
In Silico Three-Dimensional (3D) Modeling of the SecY Protein of ‘Candidatus Phytoplasma Solani’ Strains Associated with Grapevine “Bois Noir” and Its Possible Relationship with Strain Virulence



 ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Field Survey
2.2. Total Nucleic Acids Extraction and Phytoplasma Detection
2.3. SecY Gene Amplification and Nucleotide Sequence Analysis
2.4. Protein Prediction and Model Evaluation
3. Results
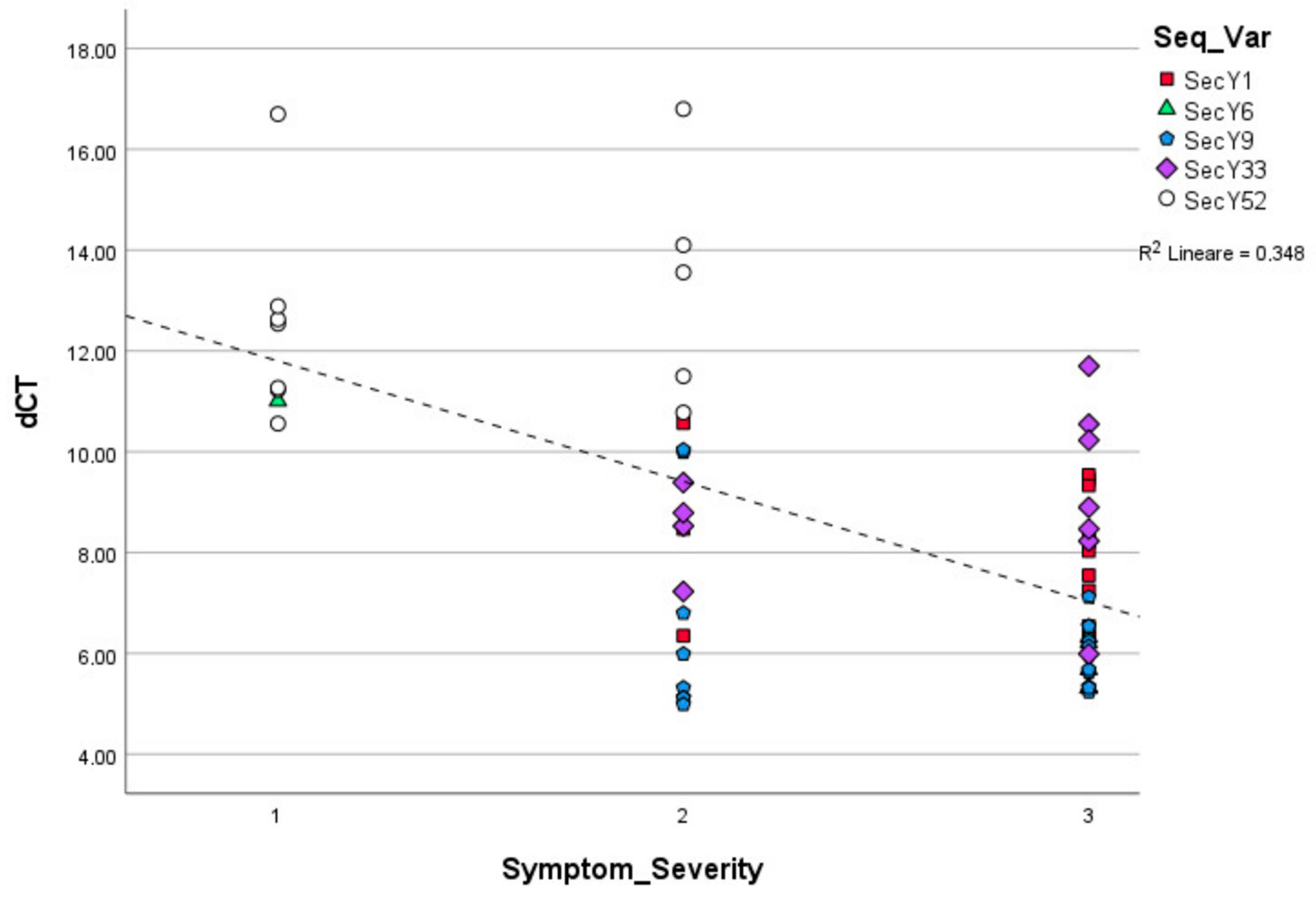
3.1. Symptom Observation and Phytoplasma Relative Quantification
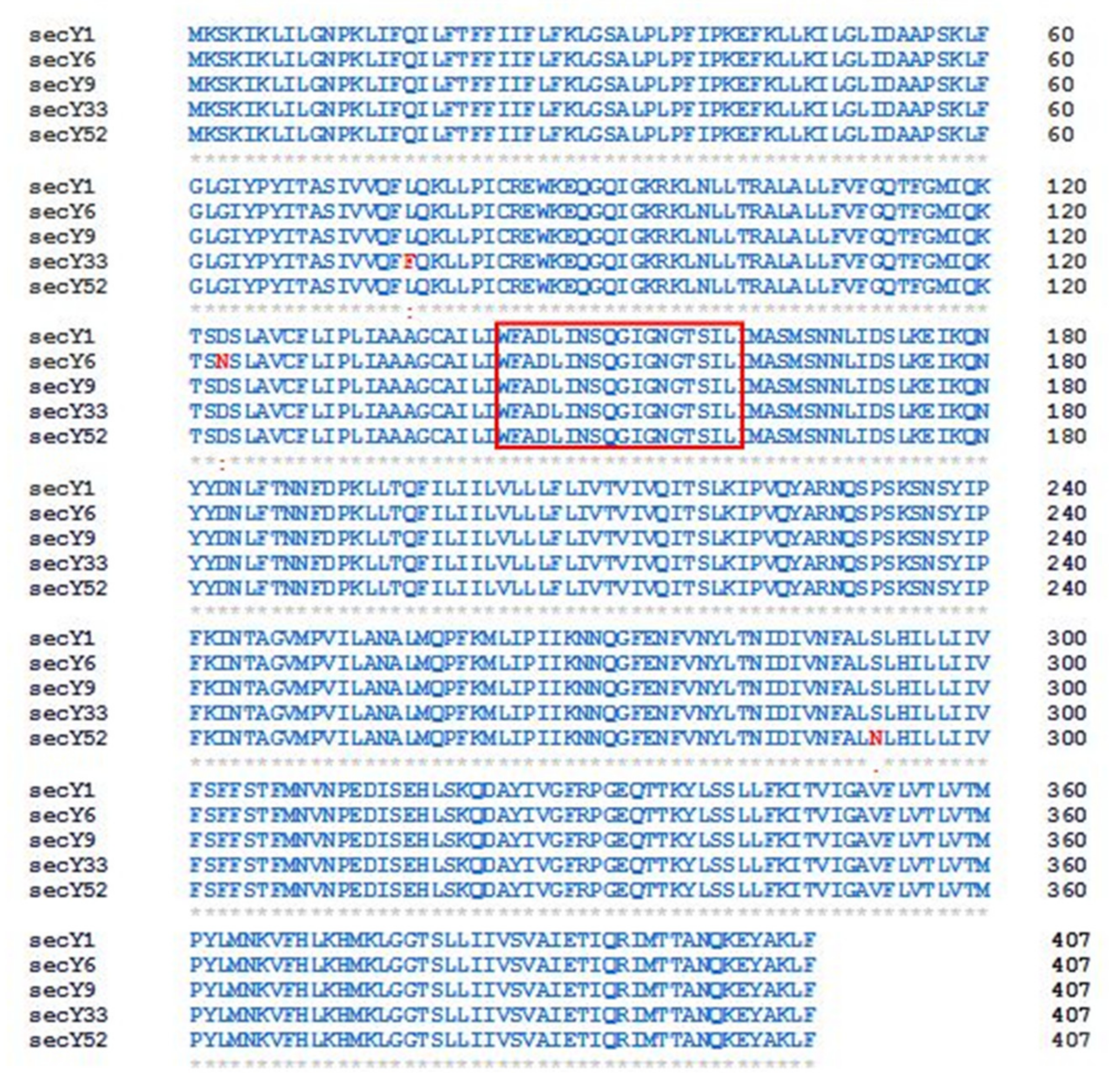
3.2. Identification of secY Sequence Variants
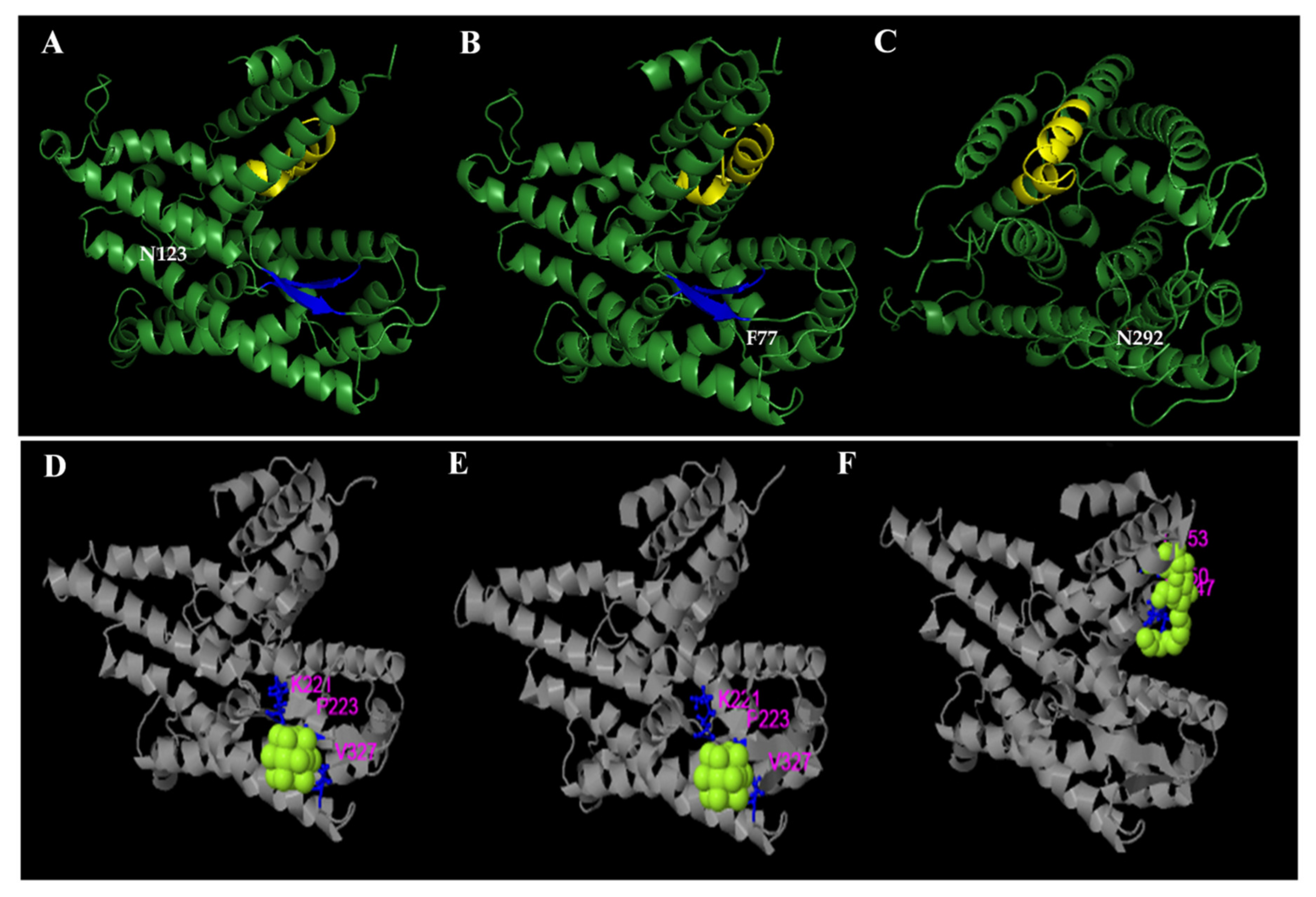
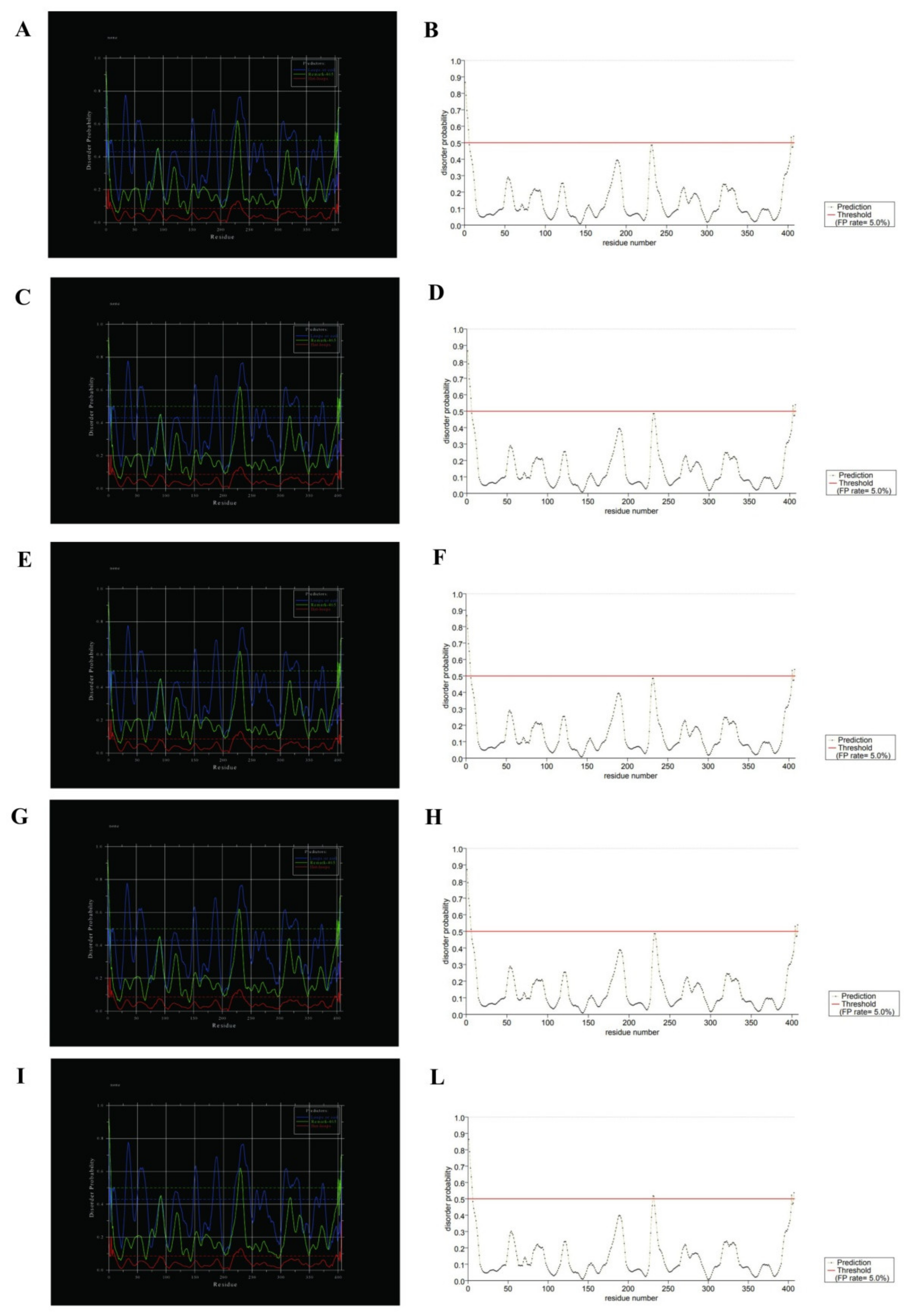
3.3. Protein Structure Predictions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Quaglino, F.; Zhao, Y.; Casati, P.; Bulgari, D.; Bianco, P.A.; Wei, W.; Davis, R.E. ‘Candidatus Phytoplasma solani’, a novel taxon associated with stolbur and Bois Noir. Related diseases of plants. Inter. J. Syst. Evol. Microb. 2013, 63, 2879–2894. [Google Scholar] [CrossRef] [PubMed]
- Bertaccini, A. Grapevine “Bois noir”: What is New Under the Sun? In Proceedings of the 5th Bois Noir Workshop, Ljubljana, Slovenia, 18–19 September 2018.
- Pierro, R.; Semeraro, T.; Luvisi, A.; Garg, H.; Vergine, M.; De Bellis, L.; Gill, H.K. The Distribution of Phytoplasmas in South and East Asia: An Emerging Threat to Grapevine Cultivation. Front. Plant Sci. 2019, 10, 1108. [Google Scholar] [CrossRef] [PubMed]
- Maixner, M. Transmission of German grapevine yellows (Vergilbungskrankheit) by the planthopper Hyalesthes obsoletus (Auchenorrhyncha: Cixiidae). Vitis 1994, 33, 103–104. [Google Scholar]
- Belli, G.; Bianco, P.A.; Conti, M. Grapevine yellows: Past, present and future. J. Plant Path. 2010, 92, 303–326. [Google Scholar]
- Kosovac, A.; Jakovljević, M.; Krstić, O.; Cvrković, T.; Mitrović, M.; Toševski, I.; Jović, J. Role of plant-specialized Hyalesthes obsoletus associated with Convolvulus arvensis and Crepis foetida in the transmission of “Candidatus Phytoplasma solani”-inflicted bois noir disease of grapevine in Serbia. Eur. J. Plant Pathol. 2019, 153, 183–195. [Google Scholar] [CrossRef]
- Batlle, A.; Altabella, N.; Sabaté, J.; Laviña, A. Study of the transmission of stolbur phytoplasma to different crop species by Macrosteles quadripunctulatus. Ann. Appl. Biol. 2008, 152, 235–242. [Google Scholar] [CrossRef]
- Cvrković, T.; Jović, J.; Mitrović, M.; Krstic, O.; Tosevski, I. Experimental and molecular evidence of Reptalus panzeri as a natural vector of “bois noir”. Plant Pathol. 2014, 63, 42–53. [Google Scholar] [CrossRef]
- Quaglino, F.; Sanna, F.; Moussa, A.; Faccincani, M.; Passera, A.; Casati, P.; Bianco, P.A.; Mori, N. Identification and ecology of alternative insect vectors of ‘Candidatus Phytoplasma solani’ to grapevine. Sci. Rep. 2019, 9, 19522. [Google Scholar] [CrossRef]
- Pacifico, D.; Alma, A.; Bagnoli, B.; Foissac, X.; Pasquini, G.; Tessitori, M.; Marzachì, C. Characterization of Bois noir isolates by restriction fragment length polymorphism of a Stolbur-specific putative membrane protein gene. Phytopathology 2009, 99, 711–715. [Google Scholar] [CrossRef][Green Version]
- Fabre, A.; Danet, J.L.; Foissac, X. The stolbur phytoplasma antigenic membrane protein gene stamp is submitted to diversifying positive selection. Gene 2011, 472, 37–41. [Google Scholar] [CrossRef]
- Quaglino, F.; Maghradze, D.; Casati, P.; Chkhaidze, N.; Lobjanidze, M.; Ravasio, A.; Passera, A.; Venturini, G.; Failla, O.; Bianco, P.A. Identification and characterization of new ‘Candidatus Phytoplasma solani’ strains associated with Bois Noir disease in Vitis vinifera L. cultivars showing a range of symptoms severity in Georgia, the Caucasus Region. Plant Dis. 2016, 100, 904–915. [Google Scholar] [CrossRef] [PubMed]
- Pierro, R.; Passera, A.; Panattoni, A.; Rizzo, D.; Stefani, L.; Bartolini, L.; Casati, P.; Luvisi, A.; Quaglino, F.; Materazzi, A. Prevalence of a ‘Candidatus Phytoplasma solani’ strain, so far associated only with otherhosts, in Bois Noir-affected grapevines within Tuscan vineyards. Ann. Appl. Biol. 2018, 173, 202–212. [Google Scholar] [CrossRef]
- Pierro, R.; Panattoni, A.; Passera, A.; Materazzi, A.; Luvisi, A.; Loni, A.; Ginanni, M.; Lucchi, A.; Bianco, P.A.; Quaglino, F. Proposal of a new Bois Noir epidemiological pattern related to ‘Candidatus Phytoplasma Solani’ strains characterized by a possible moderate virulence in Tuscany. Pathogens 2020, 9, 268. [Google Scholar] [CrossRef] [PubMed]
- Langer, M.; Maixner, M. Molecular characterisation of grapevine yellows associated phytoplasmas of the stolbur group based on RFLP analysis of non-ribosomal DNA. Vitis 2004, 43, 191–199. [Google Scholar]
- Aryan, A.; Brader, G.; Mörtel, J.; Pastar, M.; Riedle-Bauer, M. An abundant ’Candidatus Phytoplasma solani’ Stolbur tuf b phytoplasma strain is associated with grapevine, stinging nettle and Hyalesthes obsoletus. Eur. J. Plant Path. 2014, 140, 213–227. [Google Scholar] [CrossRef]
- Mori, H.; Ito, K. The Sec protein-translocation pathway. Trends Microbiol. 2001, 9, 494–500. [Google Scholar] [CrossRef]
- Lee, I.M.; Zhao, Y.; Bottner, K.D. SecY gene sequence analysis for finer differentiation of diverse strains in the aster yellows phytoplasma group. Mol. Cell. Prob. 2006, 20, 87–91. [Google Scholar] [CrossRef]
- Ma, C.; Wu, X.; Sun, D.; Park, E.; Catipovic, M.A.; Rapoport, T.A.; Gao, N.; Li, L. Structure of the substrate-engaged SecA-SecY protein translocation machine. Nat. Comm. 2019, 10, 2872. [Google Scholar] [CrossRef]
- Green, E.R.; Mecsas, J. Bacterial Secretion Systems—An overview. Microbiol. Spectr. 2016, 4, 10. [Google Scholar] [CrossRef]
- Casati, P.; Jermini, M.; Quaglino, F.; Corbani, G.; Schaerer, S.; Passera, A.; Bianco, P.A.; Rigamonti, I.E. New insights on Flavescence dorée phytoplasma ecology in the vineyard agro-ecosystem in southern Switzerland. Ann. Appl. Biol. 2017, 171, 37–51. [Google Scholar] [CrossRef]
- Li, R.; Mock, R.; Huang, Q.; Abad, J.; Hartung, J.; Kinard, G. A reliable and inexpensive method of nucleic acid extraction for the PCR-based detection of diverse plant pathogens. J. Virol. Meth. 2008, 154, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Pierro, R.; Passera, A.; Panattoni, A.; Casati, P.; Luvisi, A.; Rizzo, D.; Bianco, P.A.; Quaglino, F.; Materazzi, A. Molecular typing of ‘Bois Noir’ phytoplasma strains in the Chianti Classico area (Tuscany, Central Italy) and their association with symptom severity in Vitis vinifera L. cv. Sangiovese. Phytopathology 2018, 108, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Angelini, E.; Bianchi, G.L.; Filippin, L.; Morassutti, C.; Borgo, M. A new TaqMan method for the identification of phytoplasmas associated with grapevine yellows by real-time PCR assay. J. Microb. Meth. 2007, 68, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.M.; Bottner-Parker, K.D.; Zhao, Y.; Davis, R.E.; Harrison, N.A. Phylogenetic analysis and delineation of phytoplasmas based on secY gene sequences. Inter. J. Syst. Evol. Microb. 2010, 60, 2887–2897. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Arnold, K.; Bordoli, L.; Kopp, J.; Torsten, S. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, M.D.; Meng, E.C.; Ferrin, E.T. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, M.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511. [Google Scholar] [CrossRef]
- Linding, R.; Jensen, L.J.; Diella, F.; Bork, P.; Gibson, T.J.; Russell, R.B. Protein disorder prediction: Implications for structural proteomics. Structure 2003, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Kinoshita, K. PrDOS: Prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 2007, 35, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Pereira, G.R.C.; Tellini, G.H.A.S.; De Mesquita, J.F. In silico analysis of PFN1 related to amyotrophic lateral sclerosis. PLoS ONE 2019, 14, e0215723. [Google Scholar] [CrossRef] [PubMed]
- Bray, S.; Senapathi, T.; Barnett, C.; Grüning, B. High Throughput Molecular Dynamics and Analysis (Galaxy Training Materials). J. Cheminform 2020, 12, 54. Available online: https://training.galaxyproject.org/training-material/topics/computational-chemistry/tutorials/htmd-analysis/tutorial.html (accessed on 15 October 2021). [CrossRef]
- Bray, S. Running Molecular Dynamics Simulations Using GROMACS (Galaxy Training Materials). 2021. Available online: https://training.galaxyproject.org/training-material/topics/computational-chemistry/tutorials/md-simulation-gromacs/tutorial.html (accessed on 15 October 2021).
- Douglas, S.E. A secY homologue is found in the plastid genome of Cryptomonas phi. FEBS Lett. 1992, 298, 93. [Google Scholar] [CrossRef]
- Van den Berg, B.; Clemons, W.M.; Collinson, I.; Modis, Y.; Hartmann, E.; Harrison, S.C.; Rapoport, T.A. X-ray structure of a protein conducting channel. Nature 2004, 427, 36–44. [Google Scholar] [CrossRef]
- Jones, M.A.; Virtanen, P.; Hammarlöf, D.; Allen, J.W.; Collinson, I.; Hayes, S.C.; Low, L.D.; Koskiniemic, S. Genetic Evidence for SecY Translocon-Mediated Import of Two Contact-Dependent Growth Inhibition (CDI) Toxins. MBio 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Khor, B.Y.; Gee, J.T.; Theam, S.L.; Yee, S.C. General overview on structure prediction of twilight-zone proteins. Theor. Biol. Med. Model 2015, 12, 15. [Google Scholar] [CrossRef]
- Fiser, A. Template-based protein structure modeling. Met. Mol. Biol. 2010, 673, 73–94. [Google Scholar]
- Pereira, G.R.C.; Tavares, G.D.B.; de Freitas, M.C.; De Mesquita, J.F. In silico analysis of the tryptophan hydroxylase 2 (TPH2) protein variants related to psychiatric disorders. PLoS ONE 2020, 15, 3. [Google Scholar] [CrossRef] [PubMed]
- Pereira, G.R.C.; de Azevedo Abrahim Vieira, B.; De Mesquita, J.F. Comprehensive in silico analysis and molecular dynamics of the superoxide dismutase 1 (SOD1) variants related to amyotrophic lateral sclerosis. PLoS ONE 2021, 16, 2. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Nowich, J.S. Exploring β-Sheet Structure and Interactions with Chemical Model Systems. Acc. Chem. Res. 2008, 41, 1319–1330. [Google Scholar] [CrossRef]
- Delić, D.; Contaldo, N.; Lolić, B.; Moravčevič, D.; Miloše-Vič, D.; Bertaccini, A. Multigene Characterization of ‘Candidatus Phytoplasma solani’ strains infecting pepper, celery and maize in Bosnia and Herzegovina. Mitt. Klosterneubg. 2016, 66, 40–73. [Google Scholar]
- Crane, J.M.; Randalla, L.L. The Sec System: Protein Export in Escherichia coli. EcoSal Plus 2017, 7, 2. [Google Scholar] [CrossRef]
- Eisenberg, D.; Jucker, M. The amyloid state of proteins in human diseases. Cell 2012, 148, 1188–1203. [Google Scholar] [CrossRef]
- Knowles, T.P.J.; Vendruscolo, M.; Dobson, C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef]
- Nakajima, Y.; Suzuki, S. Environmental Stresses Induce Misfolded Protein Aggregation in Plant Cells in a Microtubule-Dependent Manner. Int J. Mol. Sci. 2013, 14, 7771–7783. [Google Scholar] [CrossRef]
- Bienkiewicz, E.A.; Adkins, J.N.; Lumb, K.J. Functional consequences of preorganized helical structure in the intrinsically disordered cell-cycle inhibitor p27(Kip1). Biochemistry 2002, 41, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model 2001, 19, 26–59. [Google Scholar] [CrossRef]
- Namba, K. Roles of partly unfolded conformations in macromolecular self-assembly. Genes Cells 2001, 6, 1–12. [Google Scholar] [CrossRef]
- Dunker, A.K.; Garner, E.; Guilliot, S.; Romero, P.; Albrecht, K.; Hart, J.; Obradovic, Z.; Kissinger, C.; Villafranca, J.E. Protein disorder and the evolution of molecular recognition: Theory, predictions and observations. Pac. Symp. Biocomput. 1998, 3, 473–484. [Google Scholar]
- Liu, J.; Tan, H.; Rost, B. Loopy proteins appear conserved in evolution. J. Mol. Biol. 2002, 322, 53–64. [Google Scholar] [CrossRef]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002; 27, 527–533. [Google Scholar]
- Dunker, A.K.; Silman, I.; Uversky, V.N.; Sussman, J.L. Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 2008, 18, 756–764. [Google Scholar] [CrossRef]
- Marín, M.; Uversky, V.N.; Ott, T. Intrinsic disorder in pathogen effectors: Protein flexibility as an evolutionary hallmark in a molecular arms race. Plant Cell 2013, 25, 3157. [Google Scholar] [CrossRef]
- Brown, C.J.; Johnson, A.K.; Daughdrill, G.W. Comparing models of evolution for ordered and disordered proteins. Mol. Biol. Evol. 2010, 27, 609–621. [Google Scholar] [CrossRef]
- Kim, D.N.; Jacobs, T.M.; Kuhlman, B. Boosting protein stability with the computational design of β-sheet surfaces. Protein Sci 2016, 25, 702–710. [Google Scholar] [CrossRef]
- Ito, K. SecY and integral membrane components of the Escherichia coli protein translocation system. Mol. Microbiol. 1992, 6, 2423–2428. [Google Scholar] [CrossRef] [PubMed]
- Tomkins, M.; Kliot, A.; Marée, A.F.M.; Hogenhout, S. A multi-layered mechanistic modelling approach to understand how effector genes extend beyond phytoplasma to modulate plant hosts, insect vectors and the environment. Curr. Opin. Plant Biol. 2018, 44, 39–48. [Google Scholar] [PubMed]
- Seemuller, E.; Sule, S.; Kube, M.; Jelkmann, W.; Schneider, B. The AAA plus ATPases and HflB/FtsH proteases of ‘Candidatus Phytoplasma mali’: Phylogenetic diversity, membrane topology, and relationship to strain virulence. Mol Plant.-Microbe. Interact. 2013, 26, 367–376. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
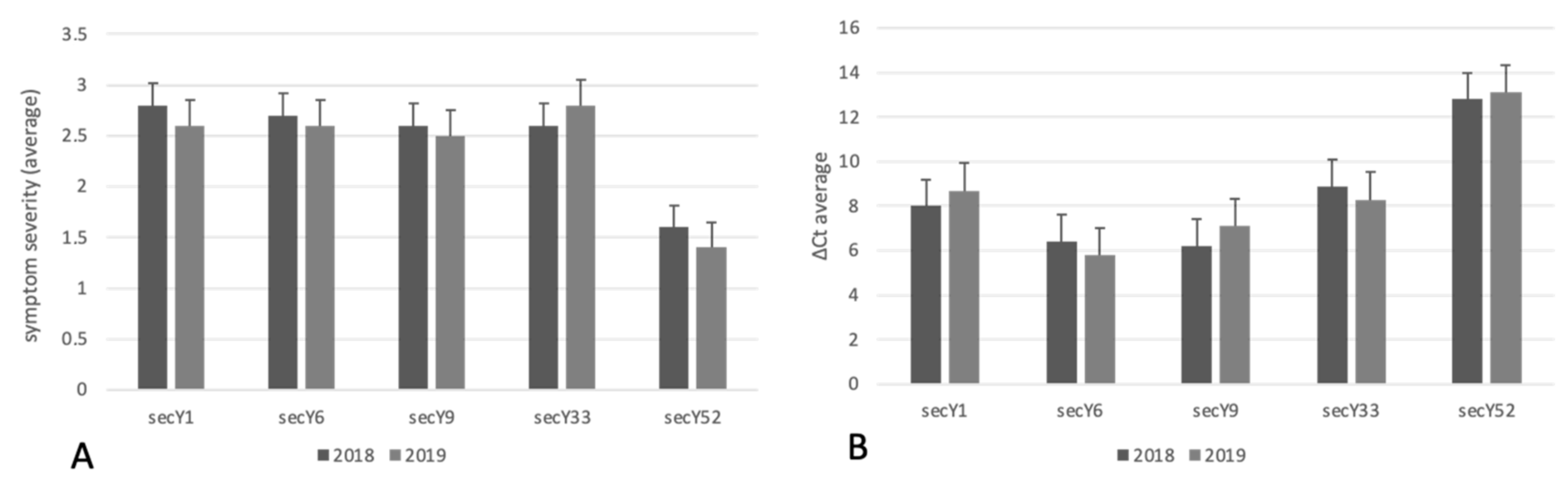
| No. of ‘Ca. P. Solani’ Strains Collected | Symptom Severity | Relative Abundance | Symptom Severity | Relative Abundance | ||
|---|---|---|---|---|---|---|
| 2018 | 2019 | Sequence Variant | (Average 2018) | (Average 2018) | (Average 2019) | (Average 2019) |
| 16 | 19 | secY1 | 2.8 | 8.0 | 2.6 | 8.7 |
| 7 | 12 | secY6 | 2.7 | 6.4 | 2.6 | 5.8 |
| 20 | 26 | secY9 | 2.6 | 6.2 | 2.5 | 7.1 |
| 11 | 7 | secY33 | 2.6 | 8.9 | 2.8 | 8.3 |
| 12 | 18 | secY52 | 1.6** | 12.8 | 1.4 ** | 13.1 |
| Validation Index | ||
|---|---|---|
| SecY Sequence Variant | PROCHECK | ERRAT |
| SecY1 | 88.8% | 86.16% |
| SecY6 | 88.5% | 85.37% |
| SecY9 | 88.8% | 86.16% |
| SecY33 | 89.1% | 82.59% |
| SecY52 | 69.8% | 59.46% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pierro, R.; De Pascali, M.; Panattoni, A.; Passera, A.; Materazzi, A.; De Bellis, L.; Luvisi, A.; Bianco, P.A.; Quaglino, F. In Silico Three-Dimensional (3D) Modeling of the SecY Protein of ‘Candidatus Phytoplasma Solani’ Strains Associated with Grapevine “Bois Noir” and Its Possible Relationship with Strain Virulence. Int. J. Plant Biol. 2022, 13, 15-30. https://doi.org/10.3390/ijpb13020004
Pierro R, De Pascali M, Panattoni A, Passera A, Materazzi A, De Bellis L, Luvisi A, Bianco PA, Quaglino F. In Silico Three-Dimensional (3D) Modeling of the SecY Protein of ‘Candidatus Phytoplasma Solani’ Strains Associated with Grapevine “Bois Noir” and Its Possible Relationship with Strain Virulence. International Journal of Plant Biology. 2022; 13(2):15-30. https://doi.org/10.3390/ijpb13020004
Chicago/Turabian StylePierro, Roberto, Mariarosaria De Pascali, Alessandra Panattoni, Alessandro Passera, Alberto Materazzi, Luigi De Bellis, Andrea Luvisi, Piero Attilio Bianco, and Fabio Quaglino. 2022. "In Silico Three-Dimensional (3D) Modeling of the SecY Protein of ‘Candidatus Phytoplasma Solani’ Strains Associated with Grapevine “Bois Noir” and Its Possible Relationship with Strain Virulence" International Journal of Plant Biology 13, no. 2: 15-30. https://doi.org/10.3390/ijpb13020004
APA StylePierro, R., De Pascali, M., Panattoni, A., Passera, A., Materazzi, A., De Bellis, L., Luvisi, A., Bianco, P. A., & Quaglino, F. (2022). In Silico Three-Dimensional (3D) Modeling of the SecY Protein of ‘Candidatus Phytoplasma Solani’ Strains Associated with Grapevine “Bois Noir” and Its Possible Relationship with Strain Virulence. International Journal of Plant Biology, 13(2), 15-30. https://doi.org/10.3390/ijpb13020004