
Chimeric Antigen Receptor T Cell Therapy in Acute Myeloid Leukemia: Trials and Tribulations


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. How Do Immune Cells Work?
3. How Do Cancer Cells Evade the Immune System?
4. What Is a Chimeric Antigen Receptor (CAR) T Cell?
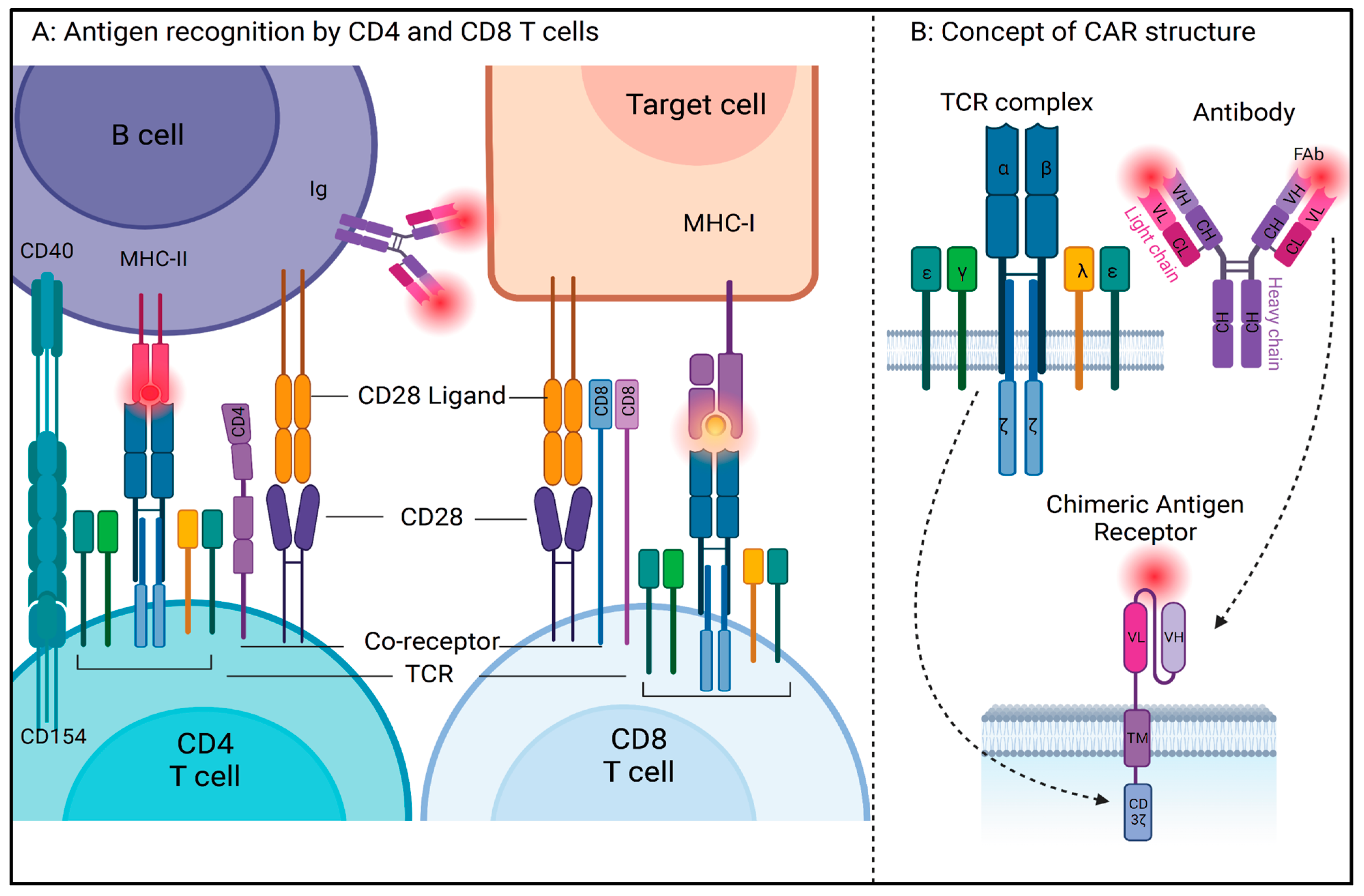
5. What Is the Structure of a CAR?
- Antigen-Binding Domain: The discovery of monoclonal antibodies in the 1970s played an important role in the conceptualization of antigen specificity using complementary determining regions of variable and constant regions of immunoglobulins [36]. In a CAR antigen, the antigen-binding domain is a single-chain variable fragment (scFv), derived from variable light (vL) and heavy chains (vH), and a flexible linker of the antigen-specific monoclonal antibody. The usage of antibody-mediated antigen recognition systems allows the CAR T cell to circumvent MHC restriction;
- Hinge Region: This region connects the antigen-binding domain to the transmembrane domain and affects the overall steric conformation of the CAR to the antigen. These can be of various lengths and are generally derived from the sequence of T cell coreceptors such as CD8, CD28, or immunoglobulins. Shorter extracellular domains increase the potential for CAR T activation, whereas lengthening the CAR antigen diminishes CAR T activation [37];
- Transmembrane Domain: This domain is the region that anchors the CAR to the T cell membrane. Generally, TM domains are derived from amino acid sequences of T cell coreceptors such as CD4, CD8, CD3zeta, and CD28 and are reported to be involved in cytokine release and cell death, apart from their overall stability. The stability of CAR and its expression on the T cell membrane is affected by the transmembrane domain, whereas the hinge domain is critical in the regulation of signaling threshold [38];
- Intracellular Signaling Domain: This domain, also called the costimulatory (CM) domain, transduces the signaling cascade and is involved in T cell activation after successful antigen recognition. This is the domain that contains the necessary ITAMs for downstream signaling cascade activation. Because of its role in cell stimulation, cytokine release, and activation-induced cell death, the intracellular domain has been the most focused of all the regions over the years through multiple generations of CAR antigens.
6. How Did the CAR Design Evolve?
7. What Are the Challenges and Strategies to Overcome Shortcomings in CAR Design?
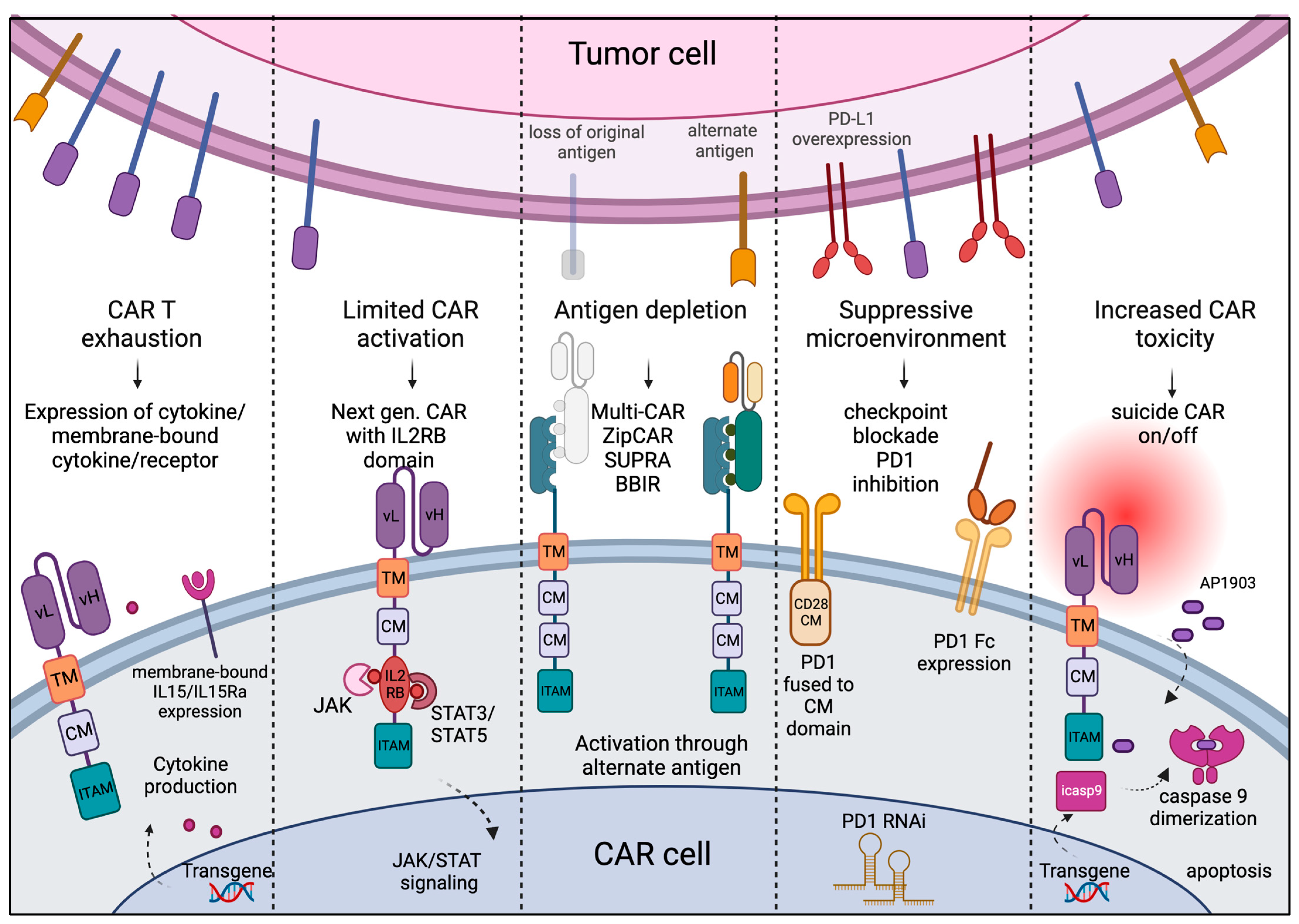
- CAR T exhaustion: Patients receiving CAR treatment may have suboptimal immune composition due to overt tumor burden or previous therapies. CAR T cells can be rapidly exhausted in vivo with limited trans-presented cytokines and a lack of helper signaling from other immune cells such as APCs and NK cells. To overcome this challenge, CAR T cells are engineered with transgenes to produce functional cytokines under the control of the Nuclear Factor for Activated T Cells (NFAT) promoter, which enables CAR T cells to recruit other immune cells. These CARs are also known as TRUCKs, or T cells Redirected for Universal Cytokine-mediated Killing, and express the cytokine transgene encoding either IL-12, IL-18, TNFRSF14 [45], or membrane-bound IL-15 [46]. Other strategies, such as pre-treatment of T cells with IL-7, IL-15, or IL-21 in culture prior to adoptive cell transfer (ACT) or in vivo inhibition of the PI-3/AKT pathway using small molecule inhibitors, are shown to prevent T cell exhaustion [47].
- CAR T-mediated toxicity and fratricide: In most cases, the antigen against which the CAR is developed is not truly a tumor-exclusive antigen and is expressed by the cells of normal tissues as well. This causes T cell-mediated on-target off-tumor toxicity. When the antigen of interest is also expressed with an activated CAR cell, there is a possibility of one CAR cell killing another, resulting in fratricide. A generalized, non-specific immune activation and acute toxicity have also been observed in several CAR T clinical trials. In fourth-generation CAR T cells, transgene coding for proteins that lead to CAR T apoptosis or shut-down in response to a specific ligand is incorporated to prevent CAR-mediated toxicity. Two frequently used suicide or off switches are herpes simplex virus thymidine kinase (HSV-TK), inducible with ganciclovir, and inducible caspase-9 (iCasp9) that dimerizes after the administration of AP1903 [48]. CRISPR-mediated deletion of the CAR target gene from the CAR cells is generally used to overcome the issue of fratricide in CAR cells.
- Suboptimal CAR activation and terminal differentiation: Optimal CAR activation is required for anti-tumor activity and longer persistence of CAR T cells in vivo. In the fifth or next-generation CAR, the intracellular CD3 zeta and CD28 costimulatory signal is accompanied by a truncated IL2 receptor beta chain cytoplasmic tail with STAT3 binding sites, which can recruit docking of transcription factors and activation of JAK/STAT signaling in response to antigen binding. This modification enhanced CAR T persistence and proliferation and prevented their terminal differentiation [49].
- Antigen escape: A common challenge in cancer is that tumor cells can shed or downregulate the expression of antigens, and after the initial clearance of the major tumor population, resistant cells without the target antigen or with an alternate antigen can outgrow. This posed a challenge in the conventional CAR design as they could only recognize one single antigen at a time. One way to overcome this challenge is a multiplexed or universal CAR strategy where the conventional single-chain variable fragment (scFv) is replaced with an adapter-specific recognition domain that binds to an adaptor that is ligated to tumor-specific antigens [50]. A split, universal, and programmable (SUPRA) is a two-component receptor system composed of a universal receptor (zipCAR) expressed on T cells and a tumor-targeting scFv adaptor (zipFv), which, when binds to tumor-specific antigens, can ligate to ZipCAR and mediate efficient tumor killing [51]. Another such strategy is to use biotin-binding immune receptor (BBIR) [52] or Bi-specific T engagers, or CART.BiTE, to target heterogenous antigen-expressing tumors [53].
- Suppressive tumor microenvironment: Tumor cells can express several inhibitory signals, such as PD-L1, that may lead to inhibitory signaling through PD1 on the engineered T cells, resulting in their rapid exhaustion. Several strategies are being used to disrupt the interaction between PD1 and PDL1, such as the expression of (a) PD1 fusion to the CD28 costimulatory domain to convert the inhibitory signal into stimulation, (b) PD1 RNA interference, and (c) the expression of a secreted PD1 Fc fragment that binds to PD-L1 on tumor cells [54,55,56]. Administration of immune checkpoint inhibitors, neutralizing monoclonal antibodies against CTLA4 and PD1, has been shown to prevent the suppression of CAR T cells in many solid tumors [57].
8. CAR Clinical Trials in AML

9. Improving CAR T Cell Therapy One Step at a Time
10. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chiba, S.; Ikushima, H.; Ueki, H.; Yanai, H.; Kimura, Y.; Hangai, S.; Nishio, J.; Negishi, H.; Tamura, T.; Saijo, S.; et al. Recognition of tumor cells by Dectin-1 orchestrates innate immune cells for anti-tumor responses. eLife 2014, 3, e04177. [Google Scholar] [CrossRef]
- Dobosz, P.; Dzieciątkowski, T. The Intriguing History of Cancer Immunotherapy. Front. Immunol. 2019, 10, 2965. [Google Scholar] [CrossRef]
- Mortaz, E.; Tabarsi, P.; Mansouri, D.; Khosravi, A.; Garssen, J.; Velayati, A.; Adcock, I.M. Cancers Related to Immunodeficiencies: Update and Perspectives. Front. Immunol. 2016, 7, 365. [Google Scholar] [CrossRef]
- Oiseth, S.J.; Aziz, M.S. Cancer immunotherapy: A brief review of the history, possibilities, and challenges ahead. J. Cancer Metastasis Treat. 2017, 3, 250. [Google Scholar] [CrossRef]
- Perica, K.; Varela, J.C.; Oelke, M.; Schneck, J.P. Adoptive T Cell Immunotherapy for Cancer. Rambam Maimonides Med. J. 2015, 6, e0004. [Google Scholar] [CrossRef]
- Kantarjian, H.; Kadia, T.; DiNardo, C.; Daver, N.; Borthakur, G.; Jabbour, E.; Garcia-Manero, G.; Konopleva, M.; Ravandi, F. Acute myeloid leukemia: Current progress and future directions. Blood Cancer J. 2021, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Hansrivijit, P.; Gale, R.P.; Barrett, J.; Ciurea, S.O. Cellular therapy for acute myeloid Leukemia–Current status and future prospects. Blood Rev. 2019, 37, 100578. [Google Scholar] [CrossRef]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; Di Nardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef] [PubMed]
- Sekeres, M.A.; Guyatt, G.; Abel, G.; Alibhai, S.; Altman, J.K.; Buckstein, R.; Choe, H.; Desai, P.; Erba, H.; Hourigan, C.S.; et al. American Society of Hematology 2020 guidelines for treating newly diagnosed acute myeloid leukemia in older adults. Blood Adv. 2020, 4, 3528–3549. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Ochando, J.; Mulder, W.J.M.; Madsen, J.C.; Netea, M.G.; Duivenvoorden, R. Trained immunity—basic concepts and contributions to immunopathology. Nat. Rev. Nephrol. 2022, 19, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Ruf, B.; Greten, T.F.; Korangy, F. Innate lymphoid cells and innate-like T cells in cancer—at the crossroads of innate and adaptive immunity. Nat. Rev. Cancer 2023, 23, 351–371. [Google Scholar] [CrossRef]
- De Maria, O.; Cornen, S.; Daëron, M.; Morel, Y.; Medzhitov, R.; Vivier, E. Harnessing innate immunity in cancer therapy. Nature 2019, 574, 45–56, Erratum in 2019, 576, E3. [Google Scholar] [CrossRef]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T Cell Activation. Annu. Rev. Immunol. 2009, 27, 591–619. [Google Scholar] [CrossRef]
- Mariuzza, R.A.; Agnihotri, P.; Orban, J. The structural basis of T-cell receptor (TCR) activation: An enduring enigma. J. Biol. Chem. 2019, 295, 914–925. [Google Scholar] [CrossRef] [PubMed]
- Van Laethem, F.; Tikhonova, A.N.; Singer, A. MHC restriction is imposed on a diverse T cell receptor repertoire by CD4 and CD8 co-receptors during thymic selection. Trends Immunol. 2012, 33, 437–441. [Google Scholar] [CrossRef]
- La Gruta, N.L.; Gras, S.; Daley, S.R.; Thomas, P.G.; Rossjohn, J. Understanding the drivers of MHC restriction of T cell receptors. Nat. Rev. Immunol. 2018, 18, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Adalia, A.; Ramirez-Santiago, G.; Osuna-Pérez, J.; Torres-Torresano, M.; Zorita, V.; Martínez-Riaño, A.; Boccasavia, V.; Borroto, A.; del Hoyo, G.M.; González-Granado, J.M.; et al. Conventional CD4+ T cells present bacterial antigens to induce cytotoxic and memory CD8+ T cell responses. Nat. Commun. 2017, 8, 1591. [Google Scholar] [CrossRef]
- Roche, P.A.; Furuta, K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat. Rev. Immunol. 2015, 15, 203–216. [Google Scholar] [CrossRef]
- Neefjes, J.; Jongsma, M.L.M.; Paul, P.; Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836. [Google Scholar] [CrossRef]
- Blees, A.; Januliene, D.; Hofmann, T.; Koller, N.; Schmidt, C.; Trowitzsch, S.; Moeller, A.; Tampé, R. Structure of the human MHC-I peptide-loading complex. Nature 2017, 551, 525–528. [Google Scholar] [CrossRef]
- Zumerle, S.; Molon, B.; Viola, A. Membrane Rafts in T Cell Activation: A Spotlight on CD28 Costimulation. Front. Immunol. 2017, 8, 1467. [Google Scholar] [CrossRef] [PubMed]
- Riley, J.L.; June, C.H. The CD28 family: A T-cell rheostat for therapeutic control of T-cell activation. Blood 2005, 105, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242, Erratum in Nat. Rev. Immunol. 2013, 13, 542. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Dhatchinamoorthy, K.; Colbert, J.D.; Rock, K.L. Cancer Immune Evasion Through Loss of MHC Class I Antigen Presentation. Front. Immunol. 2021, 12, 636568. [Google Scholar] [CrossRef]
- Beatty, G.L.; Gladney, W.L. Immune Escape Mechanisms as a Guide for Cancer Immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef]
- Wolf, Y.; Anderson, A.C.; Kuchroo, V.K. TIM3 comes of age as an inhibitory receptor. Nat. Rev. Immunol. 2019, 20, 173–185. [Google Scholar] [CrossRef]
- Shi, A.-P.; Tang, X.-Y.; Xiong, Y.-L.; Zheng, K.-F.; Liu, Y.-J.; Shi, X.-G.; Lv, Y.; Jiang, T.; Ma, N.; Zhao, J.-B. Immune Checkpoint LAG3 and Its Ligand FGL1 in Cancer. Front. Immunol. 2022, 12, 785091. [Google Scholar] [CrossRef]
- Kim, S.K.; Cho, S.W. The Evasion Mechanisms of Cancer Immunity and Drug Intervention in the Tumor Microenvironment. Front. Pharmacol. 2022, 13, 868695. [Google Scholar] [CrossRef]
- Musella, M.; Guarracino, A.; Manduca, N.; Galassi, C.; Ruggiero, E.; Potenza, A.; Maccafeo, E.; Manic, G.; Mattiello, L.; Rehim, S.S.A.; et al. Type I IFNs promote cancer cell stemness by triggering the epigenetic regulator KDM1B. Nat. Immunol. 2022, 23, 1379–1392. [Google Scholar] [CrossRef]
- Costoya, J.A.; Arce, V.M. Cancer cells escape the immune system by increasing stemness through epigenetic reprogramming. Cell. Mol. Immunol. 2022, 20, 6–7. [Google Scholar] [CrossRef]
- Styczyński, J. A brief history of CAR-T cells: From laboratory to the bedside. Acta Haematol. Pol. 2020, 51, 2–5. [Google Scholar] [CrossRef]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Alkan, S.S. Monoclonal antibodies: The story of a discovery that revolutionized science and medicine. Nat. Rev. Immunol. 2004, 4, 153–156. [Google Scholar] [CrossRef]
- Xiao, Q.; Zhang, X.; Tu, L.; Cao, J.; Hinrichs, C.S.; Su, X. Size-dependent activation of CAR-T cells. Sci. Immunol. 2022, 7, eabl3995. [Google Scholar] [CrossRef]
- Fujiwara, K.; Tsunei, A.; Kusabuka, H.; Ogaki, E.; Tachibana, M.; Okada, N. Hinge and Transmembrane Domains of Chimeric Antigen Receptor Regulate Receptor Expression and Signaling Threshold. Cells 2020, 9, 1182. [Google Scholar] [CrossRef]
- Sidaway, P. Allogeneic CAR T cells show promise. Nat. Rev. Clin. Oncol. 2022, 19, 748. [Google Scholar] [CrossRef]
- Song, F.; Hu, Y.; Zhang, Y.; Zhang, M.; Yang, T.; Wu, W.; Huang, S.; Xu, H.; Chang, A.H.; Huang, H.; et al. Safety and efficacy of autologous and allogeneic humanized CD19-targeted CAR-T cell therapy for patients with relapsed/refractory B-ALL. J. Immunother. Cancer 2023, 11, e005701. [Google Scholar] [CrossRef]
- Bedoya, D.M.; Dutoit, V.; Migliorini, D. Allogeneic CAR T Cells: An Alternative to Overcome Challenges of CAR T Cell Therapy in Glioblastoma. Front. Immunol. 2021, 12, 640082. [Google Scholar] [CrossRef] [PubMed]
- Pinto, I.S.; Cordeiro, R.A.; Faneca, H. Polymer- and lipid-based gene delivery technology for CAR T cell therapy. J. Control. Release 2023, 353, 196–215. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, S.; Schubert, M.-L.; Wang, L.; He, B.; Neuber, B.; Dreger, P.; Müller-Tidow, C.; Schmitt, M. Chimeric Antigen Receptor (CAR) T Cell Therapy in Acute Myeloid Leukemia (AML). J. Clin. Med. 2019, 8, 200. [Google Scholar] [CrossRef] [PubMed]
- Akhoundi, M.; Mohammadi, M.; Sahraei, S.S.; Sheykhhasan, M.; Fayazi, N. CAR T cell therapy as a promising approach in cancer immunotherapy: Challenges and opportunities. Cell. Oncol. 2021, 44, 495–523. [Google Scholar] [CrossRef]
- Schubert, M.-L.; Hoffmann, J.-M.; Dreger, P.; Müller-Tidow, C.; Schmitt, M. Chimeric antigen receptor transduced T cells: Tuning up for the next generation. Int. J. Cancer 2017, 142, 1738–1747. [Google Scholar] [CrossRef]
- Sallman, D.A.; Elmariah, H.; Sweet, K.; Mishra, A.; Cox, C.A.; Chakaith, M.; Semnani, R.; Shehzad, S.; Anderson, A.; Sabzevari, H.; et al. Phase 1/1b Safety Study of Prgn-3006 Ultracar-T in Patients with Relapsed or Refractory CD33-Positive Acute Myeloid Leukemia and Higher Risk Myelodysplastic Syndromes. Blood 2022, 104 (Suppl. S1), 10313–10315. [Google Scholar] [CrossRef]
- Chen, Y.-J.; Abila, B.; Kamel, Y.M. CAR-T: What Is Next? Cancers 2023, 15, 663. [Google Scholar] [CrossRef]
- Andrea, A.E.; Chiron, A.; Bessoles, S.; Hacein-Bey-Abina, S. Engineering Next-Generation CAR-T Cells for Better Toxicity Management. Int. J. Mol. Sci. 2020, 21, 8620. [Google Scholar] [CrossRef]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.-H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A novel chimeric antigen receptor containing a JAK–STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef]
- Qu, C.; Zhang, H.; Cao, H.; Tang, L.; Mo, H.; Liu, F.; Zhang, L.; Yi, Z.; Long, L.; Yan, L.; et al. Tumor buster—Where will the CAR-T cell therapy ‘missile’ go? Mol. Cancer 2022, 21, 201. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Collins, J.J.; Wong, W.W. Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses. Cell 2018, 173, 1426–1438.e11. [Google Scholar] [CrossRef]
- Urbanska, K.; Lanitis, E.; Poussin, M.; Lynn, R.C.; Gavin, B.P.; Kelderman, S.; Yu, J.; Scholler, N.; Powell, D.J., Jr. A Universal Strategy for Adoptive Immunotherapy of Cancer through Use of a Novel T-cell Antigen Receptor. Cancer Res. 2012, 72, 1844–1852. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson, R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B.; et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 2019, 37, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Roselli, E.; Faramand, R.; Davila, M.L. Insight into next-generation CAR therapeutics: Designing CAR T cells to improve clinical outcomes. J. Clin. Investig. 2021, 131, e142030. [Google Scholar] [CrossRef] [PubMed]
- Tomasik, J.; Jasiński, M.; Basak, G.W. Next generations of CAR-T cells-new therapeutic opportunities in hematology? Front. Immunol. 2022, 13, 1034707. [Google Scholar] [CrossRef] [PubMed]
- Gumber, D.; Wang, L.D. Improving CAR-T immunotherapy: Overcoming the challenges of T cell exhaustion. EBioMedicine 2022, 77, 103941. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.D.; Lai, J.; Slaney, C.Y.; Kallies, A.; Beavis, P.A.; Darcy, P.K. Cellular networks controlling T cell persistence in adoptive cell therapy. Nat. Rev. Immunol. 2021, 21, 769–784. [Google Scholar] [CrossRef]
- Cappell, K.M.; Kochenderfer, J.N. Long-term outcomes following CAR T cell therapy: What we know so far. Nat. Rev. Clin. Oncol. 2023, 20, 359–371. [Google Scholar] [CrossRef]
- Myers, R.M.; Shah, N.N.; Pulsipher, M.A. How I use risk factors for success or failure of CD19 CAR T cells to guide management of children and AYA with B-cell ALL. Blood 2023, 141, 1251–1264. [Google Scholar] [CrossRef]
- Schorr, C.; Perna, F. Targets for chimeric antigen receptor T-cell therapy of acute myeloid leukemia. Front. Immunol. 2022, 13, 1085978. [Google Scholar] [CrossRef]
- Marofi, F.; Rahman, H.S.; Al-Obaidi, Z.M.J.; Jalil, A.T.; Abdelbasset, W.K.; Suksatan, W.; Dorofeev, A.E.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; et al. Novel CAR T therapy is a ray of hope in the treatment of seriously ill AML patients. Stem Cell Res. Ther. 2021, 12, 465. [Google Scholar] [CrossRef] [PubMed]
- Koedam, J.; Wermke, M.; Ehninger, A.; Cartellieri, M.; Ehninger, G. Chimeric antigen receptor T-cell therapy in acute myeloid leukemia. Curr. Opin. Hematol. 2022, 29, 74–83. [Google Scholar] [CrossRef]
- Vishwasrao, P.; Li, G.; Boucher, J.C.; Smith, D.L.; Hui, S.K. Emerging CAR T Cell Strategies for the Treatment of AML. Cancers 2022, 14, 1241. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Ratnapriya, R.; Sosina, O.A.; Starostik, M.R.; Kwicklis, M.; Kapphahn, R.J.; Fritsche, L.G.; Walton, A.; Arvanitis, M.; Gieser, L.; Pietraszkiewicz, A.; et al. Retinal transcriptome and eQTL analyses identify genes associated with age-related macular degeneration. Nat. Genet. 2019, 51, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.X.; Son, E.W.; Yao, R. iDEP: An integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinform. 2018, 19, 1–24. [Google Scholar] [CrossRef]
- Corces, M.R.; Buenrostro, J.D.; Wu, B.; Greenside, P.G.; Chan, S.M.; Koenig, J.L.; Snyder, M.P.; Pritchard, J.K.; Kundaje, A.; Greenleaf, W.J.; et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet. 2016, 48, 1193–1203. [Google Scholar] [CrossRef]
- Liu, J.; Tong, J.; Yang, H. Targeting CD33 for acute myeloid leukemia therapy. BMC Cancer 2022, 22, 24. [Google Scholar] [CrossRef]
- Molica, M.; Perrone, S.; Mazzone, C.; Niscola, P.; Cesini, L.; Abruzzese, E.; de Fabritiis, P. CD33 Expression and Gentuzumab Ozogamicin in Acute Myeloid Leukemia: Two Sides of the Same Coin. Cancers 2021, 13, 3214. [Google Scholar] [CrossRef]
- Krupka, C.; Kufer, P.; Kischel, R.; Zugmaier, G.; Bögeholz, J.; Köhnke, T.; Lichtenegger, F.S.; Schneider, S.; Metzeler, K.; Fiegl, M.; et al. CD33 target validation and sustained depletion of AML blasts in long-term cultures by the bispecific T-cell–engaging antibody AMG 330. Blood 2014, 123, 356–365. [Google Scholar] [CrossRef]
- Tambaro, F.P.; Singh, H.; Jones, E.; Rytting, M.; Mahadeo, K.M.; Thompson, P.; Daver, N.; DiNardo, C.; Kadia, T.; Garcia-Manero, G.; et al. Autologous CD33-CAR-T cells for treatment of relapsed/refractory acute myelogenous leukemia. Leukemia 2021, 35, 3282–3286. [Google Scholar] [CrossRef]
- Albinger, N.; Pfeifer, R.; Nitsche, M.; Mertlitz, S.; Campe, J.; Stein, K.; Kreyenberg, H.; Schubert, R.; Quadflieg, M.; Schneider, D.; et al. Primary CD33-targeting CAR-NK cells for the treatment of acute myeloid leukemia. Blood Cancer J. 2022, 12, 61. [Google Scholar] [CrossRef] [PubMed]
- Sugita, M.; Galetto, R.; Zong, H.; Ewing-Crystal, N.; Trujillo-Alonso, V.; Mencia-Trinchant, N.; Yip, W.; Filipe, S.; Lebuhotel, C.; Gouble, A.; et al. Allogeneic TCRαβ deficient CAR T-cells targeting CD123 in acute myeloid leukemia. Nat. Commun. 2022, 13, 2227. [Google Scholar] [CrossRef]
- Ehninger, A.; on behalf of the Study Alliance Leukemia; Kramer, M.; Röllig, C.; Thiede, C.; Bornhäuser, M.; Von Bonin, M.; Wermke, M.; Feldmann, A.; Bachmann, M.F.; et al. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J. 2014, 4, e218. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Chen, H.; Li, W.; Huang, T.; Zhang, W.; Ling, W.; Lai, P.; Wang, Y.; Geng, S.; et al. C-Type Lectin-Like Molecule-1 as a Biomarker for Diagnosis and Prognosis in Acute Myeloid Leukemia: A Preliminary Study. BioMed Res. Int. 2021, 2021, 6643948. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, H.; Sauer, T.; Shum, T.; Parikh, K.; Mamonkin, M.; Omer, B.; Rouce, R.H.; Lulla, P.; Rooney, C.M.; Gottschalk, S.; et al. Treatment of Acute Myeloid Leukemia with T Cells Expressing Chimeric Antigen Receptors Directed to C-type Lectin-like Molecule 1. Mol. Ther. 2017, 25, 2202–2213. [Google Scholar] [CrossRef]
- Jiang, Y.-P.; Liu, B.Y.; Zheng, Q.; Panuganti, S.; Chen, R.; Zhu, J.; Mishra, M.; Huang, J.; Dao-Pick, T.; Roy, S.; et al. CLT030, a leukemic stem cell–targeting CLL1 antibody-drug conjugate for treatment of acute myeloid leukemia. Blood Adv. 2018, 2, 1738–1749. [Google Scholar] [CrossRef]
- Zheng, B.; Yu, S.-F.; Del Rosario, G.; Leong, S.R.; Lee, G.Y.; Vij, R.; Chiu, C.P.; Liang, W.-C.; Wu, Y.; Chalouni, C.; et al. An Anti–CLL-1 Antibody–Drug Conjugate for the Treatment of Acute Myeloid Leukemia. Clin. Cancer Res. 2019, 25, 1358–1368. [Google Scholar] [CrossRef]
- Kindler, T.; Lipka, D.B.; Fischer, T. FLT3 as a therapeutic target in AML: Still challenging after all these years. Blood 2010, 116, 5089–5102. [Google Scholar] [CrossRef]
- Gebru, M.T.; Wang, H.-G. Therapeutic targeting of FLT3 and associated drug resistance in acute myeloid leukemia. J. Hematol. Oncol. 2020, 13, 155. [Google Scholar] [CrossRef]
- Sommer, C.; Cheng, H.-Y.; Nguyen, D.; Dettling, D.; Yeung, Y.A.; Sutton, J.; Hamze, M.; Valton, J.; Smith, J.; Djuretic, I.; et al. Allogeneic FLT3 CAR T Cells with an Off-Switch Exhibit Potent Activity against AML and Can Be Depleted to Expedite Bone Marrow Recovery. Mol. Ther. 2020, 28, 2237–2251. [Google Scholar] [CrossRef]
- Sallman, D.A.; Brayer, J.; Sagatys, E.M.; Lonez, C.; Breman, E.; Agaugué, S.; Verma, B.; Gilham, D.E.; Lehmann, F.F.; Davila, M.L. NKG2D-based chimeric antigen receptor therapy induced remission in a relapsed/refractory acute myeloid leukemia patient. Haematologica 2018, 103, e424–e426. [Google Scholar] [CrossRef] [PubMed]
- Paczulla, A.M.; Rothfelder, K.; Raffel, S.; Konantz, M.; Steinbacher, J.; Wang, H.; Tandler, C.; Mbarga, M.; Schaefer, T.; Falcone, M.; et al. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nat. Cell Biol. 2019, 572, 254–259. [Google Scholar] [CrossRef]
- Wu, Z.; Zhang, H.; Wu, M.; Peng, G.; He, Y.; Wan, N.; Zeng, Y. Targeting the NKG2D/NKG2D-L axis in acute myeloid leukemia. Biomed. Pharmacother. 2021, 137, 111299. [Google Scholar] [CrossRef] [PubMed]
- Alkhayer, R.; Ponath, V.; Frech, M.; Adhikary, T.; Graumann, J.; Neubauer, A.; von Strandmann, E.P. KLF4-mediated upregulation of the NKG2D ligand MICA in acute myeloid leukemia: A novel therapeutic target identified by enChIP. Cell Commun. Signal. 2023, 21, 94. [Google Scholar] [CrossRef] [PubMed]
- Farber, M.; Chen, Y.; Arnold, L.; Möllmann, M.; Boog-Whiteside, E.; Lin, Y.-A.; Reinhardt, H.C.; Dührsen, U.; Hanoun, M. Targeting CD38 in acute myeloid leukemia interferes with leukemia trafficking and induces phagocytosis. Sci. Rep. 2021, 11, 22062. [Google Scholar] [CrossRef]
- Zhong, X.; Ma, H. Targeting CD38 for acute leukemia. Front. Oncol. 2022, 12, 1007783. [Google Scholar] [CrossRef]
- Cui, Q.; Qian, C.; Xu, N.; Kang, L.; Dai, H.; Cui, W.; Song, B.; Yin, J.; Li, Z.; Zhu, X.; et al. CD38-directed CAR-T cell therapy: A novel immunotherapy strategy for relapsed acute myeloid leukemia after allogeneic hematopoietic stem cell transplantation. J. Hematol. Oncol. 2021, 14, 82. [Google Scholar] [CrossRef]
- Glisovic-Aplenc, T.; Diorio, C.; Chukinas, J.A.; Veliz, K.; Shestova, O.; Shen, F.; Nunez-Cruz, S.; Vincent, T.L.; Miao, F.; Milone, M.C.; et al. CD38 as a pan-hematologic target for chimeric antigen receptor T cells. Blood Adv. 2023, 7, 4418–4430. [Google Scholar] [CrossRef]
- Raza, H.; Fatima, M.; Noor, T.; Umer, S.; Imran, A.; Malik, N.A. The Frequency of Aberrant CD7 Antigen Expression in Acute Myeloid Leukaemia Patients. Cureus 2022, 14, e22309. [Google Scholar] [CrossRef]
- Ossenkoppele, G.J.; van de Loosdrecht, A.A.; Schuurhuis, G.J. Review of the relevance of aberrant antigen expression by flow cytometry in myeloid neoplasms. Br. J. Haematol. 2011, 153, 421–436. [Google Scholar] [CrossRef]
- Ma, G.; Wang, Y.; Ahmed, T.; Zaslav, A.-L.; Hogan, L.; Avila, C.; Wada, M.; Salman, H. Anti-CD19 chimeric antigen receptor targeting of CD19 + acute myeloid leukemia. Leuk. Res. Rep. 2018, 9, 42–44. [Google Scholar] [CrossRef]
- Gomes-Silva, D.; Atilla, E.; Atilla, P.A.; Mo, F.; Tashiro, H.; Srinivasan, M.; Lulla, P.; Rouce, R.H.; Cabral, J.M.; Ramos, C.A.; et al. CD7 CAR T Cells for the Therapy of Acute Myeloid Leukemia. Mol. Ther. 2018, 27, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Dai, H.; Cui, Q.; Li, Z.; Shen, W.; Pan, J.; Shen, H.; Ma, Q.; Li, M.; Chen, S.; et al. CD7-directed CAR T-cell therapy: A potential immunotherapy strategy for relapsed/refractory acute myeloid leukemia. Exp. Hematol. Oncol. 2022, 11, 67. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, Y.; Guo, R.; Zhao, Y.; Sun, R.; Guo, S.; Lu, W.; Zhao, M. Targeted CD7 CAR T-cells for treatment of T-Lymphocyte leukemia and lymphoma and acute myeloid leukemia: Recent advances. Front. Immunol. 2023, 14, 1170968. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jin, Y.; Xia, P.; Lin, J.; Ma, J.; Li, T.; Liu, Z.; Xiang, H.; Cheng, C.; Xu, Z.; et al. Integrated analysis reveals distinct molecular, clinical, and immunological features of B7-H3 in acute myeloid leukemia. Cancer Med. 2021, 10, 7831–7846. [Google Scholar] [CrossRef] [PubMed]
- Lichtman, E.I.; Du, H.; Shou, P.; Song, F.; Suzuki, K.; Ahn, S.; Li, G.; Ferrone, S.; Su, L.; Savoldo, B.; et al. Preclinical Evaluation of B7-H3–specific Chimeric Antigen Receptor T Cells for the Treatment of Acute Myeloid Leukemia. Clin. Cancer Res. 2021, 27, 3141–3153. [Google Scholar] [CrossRef]
- Tyagi, A.; Ly, S.; El-Dana, F.; Yuan, B.; Jaggupilli, A.; Grimm, S.; Konopleva, M.Y.; Bühring, H.-J.; Battula, V.L. Evidence supporting a role for the immune checkpoint protein B7-H3 in NK cell-mediated cytotoxicity against AML. Blood 2022, 139, 2782–2796. [Google Scholar] [CrossRef]
- Shahzad, M.; Nguyen, A.; Hussain, A.; Ammad-Ud-Din, M.; Faisal, M.S.; Tariq, E.; Ali, F.; Butt, A.; Anwar, I.; Chaudhary, S.G.; et al. Outcomes with chimeric antigen receptor t-cell therapy in relapsed or refractory acute myeloid leukemia: A systematic review and meta-analysis. Front. Immunol. 2023, 14, 1152457. [Google Scholar] [CrossRef]
- Sallman, D.A.; Kerre, T.; Havelange, V.; Poiré, X.; Lewalle, P.; Wang, E.S.; Brayer, J.B.; Davila, M.L.; Moors, I.; Machiels, J.-P.; et al. CYAD-01, an autologous NKG2D-based CAR T-cell therapy, in relapsed or refractory acute myeloid leukaemia and myelodysplastic syndromes or multiple myeloma (THINK): Haematological cohorts of the dose escalation segment of a phase 1 trial. Lancet Haematol. 2023, 10, e191–e202. [Google Scholar] [CrossRef]
- Chua, C.C.; Cheok, K.P.L. Taking a step forward in CAR T-cell therapy for acute myeloid leukaemia and myelodysplastic syndrome. Lancet Haematol. 2023, 10, e161–e162. [Google Scholar] [CrossRef]
- Gottschlich, A.; Thomas, M.; Grünmeier, R.; Lesch, S.; Rohrbacher, L.; Igl, V.; Briukhovetska, D.; Benmebarek, M.-R.; Vick, B.; Dede, S.; et al. Single-cell transcriptomic atlas-guided development of CAR-T cells for the treatment of acute myeloid leukemia. Nat. Biotechnol. 2023, 41, 1618–1632. [Google Scholar] [CrossRef] [PubMed]
- Pigazzi, M.; Marini, O.; Porcù, E.; Faggin, G.; Varotto, E.; Rizzardi, P.; Buldini, B.; Locatelli, F.; Biffi, A. Preclinical Development of a CAR-T Cell Approach Targeting the CD84 Antigen Associated to Pediatric Acute Myeloid Leukemia. Blood 2022, 140 (Suppl. S1), 10247–10248. [Google Scholar] [CrossRef]
- Pérez-Amill, L.; Armand-Ugon, M.; Peña, S.; Casals, M.V.; Santos, C.; Frigola, G.; Minguela, A.; Torralba, B.; Casanovas, B.; Álamo, J.; et al. CD84: A Novel Target for CAR T-Cell Therapy for Acute Myeloid Leukemia. Blood 2022, 140 (Suppl. S1), 7379–7381. [Google Scholar] [CrossRef]
- Kirkey, D.C.; Loeb, A.M.; Castro, S.; McKay, C.N.; Perkins, L.; Pardo, L.; Leonti, A.R.; Tang, T.T.; Loken, M.R.; Brodersen, L.E.; et al. Therapeutic targeting of PRAME with mTCRCAR T cells in acute myeloid leukemia. Blood Adv. 2023, 7, 1178–1189. [Google Scholar] [CrossRef] [PubMed]
- Shao, R.; Li, Z.; Xin, H.; Jiang, S.; Zhu, Y.; Liu, J.; Huang, R.; Xu, K.; Shi, X. Biomarkers as targets for CAR-T/NK cell therapy in AML. Biomark. Res. 2023, 11, 65. [Google Scholar] [CrossRef]
- Zhang, R.; Li, Y.; Tu, S.; Huang, R.; Lai, X.; Zhang, L.; Zvonkov, E.E.; Savchenko, V.G.; Gabeeva, N.G.; Liu, Y.; et al. Improved Safety and Efficacy of a Multi-Target Chimeric Antigen Receptor Modified T Cell Therapy (4SCAR2.0) Against Relapsed or Refractory Lymphomas. Blood 2020, 136 (Suppl. S1), 47. [Google Scholar] [CrossRef]
- Atilla, E.; Benabdellah, K. The Black Hole: CAR T Cell Therapy in AML. Cancers 2023, 15, 2713. [Google Scholar] [CrossRef]
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine 2020, 59, 102975. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garg, S.; Ni, W.; Griffin, J.D.; Sattler, M. Chimeric Antigen Receptor T Cell Therapy in Acute Myeloid Leukemia: Trials and Tribulations. Hematol. Rep. 2023, 15, 608-626. https://doi.org/10.3390/hematolrep15040063
Garg S, Ni W, Griffin JD, Sattler M. Chimeric Antigen Receptor T Cell Therapy in Acute Myeloid Leukemia: Trials and Tribulations. Hematology Reports. 2023; 15(4):608-626. https://doi.org/10.3390/hematolrep15040063
Chicago/Turabian StyleGarg, Swati, Wei Ni, James D. Griffin, and Martin Sattler. 2023. "Chimeric Antigen Receptor T Cell Therapy in Acute Myeloid Leukemia: Trials and Tribulations" Hematology Reports 15, no. 4: 608-626. https://doi.org/10.3390/hematolrep15040063
APA StyleGarg, S., Ni, W., Griffin, J. D., & Sattler, M. (2023). Chimeric Antigen Receptor T Cell Therapy in Acute Myeloid Leukemia: Trials and Tribulations. Hematology Reports, 15(4), 608-626. https://doi.org/10.3390/hematolrep15040063