

Monoclonal Gammopathies and the Bone Marrow Microenvironment: From Bench to Bedside and Then Back Again

 , ,
, ,  , , ,
, , ,  , ,
, ,
Abstract
1. Introduction
2. Multiple Myeloma and Other Monoclonal Gammopathies: A Multistep Disease
Genomic Aspects
3. The Cellular and Humoral Compartment of the Bone Marrow Niche
3.1. Non-Immune Compartment
3.1.1. Stromal Cells
3.1.2. Bone Remodeling: Osteoblasts, Osteoclasts and Osteocytes
3.2. The Immune Compartment
3.2.1. Myeloid Cells
3.2.2. Lymphoid Cells
3.3. Soluble Factors Promoting Tumor Evolution
4. Bone Marrow Modulating Agents: Clinical Applications
4.1. Carfilzomib, Lenalidomide, Desametasone
4.2. Zoledronate
4.3. Curcumin
5. Targeting MM Cells to Activate the Immune System: Monoclonal Antibodies and Vaccines
5.1. Daratumumab and Isatuximab (Anti-CD38)
5.2. Vaccines
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Agarwal, A.; Ghobrial, I.M. Monoclonal gammopathy of undetermined significance and smoldering multiple myeloma: A review of the current understanding of epidemiology, biology, risk stratification, and management of myeloma precursor disease. Clin. Cancer Res. 2013, 19, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Röllig, C.; Knop, S.; Bornhäuser, M. Multiple myeloma. Lancet 2015, 385, 2197–2208. [Google Scholar] [CrossRef] [PubMed]
- Rögnvaldsson, S.; Love, T.J.; Thorsteinsdottir, S.; Reed, E.R.; Óskarsson, J.Þ.; Pétursdóttir, Í.; Sigurðardóttir, G.Á.; Viðarsson, B.; Önundarson, P.T.; Agnarsson, B.A.; et al. Iceland screens, treats, or prevents multiple myeloma (iStopMM): A population-based screening study for monoclonal gammopathy of undetermined significance and randomized controlled trial of follow-up strategies. Blood Cancer J. 2021, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Therneau, T.M.; Rajkumar, S.V.; Larson, D.R.; Plevak, M.F.; Offord, J.R.; Dispenzieri, A.; Katzmann, J.A.; Melton, L.J., III. Prevalence of monoclonal gammopathy of undetermined significance. N. Engl. J. Med. 2006, 354, 1362–1369. [Google Scholar] [CrossRef] [PubMed]
- Visram, A.; Rajkumar, S.V.; Kapoor, P.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Buadi, F.K.; Hayman, S.R.; Dingli, D.; Kourelis, T.; et al. Assessing the prognostic utility of smoldering multiple myeloma risk stratification scores applied serially post diagnosis. Blood Cancer J. 2021, 11, 186. [Google Scholar] [CrossRef]
- Tessier, C.; Allard, T.; Boudreault, J.-S.; Kaedbey, R.; Éthier, V.; Fortin, F.; Pavic, M. Testing Mayo Clinic’s New 20/20/20 Risk Model in Another Cohort of Smoldering Myeloma Patients: A Retrospective Study. Curr. Oncol. 2021, 28, 2029–2039. [Google Scholar] [CrossRef]
- Botta, C.; Mendicino, F.; Martino, E.; Vigna, E.; Ronchetti, D.; Correale, P.; Morabito, F.; Neri, A.; Gentile, M. Mechanisms of Immune Evasion in Multiple Myeloma: Open Questions and Therapeutic Opportunities. Cancers 2021, 13, 3213. [Google Scholar] [CrossRef]
- Sun, J.; Park, C.; Guenthner, N.; Gurley, S.; Zhang, L.; Lubben, B.; Adebayo, O.; Bash, H.; Chen, Y.; Maksimos, M.; et al. Tumor-associated macrophages in multiple myeloma: Advances in biology and therapy. J. Immunother. Cancer 2022, 10, e003975. [Google Scholar] [CrossRef]
- Hideshima, T.; Mitsiades, C.S.; Tonon, G.; Richardson, P.G.; Anderson, K.C. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat. Rev. Cancer 2007, 7, 585–598. [Google Scholar] [CrossRef]
- Walker, B.; Wardell, C.; Johnson, D.C.; Kaiser, M.F.; Begum, D.B.; Dahir, N.B.; Ross, F.M.; Davies, F.E.; Gonzalez, D.; Morgan, G. Characterization of IGH locus breakpoints in multiple myeloma indicates a subset of translocations appear to occur in pregerminal center B cells. Blood 2013, 121, 3413–3419. [Google Scholar] [CrossRef]
- Barwick, B.; Gupta, V.A.; Vertino, P.M.; Boise, L.H. Cell of Origin and Genetic Alterations in the Pathogenesis of Multiple Myeloma. Front. Immunol. 2019, 10, 1121. [Google Scholar] [CrossRef]
- Fonseca, R.; Blood, E.; Rue, M.; Harrington, D.; Oken, M.M.; Kyle, R.A.; Dewald, G.W.; Van Ness, B.; Van Wier, S.A.; Henderson, K.J.; et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood 2003, 101, 4569–4575. [Google Scholar] [CrossRef]
- Ziv, E.; Dean, E.; Hu, D.; Martino, A.; Serie, D.; Curtin, K.; Campa, D.; Aftab, B.; Bracci, P.; Buda, G.; et al. Genome-wide association study identifies variants at 16p13 associated with survival in multiple myeloma patients. Nat. Commun. 2015, 6, 7539. [Google Scholar] [CrossRef]
- Kuehl, W.M.; Bergsagel, P.L. Multiple myeloma: Evolving genetic events and host interactions. Nat. Rev. Cancer 2002, 2, 175–187. [Google Scholar] [CrossRef]
- Brighton, T.A.; Khot, A.; Harrison, S.J.; Ghez, D.; Weiss, B.M.; Kirsch, A.; Magen, H.; Gironella, M.; Oriol, A.; Streetly, M.; et al. Randomized, Double-Blind, Placebo-Controlled, Multicenter Study of Siltuximab in High-Risk Smoldering Multiple Myeloma. Clin. Cancer Res. 2019, 25, 3772–3775. [Google Scholar] [CrossRef]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread Genetic Heterogeneity in Multiple Myeloma: Implications for Targeted Therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef]
- Mulligan, G.; Lichter, D.I.; Di Bacco, A.; Blakemore, S.J.; Berger, A.; Koenig, E.; Bernard, H.; Trepicchio, W.; Li, B.; Neuwirth, R.; et al. Mutation of NRAS but not KRAS significantly reduces myeloma sensitivity to single-agent bortezomib therapy. Blood 2014, 123, 632–639. [Google Scholar] [CrossRef]
- Abdallah, N.; Baughn, L.B.; Rajkumar, S.V.; Kapoor, P.; Gertz, M.A.; Dispenzieri, A.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; Dingli, D.; et al. Implications of MYC Rearrangements in Newly Diagnosed Multiple Myeloma. Clin. Cancer Res. 2020, 26, 6581–6588. [Google Scholar] [CrossRef]
- Bahlis, N.J. Darwinian evolution and tiding clones in multiple myeloma. Blood 2012, 120, 927–928. [Google Scholar] [CrossRef]
- Keats, J.; Chesi, M.; Egan, J.B.; Garbitt, V.M.; Palmer, S.E.; Braggio, E.; Van Wier, S.; Blackburn, P.R.; Baker, A.S.; Dispenzieri, A.; et al. Clonal competition with alternating dominance in multiple myeloma. Blood 2012, 120, 1067–1076. [Google Scholar] [CrossRef]
- De Smedt, E.; Lui, H.; Maes, K.; De Veirman, K.; Menu, E.; Vanderkerken, K.; De Bruyne, E. The Epigenome in Multiple Myeloma: Impact on Tumor Cell Plasticity and Drug Response. Front. Oncol. 2018, 8, 566. [Google Scholar] [CrossRef] [PubMed]
- Musto, P.; Engelhardt, M.; Caers, J.; Bolli, N.; Kaiser, M.; Van de Donk, N.; Terpos, E.; Broijl, A.; De Larrea, C.F.; Gay, F.; et al. 2021 European Myeloma Network review and consensus statement on smoldering multiple myeloma: How to distinguish (and manage) Dr. Jekyll and Mr. Hyde. Haematologica 2021, 106, 2799–2812. [Google Scholar] [CrossRef] [PubMed]
- Bolli, N.; Sgherza, N.; Curci, P.; Rizzi, R.; Strafella, V.; Delia, M.; Gagliardi, V.; Neri, A.; Baldini, L.; Albano, F.; et al. What Is New in the Treatment of Smoldering Multiple Myeloma? J. Clin. Med. 2021, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.-V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef] [PubMed]
- Giannakoulas, N.; Ntanasis-Stathopoulos, I.; Terpos, E. The Role of Marrow Microenvironment in the Growth and Development of Malignant Plasma Cells in Multiple Myeloma. Int. J. Mol. Sci. 2021, 22, 4462. [Google Scholar] [CrossRef]
- Huang, M.; Wu, R.; Chen, L.; Peng, Q.; Li, S.; Zhang, Y.; Zhou, L.; Duan, L. S100A9 Regulates MDSCs-Mediated Immune Suppression via the RAGE and TLR4 Signaling Pathways in Colorectal Carcinoma. Front. Immunol. 2019, 10, 2243. [Google Scholar] [CrossRef]
- Rodríguez, P.C.; Ochoa, A.C. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: Mechanisms and therapeutic perspectives. Immunol. Rev. 2008, 222, 180–191. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Zea, A.H.; DeSalvo, J.; Culotta, K.S.; Zabaleta, J.; Quiceno, D.G.; Ochoa, J.B.; Ochoa, A.C. l-Arginine Consumption by Macrophages Modulates the Expression of CD3ζ Chain in T Lymphocytes. J. Immunol. 2003, 171, 1232–1239. [Google Scholar] [CrossRef]
- Srivastava, M.K.; Sinha, P.; Clements, V.K.; Rodriguez, P.; Ostrand-Rosenberg, S. Myeloid-Derived Suppressor Cells Inhibit T-Cell Activation by Depleting Cystine and Cysteine. Cancer Res. 2010, 70, 68–77. [Google Scholar] [CrossRef]
- Yu, J.; Du, W.; Yan, F.; Wang, Y.; Li, H.; Cao, S.; Yu, W.; Shen, C.; Liu, J.; Ren, X. Myeloid-Derived Suppressor Cells Suppress Antitumor Immune Responses through IDO Expression and Correlate with Lymph Node Metastasis in Patients with Breast Cancer. J. Immunol. 2013, 190, 3783–3797. [Google Scholar] [CrossRef]
- Lindau, D.; Gielen, P.; Kroesen, M.; Wesseling, P.; Adema, G.J. The immunosuppressive tumour network: Myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 2012, 138, 105–115. [Google Scholar] [CrossRef]
- Li, B.; Shi, M.; Li, J.; Zhang, H.; Chen, B.; Chen, L.; Gao, W.; Giuliani, N.; Zhao, R.C. Elevated Tumor Necrosis Factor-αSuppresses TAZ Expression and Impairs Osteogenic Potential of Flk-1+Mesenchymal Stem Cells in Patients with Multiple Myeloma. Stem Cells Dev. 2007, 16, 921–930. [Google Scholar] [CrossRef]
- Méndez-Ferrer, S.; Bonnet, D.; Steensma, D.P.; Hasserjian, R.P.; Ghobrial, I.M.; Gribben, J.G.; Andreeff, M.; Krause, D.S. Bone marrow niches in haematological malignancies. Nat. Rev. Cancer 2020, 20, 285–298. [Google Scholar] [CrossRef]
- Botta, C.; DI Martino, M.T.; Ciliberto, D.; Cucè, M.; Correale, P.; Rossi, M.; Tagliaferri, P.; Tassone, P. A gene expression inflammatory signature specifically predicts multiple myeloma evolution and patients survival. Blood Cancer J. 2016, 6, e511. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, Z.; Yao, C. Angiogenic Activity of Mesenchymal Stem Cells in Multiple Myeloma. Cancer Investig. 2010, 29, 37–41. [Google Scholar] [CrossRef]
- Delgado-Calle, J.; Bellido, T.; Roodman, G.D. Role of osteocytes in multiple myeloma bone disease. Curr. Opin. Support. Palliat. Care 2014, 8, 407–413. [Google Scholar] [CrossRef]
- Hameed, A.; Brady, J.J.; Dowling, P.; Clynes, M.; O’Gorman, P. Bone Disease in Multiple Myeloma: Pathophysiology and Management. Cancer Growth Metastasis 2014, 7, 33–42. [Google Scholar] [CrossRef]
- An, G.; Acharya, C.; Feng, X.; Wen, K.; Zhong, M.; Zhang, L.; Munshi, N.C.; Qiu, L.; Tai, Y.-T.; Anderson, K.C. Osteoclasts promote immune suppressive microenvironment in multiple myeloma: Therapeutic implication. Blood 2016, 128, 1590–1603. [Google Scholar] [CrossRef]
- Wang, X.; He, L.; Huang, X.; Zhang, S.; Cao, W.; Che, F.; Zhu, Y.; Dai, J. Recent Progress of Exosomes in Multiple Myeloma: Pathogenesis, Diagnosis, Prognosis and Therapeutic Strategies. Cancers 2021, 13, 1635. [Google Scholar] [CrossRef]
- Puglisi, F.; Parrinello, N.L.; Giallongo, C.; Cambria, D.; Camiolo, G.; Bellofiore, C.; Conticello, C.; Del Fabro, V.; Leotta, V.; Markovic, U.; et al. Plasticity of High-Density Neutrophils in Multiple Myeloma is Associated with Increased Autophagy Via STAT3. Int. J. Mol. Sci. 2019, 20, 3548. [Google Scholar] [CrossRef]
- Sun, J.; Muz, B.; Alhallak, K.; Markovic, M.; Gurley, S.; Wang, Z.; Guenthner, N.; Wasden, K.; Fiala, M.; King, J.; et al. Targeting CD47 as a Novel Immunotherapy for Multiple Myeloma. Cancers 2020, 12, 305. [Google Scholar] [CrossRef] [PubMed]
- Binsfeld, M.; Muller, J.; Lamour, V.; De Veirman, K.; De Raeve, H.; Bellahcène, A.; Van Valckenborgh, E.; Baron, F.; Beguin, Y.; Caers, J.; et al. Granulocytic myeloid-derived suppressor cells promote angiogenesis in the context of multiple myeloma. Oncotarget 2016, 7, 37931–37943. [Google Scholar] [CrossRef] [PubMed]
- Raje, N.; Gong, J.; Chauhan, D.; Teoh, G.; Avigan, D.; Wu, Z.; Chen, D.; Treon, S.P.; Webb, I.J.; Kufe, D.W.; et al. Bone marrow and peripheral blood dendritic cells from patients with multiple myeloma are phenotypically and functionally normal despite the detection of Kaposi’s sarcoma herpesvirus gene sequences. Blood 1999, 93, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.A.; Dhodapkar, M.V. It takes T to tango: Immunotherapy in MM. Blood 2022, 139, 1259–1260. [Google Scholar] [CrossRef] [PubMed]
- Crane, G.; Jeffery, E.; Morrison, S. Adult haematopoietic stem cell niches. Nat. Rev. Immunol. 2017, 17, 573–590. [Google Scholar] [CrossRef]
- Hoggatt, J.; Kfoury, Y.; Scadden, D.T. Hematopoietic Stem Cell Niche in Health and Disease. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 555–581. [Google Scholar] [CrossRef]
- Wei, Q.; Frenette, P.S. Niches for Hematopoietic Stem Cells and Their Progeny. Immunity 2018, 48, 632–648. [Google Scholar] [CrossRef]
- Bianco, P.; Riminucci, M.; Gronthos, S.; Robey, P.G. Bone Marrow Stromal Stem Cells: Nature, Biology, and Potential Applications. Stem Cells 2001, 19, 180–192. [Google Scholar] [CrossRef]
- Dazzi, F.; Ramasamy, R.; Glennie, S.; Jones, S.P.; Roberts, I. The role of mesenchymal stem cells in haemopoiesis. Blood Rev. 2006, 20, 161–171. [Google Scholar] [CrossRef]
- Gao, Q.; Wang, L.; Wang, S.; Huang, B.; Jing, Y.; Su, J. Bone Marrow Mesenchymal Stromal Cells: Identification, Classification, and Differentiation. Front. Cell Dev. Biol. 2022, 9, 787118. [Google Scholar] [CrossRef]
- Hideshima, T.; Anderson, K.C. Signaling Pathway Mediating Myeloma Cell Growth and Survival. Cancers 2021, 13, 216. [Google Scholar] [CrossRef]
- Neri, P. Targeting of Adhesion Molecules as a Therapeutic Strategy in Multiple Myeloma. Curr. Cancer Drug Targets 2012, 12, 776–796. [Google Scholar] [CrossRef]
- Yasui, H.; Hideshima, T.; Richardson, P.G.; Anderson, K.C. Recent advances in the treatment of Multiple Myeloma. Curr. Pharm. Biotechnol. 2006, 7, 381–393. [Google Scholar] [CrossRef]
- Maiso, P.; Mogollón, P.; Ocio, E.; Garayoa, M. Bone Marrow Mesenchymal Stromal Cells in Multiple Myeloma: Their Role as Active Contributors to Myeloma Progression. Cancers 2021, 13, 2542. [Google Scholar] [CrossRef]
- André, T.; Meuleman, N.; Stamatopoulos, B.; De Bruyn, C.; Pieters, K.; Bron, D.; Lagneaux, L. Evidences of Early Senescence in Multiple Myeloma Bone Marrow Mesenchymal Stromal Cells. PLoS ONE 2013, 8, e59756. [Google Scholar] [CrossRef]
- Corre, J.; Mahtouk, K.; Attal, M.; Gadelorge, M.; Huynh, A.; Fleury-Cappellesso, S.; Danho, C.; Laharrague, P.; Klein, B.; Rème, T.; et al. Bone marrow mesenchymal stem cells are abnormal in multiple myeloma. Leukemia 2007, 21, 1079–1088. [Google Scholar] [CrossRef]
- Li, B.; Fu, J.; Chen, P.; Zhuang, W. Impairment in Immunomodulatory Function of Mesenchymal Stem Cells from Multiple Myeloma Patients. Arch. Med Res. 2010, 41, 623–633. [Google Scholar] [CrossRef]
- Todoerti, K.; Lisignoli, G.; Storti, P.; Agnelli, L.; Novara, F.; Manferdini, C.; Codeluppi, K.; Colla, S.; Crugnola, M.; Abeltino, M.; et al. Distinct transcriptional profiles characterize bone microenvironment mesenchymal cells rather than osteoblasts in relationship with multiple myeloma bone disease. Exp. Hematol. 2010, 38, 141–153. [Google Scholar] [CrossRef]
- Xu, S.; Evans, H.; Buckle, C.; De Veirman, K.; Hu, J.; Xu, D.; Menu, E.; De Becker, A.; Vande Broek, I.; Leleu, X.; et al. Impaired osteogenic differentiation of mesenchymal stem cells derived from multiple myeloma patients is associated with a blockade in the deactivation of the Notch signaling pathway. Leukemia 2012, 26, 2546–2549. [Google Scholar] [CrossRef]
- Dimopoulos, K.; Gimsing, P.; Grønbæk, K. The role of epigenetics in the biology of multiple myeloma. Blood Cancer J. 2014, 4, e207. [Google Scholar] [CrossRef]
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Bataille, R.; Chappard, D.; Basle, M. Excessive bone resorption in human plasmacytomas: Direct induction by tumour cells in vivo. Br. J. Haematol. 1995, 90, 721–724. [Google Scholar] [CrossRef] [PubMed]
- Taube, T.; Beneton, M.N.C.; McCloskey, E.V.; Rogers, S.; Greaves, M.; Kanis, J.A. Abnormal bone remodelling in patients with myelomatosis and normal biochemical indices of bone resorption. Eur. J. Haematol. 1992, 49, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Raje, N.S.; Bhatta, S.; Terpos, E. Role of the RANK/RANKL Pathway in Multiple Myeloma. Clin. Cancer Res. 2019, 25, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Politou, M.; Terpos, E.; Anagnostopoulos, A.; Szydlo, R.; Laffan, M.; Layton, M.; Apperley, J.F.; Dimopoulos, M.; Rahemtulla, A. Role of receptor activator of nuclear factor-kappa B ligand (RANKL), osteoprotegerin and macrophage protein 1-alpha (MIP-1a) in monoclonal gammopathy of undetermined significance (MGUS). Br. J. Haematol. 2004, 126, 686–689. [Google Scholar] [CrossRef]
- Pianko, M.J.; Terpos, E.; Roodman, G.D.; Divgi, C.R.; Zweegman, S.; Hillengass, J.; Lentzsch, S. Whole-Body Low-Dose Computed Tomography and Advanced Imaging Techniques for Multiple Myeloma Bone Disease. Clin. Cancer Res. 2014, 20, 5888–5897. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Hillengass, J.; Usmani, S.; Zamagni, E.; Lentzsch, S.; Davies, F.E.; Raje, N.; Sezer, O.; Zweegman, S.; Shah, J.; et al. Role of Magnetic Resonance Imaging in the Management of Patients With Multiple Myeloma: A Consensus Statement. J. Clin. Oncol. 2015, 33, 657–664. [Google Scholar] [CrossRef]
- Tirumani, S.H.; Sakellis, C.; Jacene, H.; Shinagare, A.B.; Munshi, N.C.; Ramaiya, N.H.; Abbeele, A.D.V.D. Role of FDG-PET/CT in Extramedullary Multiple Myeloma. Clin. Nucl. Med. 2016, 41, e7–e13. [Google Scholar] [CrossRef]
- Delgado-Calle, J.; Anderson, J.; Cregor, M.D.; Hiasa, M.; Chirgwin, J.M.; Carlesso, N.; Yoneda, T.; Mohammad, K.S.; Plotkin, L.I.; Roodman, G.D.; et al. Bidirectional Notch Signaling and Osteocyte-Derived Factors in the Bone Marrow Microenvironment Promote Tumor Cell Proliferation and Bone Destruction in Multiple Myeloma. Cancer Res. 2016, 76, 1089–1100. [Google Scholar] [CrossRef]
- Zheng, Y.; Cai, Z.; Wang, S.; Zhang, X.; Qian, J.; Hong, S.; Li, H.; Wang, M.; Yang, J.; Yi, Q. Macrophages are an abundant component of myeloma microenvironment and protect myeloma cells from chemotherapy drug–induced apoptosis. Blood 2009, 114, 3625–3628. [Google Scholar] [CrossRef]
- De Beule, N.; De Veirman, K.; Maes, K.; De Bruyne, E.; Menu, E.; Breckpot, K.; De Raeve, H.; Van Rampelbergh, R.; Van Ginderachter, J.A.; Schots, R.; et al. Tumour-associated macrophage-mediated survival of myeloma cells through STAT3 activation. J. Pathol. 2017, 241, 534–546. [Google Scholar] [CrossRef]
- Papadimitriou, K.; Tsakirakis, N.; Malandrakis, P.; Vitsos, P.; Metousis, A.; Orologas-Stavrou, N.; Ntanasis-Stathopoulos, I.; Kanellias, N.; Eleutherakis-Papaiakovou, E.; Pothos, P.; et al. Deep Phenotyping Reveals Distinct Immune Signatures Correlating with Prognostication, Treatment Responses, and MRD Status in Multiple Myeloma. Cancers 2020, 12, 3245. [Google Scholar] [CrossRef]
- Wang, H.; Hu, W.-M.; Xia, Z.-J.; Liang, Y.; Lu, Y.; Lin, S.-X.; Tang, H. High numbers of CD163+ tumor-associated macrophages correlate with poor prognosis in multiple myeloma patients receiving bortezomib-based regimens. J. Cancer 2019, 10, 3239–3245. [Google Scholar] [CrossRef]
- Suyanı, E.; Sucak, G.T.; Akyürek, N.; Şahin, S.; Baysal, N.A.; Yağcı, M.; Haznedar, R. Tumor-associated macrophages as a prognostic parameter in multiple myeloma. Ann. Hematol. 2013, 92, 669–677. [Google Scholar] [CrossRef]
- Beider, K.; Bitner, H.; Leiba, M.; Gutwein, O.; Koren-Michowitz, M.; Ostrovsky, O.; Abraham, M.; Wald, H.; Galun, E.; Peled, A.; et al. Multiple myeloma cells recruit tumor-supportive macrophages through the CXCR4/CXCL12 axis and promote their polarization toward the M2 phenotype. Oncotarget 2014, 5, 11283–11296. [Google Scholar] [CrossRef]
- Berardi, S.; Ria, R.; Reale, A.; De Luisi, A.; Catacchio, I.; Moschetta, M.; Vacca, A. Multiple Myeloma Macrophages: Pivotal Players in the Tumor Microenvironment. J. Oncol. 2013, 2013, 183602. [Google Scholar] [CrossRef]
- Chen, X.; Chen, J.; Zhang, W.; Sun, R.; Liu, T.; Zheng, Y.; Wu, Y. Prognostic value of diametrically polarized tumor-associated macrophages in multiple myeloma. Oncotarget 2017, 8, 112685–112696. [Google Scholar] [CrossRef]
- Chen, H.; Li, M.; Sanchez, E.; Soof, C.M.; Bujarski, S.; Ng, N.; Cao, J.; Hekmati, T.; Zahab, B.; Nosrati, J.D.; et al. JAK1/2 pathway inhibition suppresses M2 polarization and overcomes resistance of myeloma to lenalidomide by reducing TRIB1, MUC1, CD44, CXCL12, and CXCR4 expression. Br. J. Haematol. 2019, 188, 283–294. [Google Scholar] [CrossRef]
- Zavidij, O.; Haradhvala, N.J.; Mouhieddine, T.H.; Sklavenitis-Pistofidis, R.; Cai, S.; Reidy, M.; Rahmat, M.; Flaifel, A.; Ferland, B.; Su, N.K.; et al. Single-cell RNA sequencing reveals compromised immune microenvironment in precursor stages of multiple myeloma. Nat. Cancer 2020, 1, 493–506. [Google Scholar] [CrossRef]
- Ramachandran, I.R.; Martner, A.; Pisklakova, A.; Condamine, T.; Chase, T.; Vogl, T.; Roth, J.; Gabrilovich, D.; Nefedova, Y. Myeloid-Derived Suppressor Cells Regulate Growth of Multiple Myeloma by Inhibiting T Cells in Bone Marrow. J. Immunol. 2013, 190, 3815–3823. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, L.; Wang, H.; Xiong, S.; Li, Y.; Tao, Q.; Xiao, W.; Qin, H.; Wang, Y.; Zhai, Z. Tumor-induced CD14+HLA-DR−/low myeloid-derived suppressor cells correlate with tumor progression and outcome of therapy in multiple myeloma patients. Cancer Immunol. Immunother. 2014, 64, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Görgün, G.T.; Whitehill, G.; Anderson, J.L.; Hideshima, T.; Maguire, C.; Laubach, J.; Raje, N.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. Tumor-promoting immune-suppressive myeloid-derived suppressor cells in the multiple myeloma microenvironment in humans. Blood 2013, 121, 2975–2987. [Google Scholar] [CrossRef] [PubMed]
- Favaloro, J.; Liyadipitiya, T.; Brown, R.; Yang, S.; Suen, H.; Woodland, N.; Nassif, N.; Hart, D.; Fromm, P.; Weatherburn, C.; et al. Myeloid derived suppressor cells are numerically, functionally and phenotypically different in patients with multiple myeloma. Leuk. Lymphoma 2014, 55, 2893–2900. [Google Scholar] [CrossRef] [PubMed]
- Giallongo, C.; Tibullo, D.; Parrinello, N.L.; La Cava, P.; Di Rosa, M.; Bramanti, V.; Di Raimondo, C.; Conticello, C.; Chiarenza, A.; Palumbo, G.A.; et al. Granulocyte-like myeloid derived suppressor cells (G-MDSC) are increased in multiple myeloma and are driven by dysfunctional mesenchymal stem cells (MSC). Oncotarget 2016, 7, 85764–85775. [Google Scholar] [CrossRef] [PubMed]
- Romano, A.; Parrinello, N.L.; La Cava, P.; Tibullo, D.; Giallongo, C.; Camiolo, G.; Puglisi, F.; Parisi, M.; Pirosa, M.C.; Martino, E.; et al. PMN-MDSC and arginase are increased in myeloma and may contribute to resistance to therapy. Expert Rev. Mol. Diagn. 2018, 18, 675–683. [Google Scholar] [CrossRef]
- Tian, X.; Shen, H.; Li, Z.; Wang, T.; Wang, S. Tumor-derived exosomes, myeloid-derived suppressor cells, and tumor microenvironment. J. Hematol. Oncol. 2019, 12, 84. [Google Scholar] [CrossRef]
- Wang, J.; De Veirman, K.; De Beule, N.; Maes, K.; De Bruyne, E.; Van Valckenborgh, E.; Vanderkerken, K.; Menu, E. The bone marrow microenvironment enhances multiple myeloma progression by exosome-mediated activation of myeloid-derived suppressor cells. Oncotarget 2015, 6, 43992–44004. [Google Scholar] [CrossRef]
- Wang, J.; De Veirman, K.; Faict, S.; Frassanito, M.A.; Ribatti, D.; Vacca, A.; Menu, E. Multiple myeloma exosomes establish a favourable bone marrow microenvironment with enhanced angiogenesis and immunosuppression. J. Pathol. 2016, 239, 162–173. [Google Scholar] [CrossRef]
- Favaloro, J.; Brown, R.; Aklilu, E.; Yang, S.; Suen, H.; Hart, D.; Fromm, P.; Gibson, J.; Khoo, L.; Ho, P.J.; et al. Myeloma skews regulatory T and pro-inflammatory T helper 17 cell balance in favor of a suppressive state. Leuk. Lymphoma 2013, 55, 1090–1098. [Google Scholar] [CrossRef]
- Zhuang, J.; Zhang, J.; Lwin, S.T.; Edwards, J.R.; Edwards, C.M.; Mundy, G.R.; Yang, X. Osteoclasts in Multiple Myeloma Are Derived from Gr-1+CD11b+Myeloid-Derived Suppressor Cells. PLoS ONE 2012, 7, e48871. [Google Scholar] [CrossRef]
- Perez, C.; Botta, C.; Zabaleta, A.; Puig, N.; Cedena, M.-T.; Goicoechea, I.; Alameda, D.; José-Eneriz, E.S.; Merino, J.; Rodríguez-Otero, P.; et al. Immunogenomic identification and characterization of granulocytic myeloid-derived suppressor cells in multiple myeloma. Blood 2020, 136, 199–209. [Google Scholar] [CrossRef]
- Malek, E.; de Lima, M.; Letterio, J.J.; Kim, B.G.; Finke, J.H.; Driscoll, J.J.; Giralt, S.A. Myeloid-derived suppressor cells: The green light for myeloma immune escape. Blood Rev. 2016, 30, 341–348. [Google Scholar] [CrossRef]
- Romano, A.; Parrinello, N.L.; Simeon, V.; Puglisi, F.; La Cava, P.; Bellofiore, C.; Giallongo, C.; Camiolo, G.; D’Auria, F.; Grieco, V.; et al. High-density neutrophils in MGUS and multiple myeloma are dysfunctional and immune-suppressive due to increased STAT3 downstream signaling. Sci. Rep. 2020, 10, 1983. [Google Scholar] [CrossRef]
- Romano, A.; Parrinello, N.L.; Cerchione, C.; Consoli, M.L.; Parisi, M.; Calafiore, V.; Martino, E.; Conticello, C.; Di Raimondo, F.; Palumbo, G.A. The NLR and LMR ratio in newly diagnosed MM patients treated upfront with novel agents. Blood Cancer J. 2017, 7, 649. [Google Scholar] [CrossRef]
- Shi, L.; Qin, X.; Wang, H.; Xia, Y.; Li, Y.; Chen, X.; Shang, L.; Tai, Y.-T.; Feng, X.; Acharya, P.; et al. Elevated neutrophil-to-lymphocyte ratio and monocyte-to-lymphocyte ratio and decreased platelet-to-lymphocyte ratio are associated with poor prognosis in multiple myeloma. Oncotarget 2016, 8, 18792–18801. [Google Scholar] [CrossRef]
- Ratta, M.; Fagnoni, F.; Curti, A.; Vescovini, R.; Sansoni, P.; Oliviero, B.; Fogli, M.; Ferri, E.; Della Cuna, G.R.; Tura, S.; et al. Dendritic cells are functionally defective in multiple myeloma: The role of interleukin-6. Blood 2002, 100, 230–237. [Google Scholar] [CrossRef]
- Do, T.; Johnsen, H.; Kjærsgaard, E.; Taaning, E.; Svane, I. Impaired circulating myeloid DCs from myeloma patients. Cytotherapy 2004, 6, 196–203. [Google Scholar] [CrossRef]
- Brimnes, M.K.; Svane, I.M.; Johnsen, H.E. Impaired functionality and phenotypic profile of dendritic cells from patients with multiple myeloma. Clin. Exp. Immunol. 2006, 144, 76–84. [Google Scholar] [CrossRef]
- Ray, A.; Das, D.S.; Song, Y.; Macri, V.; Richardson, P.; Brooks, C.L.; Chauhan, D.; Anderson, K.C. A novel agent SL-401 induces anti-myeloma activity by targeting plasmacytoid dendritic cells, osteoclastogenesis and cancer stem-like cells. Leukemia 2017, 31, 2652–2660. [Google Scholar] [CrossRef]
- Knight, A.; Rihova, L.; Kralova, R.; Penka, M.; Adam, Z.; Pour, L.; Piskacek, M.; Hajek, R. Plasmacytoid Dendritic Cells in Patients with MGUS and Multiple Myeloma. J. Clin. Med. 2021, 10, 3717. [Google Scholar] [CrossRef]
- Botta, C.; Cucè, M.; Pitari, M.R.; Caracciolo, D.; Gullà, A.; Morelli, E.; Riillo, C.; Biamonte, L.; Cantafio, M.E.G.; Prabhala, R.; et al. MiR-29b antagonizes the pro-inflammatory tumor-promoting activity of multiple myeloma-educated dendritic cells. Leukemia 2017, 32, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Leone, P.; Berardi, S.; Frassanito, M.A.; Ria, R.; De Re, V.; Cicco, S.; Battaglia, S.; Ditonno, P.; Dammacco, F.; Vacca, A.; et al. Dendritic cells accumulate in the bone marrow of myeloma patients where they protect tumor plasma cells from CD8+ T-cell killing. Blood 2015, 126, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Corso, A.; Castelli, G.; Pagnucco, G.; Lazzarino, M.; Bellio, L.; Klersy, C.; Bernasconi, C. Bone marrow T-cell subsets in patients with monoclonal gammopathies: Correlation with clinical stage and disease status. Haematologica 1997, 82, 43–46. [Google Scholar] [PubMed]
- Cohen, A.D.; Raje, N.; Fowler, J.A.; Mezzi, K.; Scott, E.C.; Dhodapkar, M.V. How to Train Your T Cells: Overcoming Immune Dysfunction in Multiple Myeloma. Clin. Cancer Res. 2020, 26, 1541–1554. [Google Scholar] [CrossRef] [PubMed]
- Leblay, N.; Maity, R.; Hasan, F.; Neri, P. Deregulation of Adaptive T Cell Immunity in Multiple Myeloma: Insights Into Mechanisms and Therapeutic Opportunities. Front. Oncol. 2020, 10, 636. [Google Scholar] [CrossRef]
- Gu, Y.; Jin, Y.; Ding, J.; Yujie, W.; Shi, Q.; Qu, X.; Zhao, S.; Li, J.; Lijuan, C. Low absolute CD4+ T cell counts in peripheral blood predict poor prognosis in patients with newly diagnosed multiple myeloma. Leuk. Lymphoma 2020, 61, 1869–1876. [Google Scholar] [CrossRef]
- Bae, J.; Accardi, F.; Hideshima, T.; Tai, Y.-T.; Prabhala, R.; Shambley, A.; Wen, K.; Rowell, S.; Richardson, P.G.; Munshi, N.C.; et al. Targeting LAG3/GAL-3 to overcome immunosuppression and enhance anti-tumor immune responses in multiple myeloma. Leukemia 2021, 36, 138–154. [Google Scholar] [CrossRef]
- Dhodapkar, M.V.; Krasovsky, J.; Osman, K.; Geller, M.D. Vigorous Premalignancy-specific Effector T Cell Response in the Bone Marrow of Patients with Monoclonal Gammopathy. J. Exp. Med. 2003, 198, 1753–1757. [Google Scholar] [CrossRef]
- Racanelli, V.; Leone, P.; Frassanito, M.A.; Brunetti, C.; Perosa, F.; Ferrone, S.; Dammacco, F. Alterations in the antigen processing-presenting machinery of transformed plasma cells are associated with reduced recognition by CD8+ T cells and characterize the progression of MGUS to multiple myeloma. Blood 2010, 115, 1185–1193. [Google Scholar] [CrossRef]
- Alrasheed, N.; Lee, L.; Ghorani, E.; Henry, J.Y.; Conde, L.; Chin, M.; Galas-Filipowicz, D.; Furness, A.J.; Chavda, S.J.; Richards, H.; et al. Marrow-Infiltrating Regulatory T Cells Correlate with the Presence of Dysfunctional CD4+PD-1+ Cells and Inferior Survival in Patients with Newly Diagnosed Multiple Myeloma. Clin. Cancer Res. 2020, 26, 3443–3454. [Google Scholar] [CrossRef]
- Suen, H.; Brown, R.; Yang, S.; Weatherburn, C.; Ho, P.J.; Woodland, N.; Nassif, N.; Barbaro, P.; Bryant, C.; Hart, D.; et al. Multiple myeloma causes clonal T-cell immunosenescence: Identification of potential novel targets for promoting tumour immunity and implications for checkpoint blockade. Leukemia 2016, 30, 1716–1724. [Google Scholar] [CrossRef]
- Botta, C.; Maia, C.D.S.; Garcés, J.-J.; Termini, R.; Perez, C.; Manrique, I.; Burgos, L.; Zabaleta, A.; Alignani, D.; Sarvide, S.; et al. FlowCT for the analysis of large immunophenotypic data sets and biomarker discovery in cancer immunology. Blood Adv. 2022, 6, 690–703. [Google Scholar] [CrossRef]
- Joshua, D.E.; Vuckovic, S.; Favaloro, J.; Lau, K.H.A.; Yang, S.; Bryant, C.E.; Gibson, J.; Ho, P.J. Treg and Oligoclonal Expansion of Terminal Effector CD8+ T Cell as Key Players in Multiple Myeloma. Front. Immunol. 2021, 12, 620596. [Google Scholar] [CrossRef]
- Prabhala, R.H.; Pelluru, D.; Fulciniti, M.; Prabhala, H.K.; Nanjappa, P.; Song, W.; Pai, C.; Amin, S.; Tai, Y.-T.; Richardson, P.G.; et al. Elevated IL-17 produced by Th17 cells promotes myeloma cell growth and inhibits immune function in multiple myeloma. Blood 2010, 115, 5385–5392. [Google Scholar] [CrossRef]
- Prabhala, R.H.; Neri, P.; Bae, J.E.; Tassone, P.; Shammas, M.A.; Allam, C.K.; Daley, J.F.; Chauhan, D.; Blanchard, E.; Thatte, H.S.; et al. Dysfunctional T regulatory cells in multiple myeloma. Blood 2006, 107, 301–304. [Google Scholar] [CrossRef]
- Dhodapkar, K.M.; Barbuto, S.; Matthews, P.; Kukreja, A.; Mazumder, A.; Vesole, D.; Jagannath, S.; Dhodapkar, M.V. Dendritic cells mediate the induction of polyfunctional human IL17-producing cells (Th17-1 cells) enriched in the bone marrow of patients with myeloma. Blood 2008, 112, 2878–2885. [Google Scholar] [CrossRef]
- Ma, T.; Zhang, Y.; Zhou, X.; Xie, P.; Li, J. A Unique Role of T Helper 17 Cells in Different Treatment Stages of Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2020, 20, 190–197. [Google Scholar] [CrossRef]
- Shen, C.-J.; Yuan, Z.-H.; Liu, Y.-X.; Hu, G.-Y. Increased Numbers of T Helper 17 Cells and the Correlation with Clinicopathological Characteristics in Multiple Myeloma. J. Int. Med Res. 2012, 40, 556–564. [Google Scholar] [CrossRef]
- Raja, K.R.M.; Rihova, L.; Zahradova, L.; Klincova, M.; Penka, M.; Hajek, R. Increased T Regulatory Cells Are Associated with Adverse Clinical Features and Predict Progression in Multiple Myeloma. PLoS ONE 2012, 7, e47077. [Google Scholar] [CrossRef]
- Giannopoulos, K.; Kaminska, W.; Hus, I.; Dmoszynska, A. The frequency of T regulatory cells modulates the survival of multiple myeloma patients: Detailed characterisation of immune status in multiple myeloma. Br. J. Cancer 2012, 106, 546–552. [Google Scholar] [CrossRef]
- Gupta, R.; Ganeshan, P.; Hakim, M.; Verma, R.; Sharma, A.; Kumar, L. Significantly reduced regulatory T cell population in patients with untreated multiple myeloma. Leuk. Res. 2011, 35, 874–878. [Google Scholar] [CrossRef] [PubMed]
- Foglietta, M.; Castella, B.; Mariani, S.; Coscia, M.; Godio, L.; Ferracini, R.; Ruggeri, M.; Muccio, V.; Omedé, P.; Palumbo, A.; et al. The bone marrow of myeloma patients is steadily inhabited by a normal-sized pool of functional regulatory T cells irrespectiveof the disease status. Haematologica 2014, 99, 1605–1610. [Google Scholar] [CrossRef] [PubMed]
- D’Arena, G.; Rossi, G.; Laurenti, L.; Statuto, T.; D’Auria, F.; Valvano, L.; Simeon, V.; Giudice, A.; Innocenti, I.; De Feo, V.; et al. Circulating Regulatory T-Cells in Monoclonal Gammopathies of Uncertain Significance and Multiple Myeloma: In Search of a Role. J. Immunol. Res. 2016, 2016, 9271469. [Google Scholar] [CrossRef] [PubMed]
- Hadjiaggelidou, C.; Katodritou, E. Regulatory T-Cells and Multiple Myeloma: Implications in Tumor Immune Biology and Treatment. J. Clin. Med. 2021, 10, 4588. [Google Scholar] [CrossRef] [PubMed]
- D’Arena, G.; Vitale, C.; Coscia, M.; Festa, A.; Di Minno, N.M.D.; De Feo, V.; Caraglia, M.; Calapai, G.; Laurenti, L.; Musto, P.; et al. Regulatory T Cells and Their Prognostic Relevance in Hematologic Malignancies. J. Immunol. Res. 2017, 2017, 1832968. [Google Scholar] [CrossRef]
- Braga, W.M.T.; Da Silva, B.R.; de Carvalho, A.C.; Maekawa, Y.H.; Bortoluzzo, A.; Gil Rizzatti, E.; Atanackovic, D.; Colleoni, G.W.B. FOXP3 and CTLA4 overexpression in multiple myeloma bone marrow as a sign of accumulation of CD4+ T regulatory cells. Cancer Immunol. Immunother. 2014, 63, 1189–1197. [Google Scholar] [CrossRef]
- Wang, J.-N.; Cao, X.-X.; Zhao, A.-L.; Cai, H.; Wang, X.; Li, J. Increased activated regulatory T cell subsets and aging Treg-like cells in multiple myeloma and monoclonal gammopathy of undetermined significance: A case control study. Cancer Cell Int. 2018, 18, 187. [Google Scholar] [CrossRef]
- Szudy-Szczyrek, A.; Ahern, S.; Kozioł, M.; Majowicz, D.; Szczyrek, M.; Krawczyk, J.; Hus, M. Therapeutic Potential of Innate Lymphoid Cells for Multiple Myeloma Therapy. Cancers 2021, 13, 4806. [Google Scholar] [CrossRef]
- Bailur, J.K.; Mehta, S.; Zhang, L.; Neparidze, N.; Parker, T.; Bar, N.; Anderson, T.; Xu, M.L.; Dhodapkar, K.M.; Dhodapkar, M.V. Changes in bone marrow innate lymphoid cell subsets in monoclonal gammopathy: Target for IMiD therapy. Blood Adv. 2017, 1, 2343–2347. [Google Scholar] [CrossRef]
- Österborg, A.; Nilsson, B.; Björkholm, M.; Holm, G.; Mellstedt, H. Natural killer cell activity in monoclonal gammopathies: Relation to disease activity. Eur. J. Haematol. 1990, 45, 153–157. [Google Scholar] [CrossRef]
- Pazina, T.; MacFarlane, A.; Bernabei, L.; Dulaimi, E.; Kotcher, R.; Yam, C.; Bezman, N.; Robbins, M.; Ross, E.; Campbell, K.; et al. Alterations of NK Cell Phenotype in the Disease Course of Multiple Myeloma. Cancers 2021, 13, 226. [Google Scholar] [CrossRef]
- Tienhaara, A.; Pelliniemi, T.-T. Peripheral blood lymphocyte subsets in multiple myeloma and monoclonal gammopathy of undetermined significance. Int. J. Lab. Hematol. 2008, 16, 213–223. [Google Scholar] [CrossRef]
- Famularo, G.; D’Ambrosio, A.; Quintieri, F.; Di Giovanni, S.; Parzanese, I.; Pizzuto, F.; Giacomelli, R.; Pugliese, O.; Tonietti, G. Natural killer cell frequency and function in patients with monoclonal gammopathies. J. Clin. Lab. Immunol. 1992, 37, 99–109. [Google Scholar]
- Jurisic, V.; Srdic, T.; Konjevic, G.; Markovic, O.; Colovic, M. Clinical stage-depending decrease of NK cell activity in multiple myeloma patients. Med. Oncol. 2007, 24, 312–317. [Google Scholar] [CrossRef]
- Fauriat, C.; Mallet, F.; Olive, D.; Costello, R. Impaired activating receptor expression pattern in natural killer cells from patients with multiple myeloma. Leukemia 2006, 20, 732–733. [Google Scholar] [CrossRef]
- Costello, R.T.; Boehrer, A.; Sanchez, C.; Mercier, D.; Baier, C.; Le Treut, T.; Sébahoun, G. Differential expression of natural killer cell activating receptors in blood versus bone marrow in patients with monoclonal gammopathy. Immunology 2013, 139, 338–341. [Google Scholar] [CrossRef]
- Fiegler, N.; Textor, S.; Arnold, A.; Rölle, A.; Oehme, I.; Breuhahn, K.; Moldenhauer, G.; Witzens-Harig, M.; Cerwenka, A. Downregulation of the activating NKp30 ligand B7-H6 by HDAC inhibitors impairs tumor cell recognition by NK cells. Blood 2013, 122, 684–693. [Google Scholar] [CrossRef]
- Benson, D.M., Jr.; Bakan, C.E.; Mishra, A.; Hofmeister, C.C.; Efebera, Y.; Becknell, B.; Baiocchi, R.A.; Zhang, J.; Yu, J.; Smith, M.K.; et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: A therapeutic target for CT-011, a novel monoclonal anti–PD-1 antibody. Blood 2010, 116, 2286–2294. [Google Scholar] [CrossRef]
- Giuliani, M.; Janji, B.; Berchem, G. Activation of NK cells and disruption of PD-L1/PD-1 axis: Two different ways for lenalidomide to block myeloma progression. Oncotarget 2017, 8, 24031–24044. [Google Scholar] [CrossRef]
- Swamydas, M.; Murphy, E.V.; Ignatz-Hoover, J.J.; Malek, E.; Driscoll, J.J. Deciphering mechanisms of immune escape to inform immunotherapeutic strategies in multiple myeloma. J. Hematol. Oncol. 2022, 15, 17. [Google Scholar] [CrossRef]
- de Magalhães, R.J.P.; Vidriales, M.-B.; Paiva, B.; Fernandez-Gimenez, C.; García-Sanz, R.; Mateos, M.-V.; Gutierrez, N.C.; Lecrevisse, Q.; Blanco, J.F.; Hernández, J.; et al. Analysis of the immune system of multiple myeloma patients achieving long-term disease control by multidimensional flow cytometry. Haematologica 2012, 98, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Yazdanifar, M.; Barbarito, G.; Bertaina, A.; Airoldi, I. γδ T Cells: The Ideal Tool for Cancer Immunotherapy. Cells 2020, 9, 1305. [Google Scholar] [CrossRef]
- Burjanadzé, M.; Condomines, M.; Rème, T.; Quittet, P.; Latry, P.; Lugagne, C.; Romagné, F.; Morel, Y.; Rossi, J.F.; Klein, B.; et al. In vitro expansion of gamma delta T cells with anti-myeloma cell activity by Phosphostim and IL-2 in patients with multiple myeloma. Br. J. Haematol. 2007, 139, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Uchida, R.; Ashihara, E.; Sato, K.; Kimura, S.; Kuroda, J.; Takeuchi, M.; Kawata, E.; Taniguchi, K.; Okamoto, M.; Shimura, K.; et al. γδT cells kill myeloma cells by sensing mevalonate metabolites and ICAM-1 molecules on cell surface. Biochem. Biophys. Res. Commun. 2007, 354, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Mariani, S.; Muraro, M.; Pantaleoni, F.; Fiore, F.; Nuschak, B.; Peola, S.; Foglietta, M.; Palumbo, A.; Coscia, M.; Castella, B.; et al. Effector γδ T cells and tumor cells as immune targets of zoledronic acid in multiple myeloma. Leukemia 2005, 19, 664–670. [Google Scholar] [CrossRef]
- Von Lilienfeld-Toal, M.; Nattermann, J.; Feldmann, G.; Sievers, E.; Frank, S.; Strehl, J.; Schmidt-Wolf, I.G.H. Activated γδ T cells express the natural cytotoxicity receptor natural killer p44 and show cytotoxic activity against myeloma cells. Clin. Exp. Immunol. 2006, 144, 528–533. [Google Scholar] [CrossRef]
- Niu, C.; Jin, H.; Li, M.; Zhu, S.; Zhou, L.; Jin, F.; Zhou, Y.; Xu, D.; Xu, J.; Zhao, L.; et al. Low-dose bortezomib increases the expression of NKG2D and DNAM-1 ligands and enhances induced NK and γδ T cell-mediated lysis in multiple myeloma. Oncotarget 2016, 8, 5954–5964. [Google Scholar] [CrossRef]
- Kunzmann, V.; Bauer, E.; Feurle, J.; Weissinger, F.; Tony, H.P.; Wilhelm, M. Stimulation of gammadelta T cells by aminobisphosphonates and induction of antiplasma cell activity in multiple myeloma. Blood 2000, 96, 384–392. [Google Scholar] [CrossRef]
- Girlanda, S.; Fortis, C.; Belloni, D.; Ferrero, E.; Ticozzi, P.; Sciorati, C.; Tresoldi, M.; Vicari, A.; Spies, T.; Groh, V.; et al. MICA Expressed by Multiple Myeloma and Monoclonal Gammopathy of Undetermined Significance Plasma Cells Costimulates Pamidronate-Activated γδ Lymphocytes. Cancer Res. 2005, 65, 7502–7508. [Google Scholar] [CrossRef]
- Wilhelm, M.; Kunzmann, V.; Eckstein, S.; Reimer, P.; Weissinger, F.; Ruediger, T.; Tony, H.-P. γδ T cells for immune therapy of patients with lymphoid malignancies. Blood 2003, 102, 200–206. [Google Scholar] [CrossRef]
- Abe, Y.; Muto, M.; Nieda, M.; Nakagawa, Y.; Nicol, A.; Kaneko, T.; Goto, S.; Yokokawa, K.; Suzuki, K. Clinical and immunological evaluation of zoledronate-activated Vγ9γδ T-cell-based immunotherapy for patients with multiple myeloma. Exp. Hematol. 2009, 37, 956–968. [Google Scholar] [CrossRef]
- Castella, B.; Foglietta, M.; Sciancalepore, P.; Rigoni, M.; Coscia, M.; Griggio, V.; Vitale, C.; Ferracini, R.; Saraci, E.; Omedã©, P.; et al. Anergic bone marrow Vγ9Vδ2 T cells as early and long-lasting markers of PD-1-targetable microenvironment-induced immune suppression in human myeloma. Oncoimmunology 2015, 4, e1047580. [Google Scholar] [CrossRef]
- Möller, C.; Strömberg, T.; Juremalm, M.; Nilsson, K.; Nilsson, G. Expression and function of chemokine receptors in human multiple myeloma. Leukemia 2003, 17, 203–210. [Google Scholar] [CrossRef]
- Redondo-Muñoz, J.; García-Pardo, A.; Teixidó, J. Molecular Players in Hematologic Tumor Cell Trafficking. Front. Immunol. 2019, 10, 156. [Google Scholar] [CrossRef]
- Asosingh, K.; Günthert, U.; De Raeve, H.; Van Riet, I.; Van Camp, B.; Vanderkerken, K. A unique pathway in the homing of murine multiple myeloma cells: CD44v10 mediates binding to bone marrow endothelium. Cancer Res. 2001, 61, 2862–2865. [Google Scholar]
- Neri, P.; Ren, L.; Azab, A.K.; Brentnall, M.; Gratton, K.; Klimowicz, A.C.; Lin, C.; Duggan, P.; Tassone, P.; Mansoor, A.; et al. Integrin β7-mediated regulation of multiple myeloma cell adhesion, migration, and invasion. Blood 2011, 117, 6202–6213. [Google Scholar] [CrossRef]
- De Clercq, E. Potential clinical applications of the CXCR4 antagonist bicyclam AMD3100. Mini-Rev. Med. Chem. 2005, 5, 805–824. [Google Scholar] [CrossRef]
- Alsayed, Y.; Ngo, H.; Runnels, J.; Leleu, X.; Singha, U.K.; Pitsillides, C.M.; Spencer, J.A.; Kimlinger, T.; Ghobrial, J.M.; Jia, X.; et al. Mechanisms of regulation of CXCR4/SDF-1 (CXCL12)–dependent migration and homing in multiple myeloma. Blood 2006, 109, 2708–2717. [Google Scholar] [CrossRef]
- Robak, P.; Węgłowska, E.; Dróżdż, I.; Mikulski, D.; Jarych, D.; Ferlińska, M.; Wawrzyniak, E.; Misiewicz, M.; Smolewski, P.; Fendler, W.; et al. Cytokine and Chemokine Profile in Patients with Multiple Myeloma Treated with Bortezomib. Mediat. Inflamm. 2020, 2020, 1835836. [Google Scholar] [CrossRef]
- Wang, X.-T.; He, Y.-C.; Zhou, S.-Y.; Jiang, J.-Z.; Huang, Y.-M.; Liang, Y.-Z.; Lai, Y.-R. Bone marrow plasma macrophage inflammatory protein protein-1 alpha(MIP-1 alpha) and sclerostin in multiple myeloma: Relationship with bone disease and clinical characteristics. Leuk. Res. 2014, 38, 525–531. [Google Scholar] [CrossRef]
- Xu, R.; Li, Y.; Yan, H.; Zhang, E.; Huang, X.; Chen, Q.; Chen, J.; Qu, J.; Liu, Y.; He, J.; et al. CCL2 promotes macrophages-associated chemoresistance via MCPIP1 dual catalytic activities in multiple myeloma. Cell Death Dis. 2019, 10, 781. [Google Scholar] [CrossRef] [PubMed]
- Zahoor, M.; Westhrin, M.; Aass, K.R.; Moen, S.H.; Misund, K.; Psonka-Antonczyk, K.M.; Giliberto, M.; Buene, G.; Sundan, A.; Waage, A.; et al. Hypoxia promotes IL-32 expression in myeloma cells, and high expression is associated with poor survival and bone loss. Blood Adv. 2017, 1, 2656–2666. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Ning, F.-Y.; Wang, J.-H.; Yan, H.-M.; Kong, H.-W.; Zhang, Y.-T.; Shen, Q. Expression of interleukin-32 in bone marrow of patients with myeloma and its prognostic significance. World J. Clin. Cases 2019, 7, 4234–4244. [Google Scholar] [CrossRef] [PubMed]
- Musolino, C.; Allegra, A.; Innao, V.; Allegra, A.G.; Pioggia, G.; Gangemi, S. Inflammatory and Anti-Inflammatory Equilibrium, Proliferative and Antiproliferative Balance: The Role of Cytokines in Multiple Myeloma. Mediat. Inflamm. 2017, 2017, 1852517. [Google Scholar] [CrossRef] [PubMed]
- Lauta, V.M. A review of the cytokine network in multiple myeloma. Cancer 2003, 97, 2440–2452. [Google Scholar] [CrossRef]
- Podar, K.; Tai, Y.-T.; Davies, F.; Lentzsch, S.; Sattler, M.; Hideshima, T.; Lin, B.K.; Gupta, D.; Shima, Y.; Chauhan, D.; et al. Vascular endothelial growth factor triggers signaling cascades mediating multiple myeloma cell growth and migration. Blood 2001, 98, 428–435. [Google Scholar] [CrossRef]
- Palano, M.T.; Giannandrea, D.; Platonova, N.; Gaudenzi, G.; Falleni, M.; Tosi, D.; Lesma, E.; Citro, V.; Colombo, M.; Saltarella, I.; et al. Jagged Ligands Enhance the Pro-Angiogenic Activity of Multiple Myeloma Cells. Cancers 2020, 12, 2600. [Google Scholar] [CrossRef]
- Korde, N.; Kristinsson, S.Y.; Landgren, O. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM): Novel biological insights and development of early treatment strategies. Blood 2011, 117, 5573–5581. [Google Scholar] [CrossRef]
- Rajkumar, S.V. Prevention of Progression in Monoclonal Gammopathy of Undetermined Significance. Clin. Cancer Res. 2009, 15, 5606–5608. [Google Scholar] [CrossRef]
- Hjorth, M.; Hellquist, L.; Holmberg, E.; Magnusson, B.; Rödjer, S.; Westin, J. Initial versus deferred melphalan-prednisone therapy for asymptomatic multiple myeloma stage I—A randomized study. Eur. J. Haematol. 2009, 50, 95–102. [Google Scholar] [CrossRef]
- Riccardi, A.; Ucci, G.; Luoni, R.; Brugnatelli, S.; Mora, O.; Spanedda, R.; De Paoli, A.; Barbarano, L.; Di Stasi, M.; Alberio, F.; et al. Treatment of multiple myeloma according to the extension of the disease: A prospective, randomised study comparing a less with a more aggressive cytostatic policy. Br. J. Cancer 1994, 70, 1203–1210. [Google Scholar] [CrossRef]
- Riccardi, A.; Mora, O.; Tinelli, C.; Valentini, D.; Brugnatelli, S.; Spanedda, R.; De Paoli, A.; Barbarano, L.; Di Stasi, M.; Giordano, M.; et al. Long-term survival of stage I multiple myeloma given chemotherapy just after diagnosis or at progression of the disease: A multicentre randomized study. Br. J. Cancer 2000, 82, 1254–1260. [Google Scholar] [CrossRef]
- Musto, P.; Falcone, A.; Sanpaolo, G.; Bodenizza, C.; Cascavilla, N.; Melillo, L.; Scalzulli, P.R.; Del L’Olio, M.; La Sala, A.; Mantuano, S.; et al. Pamidronate Reduces Skeletal Events but does not Improve Progression-free Survival in Early-stage Untreated Myeloma: Results of a Randomized Trial. Leuk. Lymphoma 2003, 44, 1545–1548. [Google Scholar] [CrossRef]
- d’Arena, G.; Gobbi, P.G.; Broglia, C.; Sacchi, S.; Quarta, G.; Baldini, L.; Iannitto, E.; Falcone, A.; Guariglia, R.; Pietrantuono, G.; et al. Pamidronate versus observation in asymptomatic myeloma: Final results with long-term follow-up of a randomized study. Leuk. Lymphoma 2011, 52, 771–775. [Google Scholar] [CrossRef]
- Musto, P.; Petrucci, M.T.; Bringhen, S.; Guglielmelli, T.; Caravita, T.; Bongarzoni, V.; Andriani, A.; D’Arena, G.; Balleari, E.; Pietrantuono, G.; et al. A multicenter, randomized clinical trial comparing zoledronic acid versus observation in patients with asymptomatic myeloma. Cancer 2008, 113, 1588–1595. [Google Scholar] [CrossRef]
- Pozzi, S.; Raje, N. The Role of Bisphosphonates in Multiple Myeloma: Mechanisms, Side Effects, and the Future. Oncologist 2011, 16, 651–662. [Google Scholar] [CrossRef]
- Mateos, M.V.; Hernandez, M.T.; Giraldo, P.; de la Rubia, J.; de Arriba, F.; Lopez Corral, L.; Rosinol, L.; Paiva, B.; Palomera, L.; Bargay, J.; et al. Lenalidomide plus Dexamethasone for High-Risk Smoldering Multiple Myeloma. N. Engl. J. Med. 2013, 369, 438–447. [Google Scholar] [CrossRef]
- Mateos, M.-V.; Hernández, M.-T.; Giraldo, P.; de la Rubia, J.; de Arriba, F.; Corral, L.L.; Rosiñol, L.; Paiva, B.; Palomera, L.; Bargay, J.; et al. Lenalidomide plus dexamethasone versus observation in patients with high-risk smouldering multiple myeloma (QuiRedex): Long-term follow-up of a randomised, controlled, phase 3 trial. Lancet Oncol. 2016, 17, 1127–1136. [Google Scholar] [CrossRef]
- Paiva, B.; Mateos, M.V.; Sanchez-Abarca, L.I.; Puig, N.; Vidriales, M.-B.; López-Corral, L.; Corchete, L.A.; Hernandez, M.T.; Bargay, J.; de Arriba, F.; et al. Immune status of high-risk smoldering multiple myeloma patients and its therapeutic modulation under LenDex: A longitudinal analysis. Blood 2016, 127, 1151–1162. [Google Scholar] [CrossRef]
- Lonial, S.; Jacobus, S.; Fonseca, R.; Weiss, M.; Kumar, S.; Orlowski, R.Z.; Kaufman, J.L.; Yacoub, A.M.; Buadi, F.K.; O’Brien, T.; et al. Randomized Trial of Lenalidomide Versus Observation in Smoldering Multiple Myeloma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2019, 38, 1126–1137. [Google Scholar] [CrossRef]
- Kuhn, D.J.; Chen, Q.; Voorhees, P.M.; Strader, J.S.; Shenk, K.D.; Sun, C.M.; Demo, S.D.; Bennett, M.K.; van Leeuwen, F.; Chanan-Khan, A.A.; et al. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood 2007, 110, 3281–3290. [Google Scholar] [CrossRef] [PubMed]
- Mailankody, S.; Kazandjian, D.; Korde, N.; Roschewski, M.; Manasanch, E.; Bhutani, M.; Tageja, N.; Kwok, M.; Zhang, Y.; Zingone, A.; et al. Baseline mutational patterns and sustained MRD negativity in patients with high-risk smoldering myeloma. Blood Adv. 2017, 1, 1911–1918. [Google Scholar] [CrossRef] [PubMed]
- Kazandjian, D.; Hill, E.; Dew, A.; Morrison, C.; Roswarski, J.; Korde, N.; Emanuel, M.; Petrosyan, A.; Bhutani, M.; Calvo, K.R.; et al. Carfilzomib, Lenalidomide, and Dexamethasone Followed by Lenalidomide Maintenance for Prevention of Symptomatic Multiple Myeloma in Patients with High-risk Smoldering Myeloma. JAMA Oncol. 2021, 7, 1678. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.B.; Yee, A.J.; Raje, N. Treatment of Smoldering Multiple Myeloma: Ready for Prime Time? Cancers 2020, 12, 1223. [Google Scholar] [CrossRef] [PubMed]
- Puíg, N.; Contreras, T.; Paiva, B.; Cedena, M.T.; Martinez-Lopez, J.; Oriol, A.; Gutiérrez, N.; Ríos-Tamayo, R.; Rosiñol, L.; Calasanz, M.J.; et al. Analysis of treatment efficacy in the GEM-CESAR trial for high-risk smoldering multiple myeloma patients: Comparison between the standard and IMWG MRD criteria and QIP-MS including FLC (QIP-FLC-MS). J. Clin. Oncol. 2020, 38, 8512. [Google Scholar] [CrossRef]
- Goodman, A.M.; Kim, M.S.; Prasad, V. Persistent challenges with treating multiple myeloma early. Blood 2021, 137, 456–458. [Google Scholar] [CrossRef]
- Fazzi, R.; Petrini, I.; Giuliani, N.; Morganti, R.; Carulli, G.; Palma, B.D.; Notarfranchi, L.; Galimberti, S.; Buda, G. Phase II Trial of Maintenance Treatment With IL2 and Zoledronate in Multiple Myeloma After Bone Marrow Transplantation: Biological and Clinical Results. Front. Immunol. 2021, 11, 573156. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Kumar, A.; Aggarwal, M.S.; Shishodia, S. Curcumin Derived from Turmeric (Curcuma longa): A Spice for All Seasons. Phytopharm. Cancer Chemoprev. 2005, 23, 351–387. [Google Scholar]
- Bharti, A.C.; Donato, N.; Singh, S.; Aggarwal, B.B. Curcumin (diferuloylmethane) down-regulates the constitutive activation of nuclear factor–κB and IκBα kinase in human multiple myeloma cells, leading to suppression of proliferation and induction of apoptosis. Blood 2003, 101, 1053–1062. [Google Scholar] [CrossRef]
- Bharti, A.C.; Takada, Y.; Aggarwal, B.B. Curcumin (Diferuloylmethane) Inhibits Receptor Activator of NF-κB Ligand-Induced NF-κB Activation in Osteoclast Precursors and Suppresses Osteoclastogenesis. J. Immunol. 2004, 172, 5940–5947. [Google Scholar] [CrossRef]
- Golombick, T.; Diamond, T.H.; Badmaev, V.; Manoharan, A.; Ramakrishna, R. The Potential Role of Curcumin in Patients with Monoclonal Gammopathy of Undefined Significance—Its Effect on Paraproteinemia and the Urinary N-Telopeptide of Type I Collagen Bone Turnover Marker. Clin. Cancer Res. 2009, 15, 5917–5922. [Google Scholar] [CrossRef]
- International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: A report of the International Myeloma Working Group. Br. J. Haematol. 2003, 121, 749–757. [Google Scholar] [CrossRef]
- Sharma, R.A.; McLelland, H.R.; Hill, K.A.; Ireson, C.R.; Euden, S.A.; Manson, M.M.; Pirmohamed, M.; Marnett, L.J.; Gescher, A.J.; Steward, W.P. Pharmacodynamic and pharmacokinetic study of oral Curcuma extract in patients with colorectal cancer. Clin. Cancer Res. 2001, 7, 1894–1900. [Google Scholar]
- Sharma, R.A.; Euden, S.A.; Platton, S.L.; Cooke, D.N.; Shafayat, A.; Hewitt, H.R.; Marczylo, T.H.; Morgan, B.; Hemingway, D.; Plummer, S.M.; et al. Phase I Clinical Trial of Oral Curcumin. Clin. Cancer Res. 2004, 10, 6847–6854. [Google Scholar] [CrossRef]
- Lao, C.D.; Ruffin, M.T.; Normolle, D.; Heath, D.D.; Murray, S.I.; Bailey, J.M.; Boggs, M.E.; Crowell, J.; Rock, C.L.; Brenner, D.E. Dose escalation of a curcuminoid formulation. BMC Complement. Altern. Med. 2006, 6, 10. [Google Scholar] [CrossRef]
- Aaronson, N.K.; Ahmedzai, S.; Bergman, B.; Bullinger, M.; Cull, A.; Duez, N.J.; Filiberti, A.; Flechtner, H.; Fleishman, S.B.; De Haes, J.C.J.M.; et al. The European Organization for Research and Treatment of Cancer QLQ-C30: A Quality-of-Life Instrument for Use in International Clinical Trials in Oncology. J. Natl. Cancer Inst. 1993, 85, 365–376. [Google Scholar] [CrossRef]
- Suzuki, K.; Nishiwaki, K.; Yano, S. Treatment Strategies Considering Micro-Environment and Clonal Evolution in Multiple Myeloma. Cancers 2021, 13, 215. [Google Scholar] [CrossRef]
- Landgren, C.O.; Chari, A.; Cohen, Y.C.; Spencer, A.; Voorhees, P.; Estell, J.A.; Sandhu, I.; Jenner, M.W.; Williams, C.; Cavo, M.; et al. Daratumumab monotherapy for patients with intermediate-risk or high-risk smoldering multiple myeloma: A randomized, open-label, multicenter, phase 2 study (CENTAURUS). Leukemia 2020, 34, 1840–1852. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Voorhees, P.M.; Goldschmidt, H.; Baker, R.I.; Shi, Y.; Rousseau, E.; Dennis, R.M.; Carson, R.L.; Rajkumar, S.V. Subcutaneous daratumumab (DARA SC) versus active monitoring in patients (pts) with high-risk smoldering multiple myeloma (SMM): Randomized, open-label, phase 3 AQUILA study. J. Clin. Oncol. 2022, 40, TPS8075. [Google Scholar] [CrossRef]
- Zamagni, Z.; Tacchetti, T.; Deias, D.; Patriarca, P. The Role of Monoclonal Antibodies in Smoldering and Newly Diagnosed Transplant-Eligible Multiple Myeloma. Pharmaceuticals 2020, 13, 451. [Google Scholar] [CrossRef]
- Moreno, L.; Perez, C.; Zabaleta, A.; Manrique, I.; Alignani, D.; Ajona, D.; Blanco, L.; Lasa, M.; Maiso, P.; Rodriguez, I.; et al. The Mechanism of Action of the Anti-CD38 Monoclonal Antibody Isatuximab in Multiple Myeloma. Clin. Cancer Res. 2019, 25, 3176–3187. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.; Bringhen, S.; Anttila, P.; Capra, M.; Cavo, M.; Cole, C.; Gasparetto, C.; Hungria, V.; Jenner, M.; Vorobyev, V.; et al. Isatuximab as monotherapy and combined with dexamethasone in patients with relapsed/refractory multiple myeloma. Blood 2021, 137, 1154–1165. [Google Scholar] [CrossRef] [PubMed]
- Manasanch, E.E.; Jagannath, S.; Lee, H.C.; Patel, K.K.; Graham, C.; Kaufman, G.P.; Thomas, S.K.; Iyer, S.; Mailankody, S.; Korde, N.; et al. A Multicenter Phase II Single Arm Trial of Isatuximab in Patients with High Risk Smoldering Multiple Myeloma (HRSMM). Blood 2019, 134, 3116. [Google Scholar] [CrossRef]
- Nooka, A.K.; Wang, M.L.; Yee, A.J.; Kaufman, J.L.; Bae, J.; Peterkin, D.; Richardson, P.G.; Raje, N.S. Assessment of Safety and Immunogenicity of PVX-410 Vaccine With or Without Lenalidomide in Patients With Smoldering Multiple Myeloma: A Nonrandomized Clinical Trial. JAMA Oncol. 2018, 4, e183267. [Google Scholar] [CrossRef]
- Padala, S.A.; Barsouk, A.; Barsouk, A.; Rawla, P.; Vakiti, A.; Kolhe, R.; Kota, V.; Ajebo, G.H. Epidemiology, Staging, and Management of Multiple Myeloma. Med. Sci. 2021, 9, 3. [Google Scholar] [CrossRef]
- Jagannath, S.; Laubach, J.; Wong, E.; Stockerl-Goldstein, K.; Rosenbaum, C.; Dhodapkar, M.; Jou, Y.-M.; Lynch, M.; Robbins, M.; Shelat, S.; et al. Elotuzumab monotherapy in patients with smouldering multiple myeloma: A phase 2 study. Br. J. Haematol. 2018, 182, 495–503. [Google Scholar] [CrossRef]
- Kyle, R.A.; Durie, B.G.; Rajkumar, S.V.; Landgren, O.; Blade, J.; Merlini, G.; Kroger, N.; Einsele, H.; Vesole, D.H.; Dimopoulos, M.; et al. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia 2010, 24, 1121–1127. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Plasma Cell Disorder | Diagnosis | Note |
|---|---|---|
| Non-IgM MGUS (IgG, IgA, IgD) | Serum monoclonal protein < 3 g/dL) Clonal plasma cells in the bone marrow < 10% * Absence of end-organ damage (CRAB symptoms) or amyloidosis that can be attributed to the plasma cell proliferative disorder | Up to 85% of MGUS cases Annual risk of progression of 1% * Bone marrow can be deferred in patients with low-risk MGUS (IgG type, M protein < 15 gm/L, normal free light chain ratio) in whom there are no clinical features concerning myeloma |
| IgM MGUS | Serum IgM monoclonal protein < 3 g/dL) Clonal plasma cells in the bone marrow < 10% No evidence of anemia, constitutional symptoms, hyperviscosity, lymphadenopathy, hepatosplenomegaly, or other end-organ damage that can be attributed to the plasma cell proliferative disorder | 15% of MGUS cases |
| Light chain MGUS | Abnormal FCL ratio Increased level of involved FLC No immunoglobulin heavy chain expression on immunofissation Absence of CRAB symptoms or amyloidosis that can be attributed to the plasma cell proliferative disorder Clonal plasma cells in the bone marrow < 10% Urinary monoclonal protein < 500 mg/24 h | |
| Monoclonal gammopathy of renal significance (MGRS) | One or more renal lesions related to the monoclonal immunoglobulin produced The underlying B-cell or plasma cell clone neither causes tumor complications nor meets any current hematologic criteria for specific therapy The diagnosis of MGRS can only be established with renal biopsy | Based on consensus report of the International Kidney and Monoclonal Gammopathy Research Group [6] |
| Monoclonal gammopathy of neurologic significance (MGNS) | Chronic neuropathy with sensory ataxia, ocular, and/or bulbar motor weakness in the presence of a monoclonal IgM reacting against gangliosides containing disialosyl epitopes The diagnosis of MGNS is one of exclusion | |
| Smoldering Multiple Myeloma | Serum monoclonal protein (IgM or IgA) ≥ 3 g/dL) or urinary monoclonal protein ≥ 500 mg/24 h Clonal plasma cells in the bone marrow 10–60% Absence of MDE or amyloidosis | Based on Mayo 2018 criteria [7] there are three groups of patients:
|
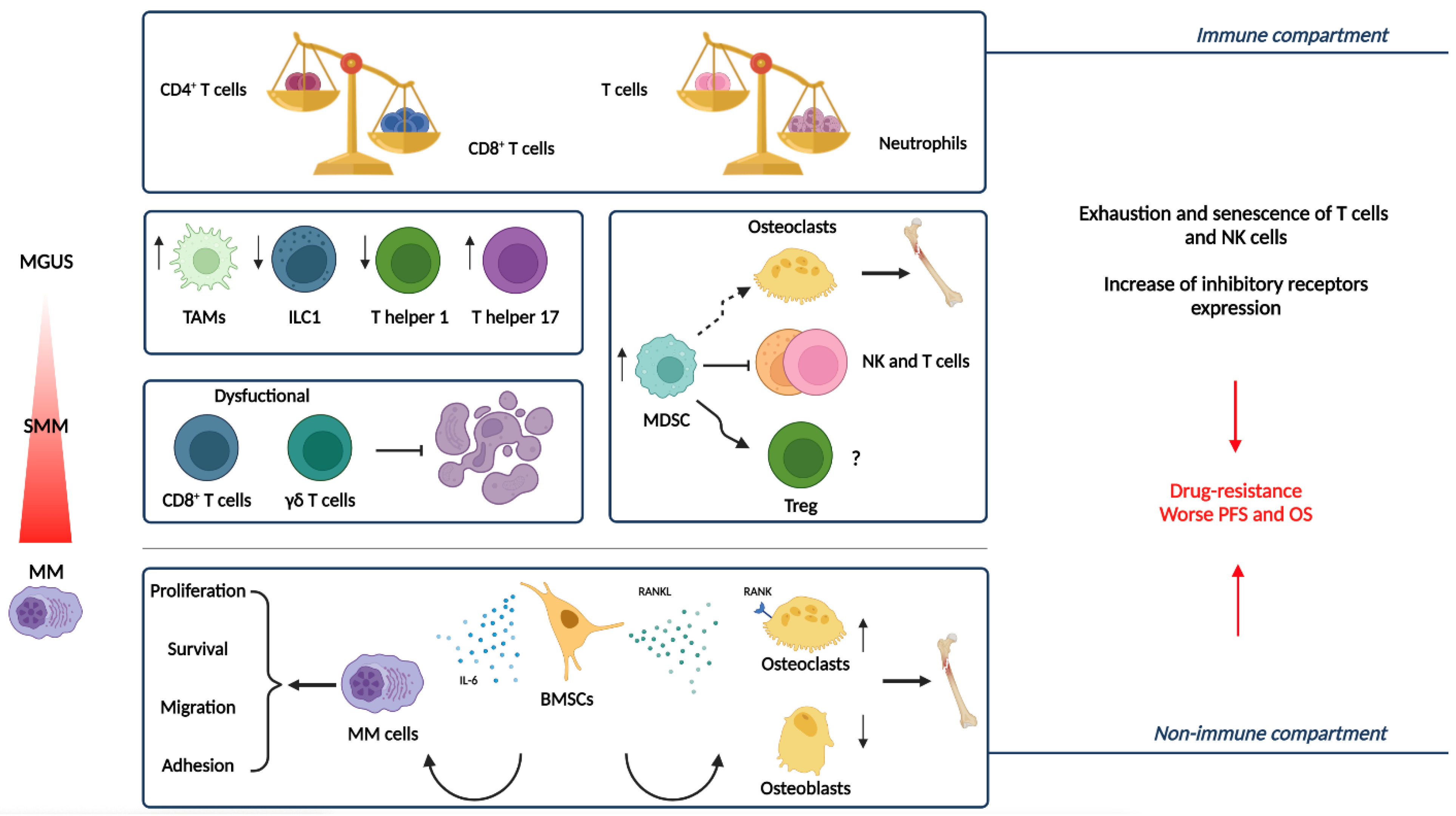
| Cellular Component | Function | References |
|---|---|---|
| MDSCs | Hamper the anti-tumor immune response by multiple mechanisms dependent on:
Induce Treg development Differentiate into osteoclasts, contributing to the formation of osteolytic lesions | [27,28,29,30,31,32] |
| BMSCs | Generate premetastatic niche:
| [8,33,34,35,36] |
| Osteoclasts | Create an immunosuppressive microenvironment:
| [37,38,39] |
| Dendritic cells | Immunosuppressive and tumor-promoting actions:
| [40,41] |
| TAMs | Support MM cells proliferation and survival through activation of the IL-6/JAK/STAT3 pathway. Decrease T cell proliferation and activation through the downregulation of IFN-γ, IL-2, and TNF-α secretion. Immune suppressive activity mediated by their secretion of IL-6, IL-10, activating Tregs and M2 macrophages, and TGF-β, inhibiting both cytotoxic T-cells and NK cells Angiogenic and vasculogenic activities:
| [25,42] |
| Neutrophils | Overexpress IFN-γ, resulting in increased autophagy flux and JAK-2/STAT3 pathway activation, which supports their promotion of pro-inflammatory and survival signals within MM niches. Produce arginase that inhibits T cell activation and proliferation. Reduced lysozyme activity and increased secretion of the amino acid degrading enzyme. | [9,43] |
| T cells | Th17 cells:
| [44,45] |
| Clinical Trial | Phase | Therapeutic Regimen | Patients | Follow-Up and Results | |
|---|---|---|---|---|---|
| Lenalidomide-Based Treatments | QuiRedex- NCT00480363 | III | RD +/− R maintenance for 2 years | 119 High-risk SMM | Median FU: 10.8 years HR OS: 46% HR PFS: 73% median TTP: 9.0 years (treatment arm) vs. 2.1 years (control arm) median OS: not reached (treatment arm) vs. 7.8 years (control arm) |
| ECOG E3A06-NCT01169337 | III | R | 182 Intermediate/high-risk SMM | Median FU: 35 months HR PFS: 72% 3 years PFS: 91% (treatment arm) vs. 66% (control group) | |
| Proteasome Inhibitor-Based Treatments | NCT01572480 | II | KRd + R maintenance | 18 High-Risk SMM | Median FU: 43.3 months MRD-negative: 63% Estimated 4 years PFS: 71% Estimated 4 years OS: 100% |
| GEM-CESARNCT02415413 | II | KRd + high-dose Melphalan and ASCT + Rd maintenance for up to 2 years | 90 High/ultra high-risk SMM | Median FU: 32 months OS: 98% PFS: 93% biochemical relapses: 5 patients ORR after induction: 98% ORR after ASCT: 98% ORR after consolidation: 100% CR: 68.6% (55% of them achieving MRD negativity) | |
| NCT02916771 | II | IxRd + IxR mainteinance | 26 (56 planned) High-risk SMM | ORR: 89% (after at least 3 cycles of treatment) CR: 19.2% No progression to active MM. | |
| Monoclonal antibody-Based Treatments | CENTAURUS NCT02316106 | II | Dara (three different treatment schedules: extended intense, extended intermediate and short dosing) | 123 Intermediate/high-risk SMM | Median FU: 26 months CR: 4.9%, 9.8%, 0% (respectively, in the three treatment schedules) 2 years PFS: 89.9%, 82%, 75,3% (respectively, in the three treatment schedules) |
| NCT03236428 | II | Dara | 28 Lower-risk SMM | PR: 53% VGPR: 20% | |
| NCT02960555 | II | Isatuximab | 24 High-risk SMM | PR: 42% VGPR:17% CR with MRD negativity: 5% | |
| NCT01441973 | II | Elo | 31 | FU > 28 months Modest activity of Elo monotherapy. ORR: 10% 2-year PFS: 69% | |
| NCT02279394 | II | EloRd + EloR | 50 High-risk SMM | PR: 84% No progression to active MM. | |
| NCT02603887 | Pilot study | Pembrolizumab | 13 Intermediate/high-risk SMM | After a median of 8 cycles: 85% CR, 15% progressed to active MM, 8% MRD negativity (for up 27 months) | |
| NCT01484275 | Pilot study | Siltuximab | 85 High-risk SMM | Median FU: 29.2 months 1-year PFS: 84.5% (siltuximab) vs. 74.4% (placebo) median PFS: not reached (siltuximab) vs. 23.5 months (placebo) OS: not reached in both arms | |
| Vaccines | NCT01718899 | I/IIa | PVX410 +/− R | 20 Intermediate/high-risk SMM | progressions: 3 (PVX-410-alone) vs. 1 (PVX-410 + R) median TTP: 36 weeks (PVX-410-alone) vs. not reached (PVX-410 + R) |
| Ibrutinib | NCT02943473 | II | Ibrutinib | 9 High-risk SMM | poor efficacy unfavorable risk/benefit ratio |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plano, F.; Corsale, A.M.; Gigliotta, E.; Camarda, G.; Vullo, C.; Di Simone, M.; Shekarkar Azgomi, M.; Speciale, M.; Carlisi, M.; Caccamo, N.; et al. Monoclonal Gammopathies and the Bone Marrow Microenvironment: From Bench to Bedside and Then Back Again. Hematol. Rep. 2023, 15, 23-49. https://doi.org/10.3390/hematolrep15010004
Plano F, Corsale AM, Gigliotta E, Camarda G, Vullo C, Di Simone M, Shekarkar Azgomi M, Speciale M, Carlisi M, Caccamo N, et al. Monoclonal Gammopathies and the Bone Marrow Microenvironment: From Bench to Bedside and Then Back Again. Hematology Reports. 2023; 15(1):23-49. https://doi.org/10.3390/hematolrep15010004
Chicago/Turabian StylePlano, Federica, Anna Maria Corsale, Emilia Gigliotta, Giulia Camarda, Candida Vullo, Marta Di Simone, Mojtaba Shekarkar Azgomi, Maria Speciale, Melania Carlisi, Nadia Caccamo, and et al. 2023. "Monoclonal Gammopathies and the Bone Marrow Microenvironment: From Bench to Bedside and Then Back Again" Hematology Reports 15, no. 1: 23-49. https://doi.org/10.3390/hematolrep15010004
APA StylePlano, F., Corsale, A. M., Gigliotta, E., Camarda, G., Vullo, C., Di Simone, M., Shekarkar Azgomi, M., Speciale, M., Carlisi, M., Caccamo, N., Dieli, F., Meraviglia, S., Siragusa, S., & Botta, C. (2023). Monoclonal Gammopathies and the Bone Marrow Microenvironment: From Bench to Bedside and Then Back Again. Hematology Reports, 15(1), 23-49. https://doi.org/10.3390/hematolrep15010004