
Acquired Hemophilia A Treated with Recombinant Porcine Factor VIII: Case Report and Literature Review on Its Efficacy

 ,
, {kind=link}
{kind=link}
Abstract
1. Introduction
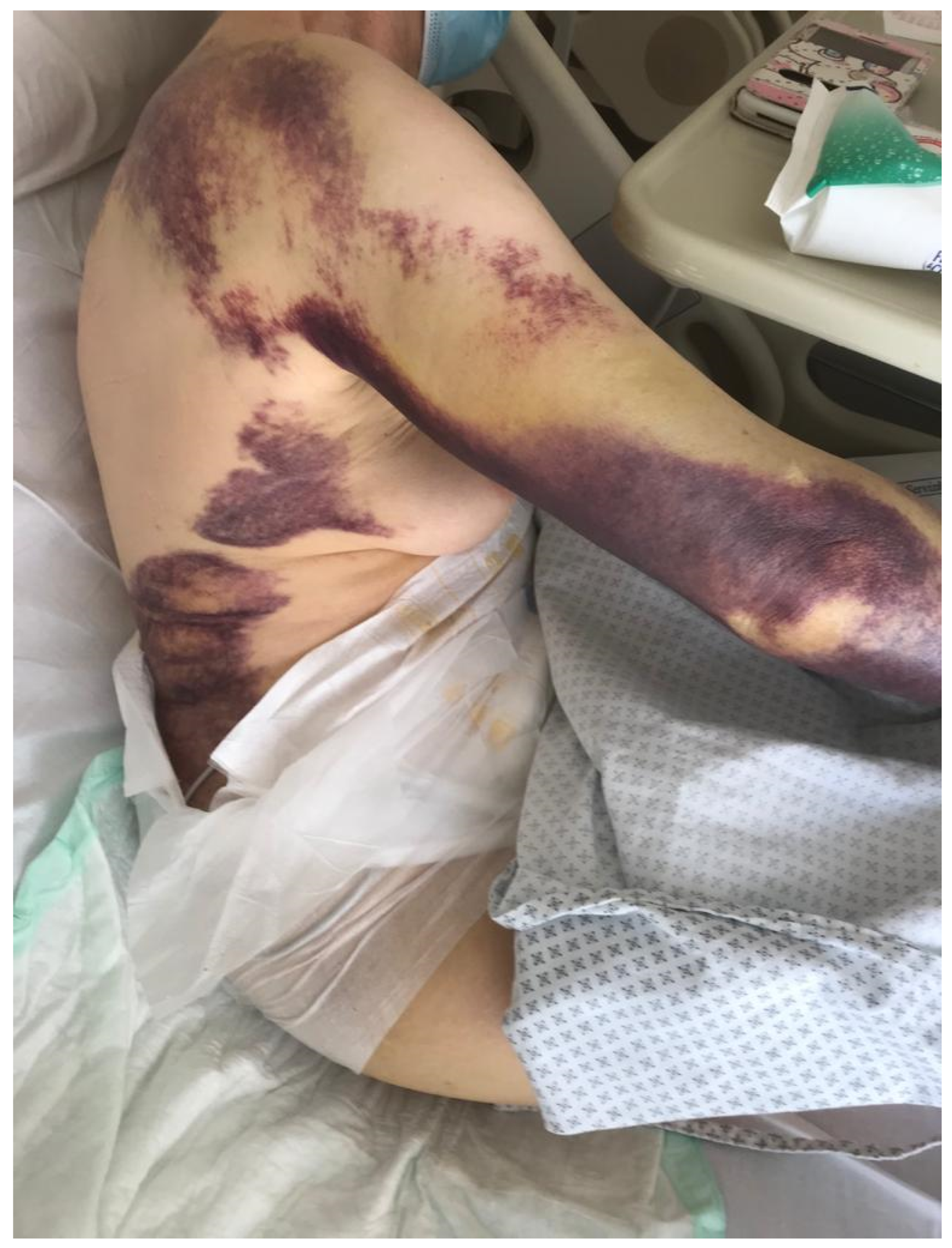
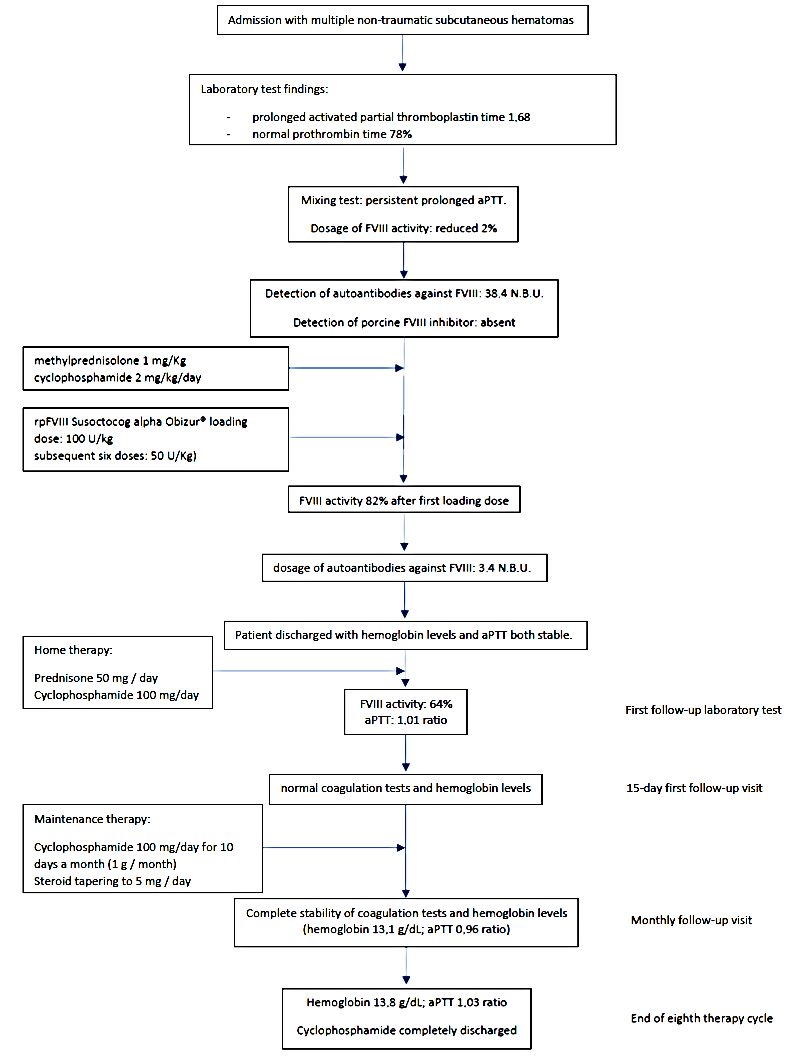
2. Case Presentation
3. Discussion and Literature Review
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tiede, A.; Collins, P.; Knoebl, P.; Teitel, J.; Kessler, C.; Shima, M.; Di Minno, G.; Oiron, R.D.; Salaj, P.; Jiménez-Yuste, V.; et al. International recommendations on the diagnosis and treatment of acquired hemophilia A. Haematologica 2020, 105, 1791–1801. [Google Scholar] [CrossRef] [PubMed]
- Knoebl, P.; Marco, P.; Baudo, F.; Collins, P.; Huth-Kühne, A.; Nemes, L.; Pellegrini, F.; Tengborn, L.; Lévesque, H.; Contributors, O.B.O.T.E.R. Demographic and clinical data in acquired hemophilia A: Results from the European Acquired Haemophilia Registry (EACH2). J. Thromb. Haemost. 2012, 10, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, M.; Siragusa, S.; Mancuso, S.; Kessler, C.M. Acquired haemophilia in cancer: A systematic and critical literature review. Haemophilia 2018, 24, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Kruse-Jarres, R.; Kempton, C.L.; Baudo, F.; Collins, P.W.; Knoebl, P.; Leissinger, C.A.; Tiede, A.; Kessler, C.M. Acquired hemophilia A: Updated review of evidence and treatment guidance. Am. J. Hematol. 2017, 92, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Tiede, A.; Klamroth, R.; Scharf, R.E.; Trappe, R.U.; Holstein, K.; Huth-Kühne, A.; Gottstein, S.; Geisen, U.; Schenk, J.; Scholz, U.; et al. Prognostic factors for remission of and survival in acquired hemophilia A (AHA): Results from the GTH-AH 01/2010 study. Blood 2015, 125, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Huth-Kühne, A.; Baudo, F.; Collins, P.; Ingerslev, J.; Kessler, C.M.; Lévesque, H.; Castellano, M.E.M.; Shima, M.; St-Louis, J. International recommendations on the diagnosis and treatment of patients with acquired hemophilia A. Haematologica 2009, 94, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Holstein, K.; Liu, X.; Smith, A.; Knöbl, P.; Klamroth, R.; Geisen, U.; Eichler, H.; Miesbach, W.; Tiede, A. Bleeding and response to hemostatic therapy in acquired hemophilia A: Results from the GTH-AH 01/2010 study. Blood 2020, 136, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Tiede, A. Critical Bleeding in Acquired Hemophilia A: Bypassing Agents or Recombinant Porcine Factor VIII? Hamostaseologie 2020, 41, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Aledort, L.; Ljung, R.; Mann, K.; Pipe, S. Factor VIII therapy for hemophilia A: Current and future issues. Expert Rev. Hematol. 2014, 7, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Kruse-Jarres, R.; St-Louis, J.; Greist, A.; Shapiro, A.; Smith, H.; Chowdary, P.; Drebes, A.; Gomperts, E.; Bourgeois, C.; Mo, M.; et al. Efficacy and safety of OBI-1, an antihaemophilic factor VIII (recombinant), porcine sequence, in subjects with acquired haemophilia A. Haemophilia 2015, 21, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, M.D.; Cuker, A.; Hardesty, B.; Roberts, J.C.; Sholzberg, M. Recombinant porcine sequence factor VIII (rpFVIII) for acquired haemophilia A: Practical clinical experience of its use in seven patients. Haemophilia 2017, 23, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.; Kasthuri, R.; Mooberry, M.J.; Chen, S.L.; Key, N.S.; Ma, A.D. Lower doses of recombinant porcine factor VIII maintain excellent haemostatic efficacy. Haemophilia 2016, 22, e549–e551. [Google Scholar] [CrossRef] [PubMed]
- Stemberger, M.; Mohnle, P.; Tschop, J.; Ney, L.; Spannagl, M.; Reincke, M. Successful bleeding control with recombinant porcine factor VIII in reduced loading doses in two patients with acquired haemophilia A and failure of bypassing agent therapy. Haemophilia 2016, 22, e472–e474. [Google Scholar] [CrossRef]
- Zanon, E.; Pasca, S.; Borchiellini, A.; Lodigiani, C.; Molinari, A.C.; Ambaglio, C.; Valeri, F.; Preti, P.S.; Moscatelli, P.; Simioni, P. Susoctocog-alfa (Obizur((R))) in the treatment of nine elderly patients with acquired haemophilia A: An Italian multicentre real world experience. Blood Transfus. 2020, 18, 312–321. [Google Scholar]
- Burness, C.B.; Scott, L.J. Susoctocog Alfa: A Review in Acquired Haemophilia, A. Drugs 2016, 76, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Tiede, A.; Worster, A. Lessons from a systematic literature review of the effectiveness of recombinant factor VIIa in acquired haemophilia. Ann. Hematol. 2018, 97, 1889–1901. [Google Scholar] [CrossRef] [PubMed]
- Baudo, F.; Collins, P.; Huth-Kühne, A.; Levesque, H.; Marco, P.; Nemes, L.; Pellegrini, F.; Tengborn, L.; Knoebl, P. Management of bleeding in acquired hemophilia A: Results from the European Acquired Haemophilia (EACH2) Registry. Blood 2012, 120, 39–46. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borro, M.; Tassara, R.; Paris, L.; Artom, N.; Brignone, M.; Rebella, L.; Tassara, R. Acquired Hemophilia A Treated with Recombinant Porcine Factor VIII: Case Report and Literature Review on Its Efficacy. Hematol. Rep. 2023, 15, 17-22. https://doi.org/10.3390/hematolrep15010003
Borro M, Tassara R, Paris L, Artom N, Brignone M, Rebella L, Tassara R. Acquired Hemophilia A Treated with Recombinant Porcine Factor VIII: Case Report and Literature Review on Its Efficacy. Hematology Reports. 2023; 15(1):17-22. https://doi.org/10.3390/hematolrep15010003
Chicago/Turabian StyleBorro, Matteo, Riccardo Tassara, Luca Paris, Nathan Artom, Marcello Brignone, Lara Rebella, and Rodolfo Tassara. 2023. "Acquired Hemophilia A Treated with Recombinant Porcine Factor VIII: Case Report and Literature Review on Its Efficacy" Hematology Reports 15, no. 1: 17-22. https://doi.org/10.3390/hematolrep15010003
APA StyleBorro, M., Tassara, R., Paris, L., Artom, N., Brignone, M., Rebella, L., & Tassara, R. (2023). Acquired Hemophilia A Treated with Recombinant Porcine Factor VIII: Case Report and Literature Review on Its Efficacy. Hematology Reports, 15(1), 17-22. https://doi.org/10.3390/hematolrep15010003