Metab. Agents Med. Chem. Former. Curr. Med. Chem. Immunol. Endocr. Metab. Agents 2017, 17, 15–31. [Google Scholar] [CrossRef]
- Sinowatz, F.; Wrobel, K.H.; Sinowatz, S.; Kugler, P. Ultrastructural evidence for phagocytosis of spermatozoa in the bovine rete testis and testicular straight tubules. J. Reprod. Fertil. 1979, 57, 1–4. [Google Scholar] [CrossRef]
- Wysocki, P.; Strzezek, J. Isolation and biochemical characteristics of a molecular form of epididymal acid phosphatase of boar seminal plasma. Theriogenology 2006, 66, 2152–2159. [Google Scholar] [CrossRef]
- Kumi-Diaka, J.; Townsend, J. Effects of Genistein Isoflavone (4′,5′,7-Trihydroxyisoflavone) and Dexamethasone on Functional Characteristics of Spermatozoa. J. Med. Food 2001, 4, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.J.; Jung, E.Y.; Baek, I.J.; Lee, Y.B.; Lee, J.Y.; Nam, S.Y. Exposure to genistein does not adversely affect the reproductive system in adult male mice adapted to a soy-based commercial diet. J. Vet. Sci. 2004, 5, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.; Afeiche, M.; Gaskins, A.; Williams, P.; Petrozza, J.; Tanrikut, C.; Hauser, R.; Chavarro, J. Fruit and vegetable intake and their pesticide residues in relation to semen quality among men from a fertility clinic. Hum. Reprod. 2015, 30, 1342–1351. [Google Scholar] [CrossRef] [PubMed]
- Morán-Martínez, J.; Carranza-Rosales, P.; Morales-Vallarta, M.; Heredia-Rojas, J.A.; Bassol-Mayagoitia, S.; Betancourt-Martínez, N.D.; Cerda-Flores, R.M. Chronic environmental exposure to lead affects semen quality in a Mexican men population. Iran. J. Reprod. Med. 2013, 11, 267–274. [Google Scholar]
- Wang, C.; Yang, L.; Wang, S.; Zhang, Z.; Yu, Y.; Wang, M.; Cromie, M.; Gao, W.; Wang, S.L. The classic EDCs, phthalate esters and organochlorines, in relation to abnormal sperm quality: A systematic review with meta-analysis. Sci. Rep. 2016, 6, 19982. [Google Scholar] [CrossRef] [PubMed]
- Lassen, T.H.; Frederiksen, H.; Jensen, T.K.; Petersen, J.H.; Joensen, U.N.; Main, K.M.; Skakkebaek, N.E.; Juul, A.; Jorgensen, N.; Andersson, A.M. Urinary bisphenol A levels in young men: Association with reproductive hormones and semen quality. Environ. Health Perspect. 2014, 122, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Vilela, J.; Hartmann, A.; Silva, E.F.; Cardoso, T.; Corcini, C.D.; Varela-Junior, A.S.; Martinez, P.E.; Colares, E.P. Sperm impairments in adult vesper mice (Calomys laucha) caused by in utero exposure to bisphenol A. Andrologia 2014, 46, 971–978. [Google Scholar] [CrossRef]
- Rahman, M.S.; Kwon, W.S.; Lee, J.S.; Yoon, S.J.; Ryu, B.Y.; Pang, M.G. Bisphenol-A affects male fertility via fertility-related proteins in spermatozoa. Sci. Rep. 2015, 5, 9169. [Google Scholar] [CrossRef]
- Miki, K.; Qu, W.; Goulding, E.H.; Willis, W.D.; Bunch, D.O.; Strader, L.F.; Perreault, S.D.; Eddy, E.M.; O’Brien, D.A. Glyceraldehyde 3-phosphate dehydrogenase-S, a sperm-specific glycolytic enzyme, is required for sperm motility and male fertility. Proc. Natl. Acad. Sci. USA. 2004, 101, 16501–16506. [Google Scholar] [CrossRef] [PubMed]
- Nakada, K.; Sato, A.; Yoshida, K.; Morita, T.; Tanaka, H.; Inoue, S.; Yonekawa, H.; Hayashi, J. Mitochondria-related male infertility. Proc. Natl. Acad. Sci. USA 2006, 103, 15148–15153. [Google Scholar] [CrossRef]
- Sutovsky, P.; Manandhar, G. Mammalian spermatogenesis and sperm structure: Anatomical and compartmental analysis. Sperm Cell Prod. Maturation Fertil. Regen. 2006, 1–30. [Google Scholar]
- Ramalho-Santos, J.; Varum, S.; Amaral, S.; Mota, P.C.; Sousa, A.P.; Amaral, A. Mitochondrial functionality in reproduction: From gonads and gametes to embryos and embryonic stem cells. Hum. Reprod. Update 2009, 15, 553–572. [Google Scholar] [CrossRef] [PubMed]
- Barroso, G.; Taylor, S.; Morshedi, M.; Manzur, F.; Gavino, F.; Oehninger, S. Mitochondrial membrane potential integrity and plasma membrane translocation of phosphatidylserine as early apoptotic markers: A comparison of two different sperm subpopulations. Fertil. Steril. 2006, 85, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Paoli, D.; Gallo, M.; Rizzo, F.; Baldi, E.; Francavilla, S.; Lenzi, A.; Lombardo, F.; Gandini, L. Mitochondrial membrane potential profile and its correlation with increasing sperm motility. Fertil. Steril. 2011, 95, 2315–2319. [Google Scholar] [CrossRef]
- Troiano, L.; Granata, A.R.; Cossarizza, A.; Kalashnikova, G.; Bianchi, R.; Pini, G.; Tropea, F.; Carani, C.; Franceschi, C. Mitochondrial membrane potential and DNA stainability in human sperm cells: A flow cytometry analysis with implications for male infertility. Exp. Cell Res. 1998, 241, 384–393. [Google Scholar] [CrossRef]
- Wang, M.-J.; Ou, J.-X.; Chen, G.-W.; Wu, J.-P.; Shi, H.-J.; O, W.-S.; Martin-DeLeon, P.A.; Chen, H. Does Prohibitin Expression Regulate Sperm Mitochondrial Membrane Potential, Sperm Motility, and Male Fertility? Mary Ann Liebert, Inc.: Larchmont, NY, USA, 2012. [Google Scholar]
- Gallon, F.; Marchetti, C.; Jouy, N.; Marchetti, P. The functionality of mitochondria differentiates human spermatozoa with high and low fertilizing capability. Fertil. Steril. 2006, 86, 1526–1530. [Google Scholar] [CrossRef]
- Marchetti, C.; Obert, G.; Deffosez, A.; Formstecher, P.; Marchetti, P. Study of mitochondrial membrane potential, reactive oxygen species, DNA fragmentation and cell viability by flow cytometry in human sperm. Hum. Reprod. 2002, 17, 1257–1265. [Google Scholar] [CrossRef]
- Mayorga-Torres, B.J.; Cardona-Maya, W.; Cadavid, A.; Camargo, M. Evaluation of sperm functional parameters in normozoospermic infertile individuals. Actas. Urol. Esp. 2013, 37, 221–227. [Google Scholar] [CrossRef]
- Wang, X.; Sharma, R.K.; Gupta, A.; George, V.; Thomas, A.J.; Falcone, T.; Agarwal, A. Alterations in mitochondria membrane potential and oxidative stress in infertile men: A prospective observational study. Fertil. Steril. 2003, 80, 844–850. [Google Scholar] [CrossRef]
- Caito, S.W.; Aschner, M. Mitochondrial Redox Dysfunction and Environmental Exposures. Antioxid Redox Signal 2015, 23, 578–595. [Google Scholar] [CrossRef]
- Skibinska, I.; Jendraszak, M.; Borysiak, K.; Jedrzejczak, P.; Kotwicka, M. 17beta-estradiol and xenoestrogens reveal synergistic effect on mitochondria of human sperm. Ginekol. Pol. 2016, 87, 360–366. [Google Scholar] [CrossRef]
- Aly, H.A. Aroclor 1254 induced oxidative stress and mitochondria mediated apoptosis in adult rat sperm in vitro. Environ. Toxicol. Pharmacol. 2013, 36, 274–283. [Google Scholar] [CrossRef]
- Grizard, G.; Ouchchane, L.; Roddier, H.; Artonne, C.; Sion, B.; Vasson, M.P.; Janny, L. In vitro alachlor effects on reactive oxygen species generation, motility patterns and apoptosis markers in human spermatozoa. Reprod. Toxicol. 2007, 23, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Pant, N.; Shukla, M.; Kumar Patel, D.; Shukla, Y.; Mathur, N.; Kumar Gupta, Y.; Saxena, D.K. Correlation of phthalate exposures with semen quality. Toxicol. Appl. Pharmacol. 2008, 231, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, D.; Vanage, G. Mutagenic effect of Bisphenol A on adult rat male germ cells and their fertility. Reprod. Toxicol. 2013, 40, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Hulak, M.; Gazo, I.; Shaliutina, A.; Linhartova, P. In vitro effects of bisphenol A on the quality parameters, oxidative stress, DNA integrity and adenosine triphosphate content in sterlet (Acipenser ruthenus) spermatozoa. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2013, 158, 64–71. [Google Scholar] [CrossRef]
- Chitra, K.C.; Sujatha, R.; Latchoumycandane, C.; Mathur, P.P. Effect of lindane on antioxidant enzymes in epididymis and epididymal sperm of adult rats. Asian. J. Androl. 2001, 3, 205–208. [Google Scholar]
- Latchoumycandane, C.; Chitra, C.; Mathur, P. Induction of oxidative stress in rat epididymal sperm after exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Arch. Toxicol. 2002, 76, 113–118. [Google Scholar] [CrossRef]
- Chitra, K.C.; Latchoumycandane, C.; Mathur, P.P. Induction of oxidative stress by bisphenol A in the epididymal sperm of rats. Toxicology 2003, 185, 119–127. [Google Scholar] [CrossRef]
- Palleschi, S.; Rossi, B.; Diana, L.; Silvestroni, L. Di (2-ethylhexyl) phthalate stimulates Ca2+ entry, chemotaxis and ROS production in human granulocytes. Toxicol. Lett. 2009, 187, 52–57. [Google Scholar] [CrossRef]
- Drbohlav, P.; Jirsová, S.; Masata, J.; Jech, L.; Bencko, V.; Omelka, M.; Zvárová, J. Relationship between the levels of toxic polychlorinated biphenyls in blood and follicular fluid of sterile women. Ceska Gynekol. 2005, 70, 377–383. [Google Scholar]
- Tsutsumi, O.; Uechi, H.; Sone, H.; Yonemoto, J.; Takai, Y.; Momoeda, M.; Tohyama, C.; Hashimoto, S.; Morita, M.; Taketani, Y. Presence of dioxins in human follicular fluid: Their possible stage-specific action on the development of preimplantation mouse embryos. Biochem. Biophys. Res. Commun. 1998, 250, 498–501. [Google Scholar] [CrossRef]
- Sofikitis, N.V.; Miyagawa, I.; Zavos, P.M.; Toda, T.; Iino, A.; Terakawa, N. Confocal scanning laser microscopy of morphometric human sperm parameters: Correlation with acrosin profiles and fertilizing capacity. Fertil. Steril. 1994, 62, 376–386. [Google Scholar] [CrossRef]
- Langlois, M.R.; Oorlynck, L.; Vandekerckhove, F.; Criel, A.; Bernard, D.; Blaton, V. Discrepancy between sperm acrosin activity and sperm morphology: Significance for fertilization in vitro. Clin. Chim. Acta 2005, 351, 121–129. [Google Scholar] [CrossRef]
- Reichart, M.; Lederman, H.; Har-Even, D.; Kedem, P.; Bartoov, B. Human sperm acrosin activity with relation to semen parameters and acrosomal ultrastructure. Andrologia 1993, 25, 59–66. [Google Scholar] [CrossRef]
- Breitbart, H.; Cohen, G.; Rubinstein, S. Role of actin cytoskeleton in mammalian sperm capacitation and the acrosome reaction. Reproduction 2005, 129, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Aitken, R.J. Sperm function tests and fertility. Int. J. Androl. 2006, 29, 69–75. [Google Scholar] [CrossRef]
- Rijsselaere, T.; Van Soom, A.; Tanghe, S.; Coryn, M.; Maes, D.; de Kruif, A. New techniques for the assessment of canine semen quality: A review. Theriogenology 2005, 64, 706–719. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Martinez, H. Laboratory semen assessment and prediction of fertility: Still utopia? Reprod. Domest. Anim. 2003, 38, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Knez, J.; Kranvogl, R.; Breznik, B.P.; Vončina, E.; Vlaisavljević, V. Are urinary bisphenol A levels in men related to semen quality and embryo development after medically assisted reproduction? Fertil. Steril. 2014, 101, 215–221.e215. [Google Scholar] [CrossRef] [PubMed]
- Li, D.-K.; Zhou, Z.; Miao, M.; He, Y.; Wang, J.; Ferber, J.; Herrinton, L.J.; Gao, E.; Yuan, W. Urine bisphenol-A (BPA) level in relation to semen quality. Fertil. Steril. 2011, 95, 625–630.e624. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Kwon, W.; Lee, J.; Yoon, S.; Ryu, B.; Pang, M.; Rahman, M.; Kwon, W.; Lee, J.; Yoon, S.; et al. Bisphenol-A affects male fertility via fertility-related proteins in spermatozoa. Sci. Rep. 2015, 5, 9169. [Google Scholar] [CrossRef]
- Axelsson, J.; Rylander, L.; Rignell-Hydbom, A.; Jönsson, B.A.; Lindh, C.H.; Giwercman, A. Phthalate exposure and reproductive parameters in young men from the general Swedish population. Environ. Int. 2015, 85, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Duty, S.M.; Silva, M.J.; Barr, D.B.; Brock, J.W.; Ryan, L.; Chen, Z.; Herrick, R.F.; Christiani, D.C.; Hauser, R. Phthalate exposure and human semen parameters. Epidemiology 2003, 269–277. [Google Scholar] [CrossRef]
- Lenters, V.; Portengen, L.; Smit, L.A.; Jönsson, B.A.; Giwercman, A.; Rylander, L.; Lindh, C.H.; Spanò, M.; Pedersen, H.S.; Ludwicki, J.K. Phthalates, perfluoroalkyl acids, metals and organochlorines and reproductive function: A multipollutant assessment in Greenlandic, Polish and Ukrainian men. Occup. Environ. Med. 2015, 72, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Hauser, R.; Altshul, L.; Chen, Z.; Ryan, L.; Overstreet, J.; Schiff, I.; Christiani, D.C. Environmental organochlorines and semen quality: Results of a pilot study. Environ. Health Perspect. 2002, 110, 229. [Google Scholar] [CrossRef] [PubMed]
- Hauser, R.; Chen, Z.; Pothier, L.; Ryan, L.; Altshul, L. The relationship between human semen parameters and environmental exposure to polychlorinated biphenyls and p, p’-DDE. Environ. Health Perspect. 2003, 111, 1505. [Google Scholar] [CrossRef]
- Hauser, R.; Williams, P.; Altshul, L.; Calafat, A.M. Evidence of interaction between polychlorinated biphenyls and phthalates in relation to human sperm motility. Environ. Health Perspect. 2005, 113, 425. [Google Scholar] [CrossRef]
- Richthoff, J.; Rylander, L.; Jönsson, B.A.; Akesson, H.; Hagmar, L.; Nilsson-Ehle, P.; Stridsberg, M.; Giwercman, A. Serum levels of 2, 2′, 4, 4′, 5, 5′-hexachlorobiphenyl (CB-153) in relation to markers of reproductive function in young males from the general Swedish population. Environ. Health Perspect. 2003, 111, 409. [Google Scholar] [CrossRef]
- Toft, G.; Rignell-Hydbom, A.; Tyrkiel, E.; Shvets, M.; Giwercman, A.; Lindh, C.H.; Pedersen, H.S.; Ludwicki, J.K.; Lesovoy, V.; Hagmar, L. Semen quality and exposure to persistent organochlorine pollutants. Epidemiology 2006, 450–458. [Google Scholar] [CrossRef]
- Aneck-Hahn, N.; Schulenburg, G.; Bornman, M.; Farias, P.; De Jager, C.; Aneck-Hahn, N.; Schulenburg, G.; Bornman, M.; Farias, P.; De Jager, C. Impaired semen quality associated with environmental DDT exposure in young men living in a malaria area in the Limpopo Province, South Africa. J. Androl. 2007, 28, 423–434. [Google Scholar] [CrossRef]
- De Jager, C.; Farias, P.; Barraza-Villarreal, A.; Avila, M.H.; Ayotte, P.; Dewailly, E.; Dombrowski, C.; Rousseau, F.; Sanchez, V.D.; Bailey, J.L. Reduced seminal parameters associated with environmental DDT exposure and p, p′-DDE concentrations in men in Chiapas, Mexico: A cross-sectional study. J. Androl. 2006, 27, 16–27. [Google Scholar] [CrossRef]
- Pant, N.; Pant, A.; Chaturvedi, P.; Shukla, M.; Mathur, N.; Gupta, Y.; Saxena, D.; Pant, N.; Pant, A.; Chaturvedi, P.; et al. Semen quality of environmentally exposed human population: The toxicological consequence. Environ. Sci. Pollut. Res. Int. 2013, 20, 8274–8281. [Google Scholar] [CrossRef]
- Tavares, R.; Amaral, S.; Paiva, C.; Baptista, M.; Ramalho-Santos, J.; Tavares, R.; Amaral, S.; Paiva, C.; Baptista, M.; Ramalho-Santos, J. In vitro exposure to the organochlorine p,p’-DDE affects functional human sperm parameters. Chemosphere 2015, 120, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-X.; You, L.; Zeng, Q.; Sun, Y.; Huang, Y.-H.; Wang, C.; Wang, P.; Cao, W.-C.; Yang, P.; Li, Y.-F. Phthalate exposure and human semen quality: Results from an infertility clinic in China. Environ. Res. 2015, 142, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Joensen, U.N.; Bossi, R.; Leffers, H.; Jensen, A.A.; Skakkebæk, N.E.; Jørgensen, N. Do perfluoroalkyl compounds impair human semen quality? Environ. Health Perspect. 2009, 117, 923. [Google Scholar] [CrossRef] [PubMed]
- Vested, A.; Ramlau-Hansen, C.H.; Olsen, S.F.; Bonde, J.P.; Kristensen, S.L.; Halldorsson, T.I.; Becher, G.; Haug, L.S.; Ernst, E.H.; Toft, G. Associations of in utero exposure to perfluorinated alkyl acids with human semen quality and reproductive hormones in adult men. Environ. Health Perspect. 2013, 121, 453. [Google Scholar] [CrossRef]
- Dallinga, J.W.; Moonen, E.J.; Dumoulin, J.C.; Evers, J.L.; Geraedts, J.P.; Kleinjans, J.C. Decreased human semen quality and organochlorine compounds in blood. Hum. Reprod. 2002, 17, 1973–1979. [Google Scholar] [CrossRef]
- Padungtod, C.; Savitz, D.A.; Overstreet, J.W.; Christiani, D.C.; Ryan, L.M.; Xu, X. Occupational pesticide exposure and semen quality among Chinese workers. J. Occup. Environ. Med. 2000, 42, 982–992. [Google Scholar] [CrossRef]
- Pant, N.; Kumar, G.; Upadhyay, A.; Patel, D.; Gupta, Y.; Chaturvedi, P. Reproductive toxicity of lead, cadmium, and phthalate exposure in men. Environ. Sci. Pollut. Res. 2014, 21, 11066–11074. [Google Scholar] [CrossRef] [PubMed]
- Tavares, R.; Mansell, S.; Barratt, C.; Wilson, S.; Publicover, S.; RamalhoSantos, J.; Tavares, R.; Mansell, S.; Barratt, C.; Wilson, S.; et al. p’-DDE activates CatSper and compromises human sperm function at environmentally relevant concentrations. Hum. Reprod. 2013, 28, 3167–3177. [Google Scholar] [CrossRef] [PubMed]
- Pant, N.; Pant, A.; Shukla, M.; Mathur, N.; Gupta, Y.; Saxena, D.; Pant, N.; Pant, A.; Shukla, M.; Mathur, N.; et al. Environmental and experimental exposure of phthalate esters: The toxicological consequence on human sperm. Hum. Exp. Toxicol. 2011, 30, 507–514. [Google Scholar] [CrossRef]
- Russo, A.; Troncoso, N.; Sanchez, F.; Garbarino, J.; Vanella, A.; Russo, A.; Troncoso, N.; Sanchez, F.; Garbarino, J.; Vanella, A. Propolis protects human spermatozoa from DNA damage caused by benzo[a]pyrene and exogenous reactive oxygen species. Life Sci. 2006, 78, 1401–1406. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, D.; Nandi, P.; Varghese, A.; Gutgutia, R.; Banerjee, S.; Bhattacharyya, A.; Mukhopadhyay, D.; Nandi, P.; Varghese, A.; Gutgutia, R.; et al. The in vitro effect of benzo[a]pyrene on human sperm hyperactivation and acrosome reaction. Fertil. Steril. 2010, 94, 595–598. [Google Scholar] [CrossRef]
- Fraser, L.; Beyret, E.; Milligan, S.; Adeoya-Osiguwa, S.; Fraser, L.; Beyret, E.; Milligan, S.; Adeoya-Osiguwa, S. Effects of estrogenic xenobiotics on human and mouse spermatozoa. Hum. Reprod. 2006, 21, 1184–1193. [Google Scholar] [CrossRef]
- Mohamed, E.-S.A.; Park, Y.-J.; Song, W.-H.; Shin, D.-H.; You, Y.-A.; Ryu, B.-Y.; Pang, M.-G. Xenoestrogenic compounds promote capacitation and an acrosome reaction in porcine sperm. Theriogenology 2011, 75, 1161–1169. [Google Scholar] [CrossRef]
- Adeoya-Osiguwa, S.; Markoulaki, S.; Pocock, V.; Milligan, S.; Fraser, L.; Adeoya-Osiguwa, S.; Markoulaki, S.; Pocock, V.; Milligan, S.; Fraser, L. 17β-Estradiol and environmental estrogens significantly affect mammalian sperm function. Hum. Reprod. 2003, 18, 100–107. [Google Scholar] [CrossRef]
- Kholkute, S.; Rodriguez, J.; Dukelow, W.; Kholkute, S.; Rodriguez, J.; Dukelow, W. Effects of polychlorinated biphenyls (PCBs) on in vitro fertilization in the mouse. Reprod. Toxicol. 1994, 8, 69–73. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).