
Unusual Cardiac Manifestations of a Pheochromocytoma in a Girl

 ,
,
Abstract
1. Introduction
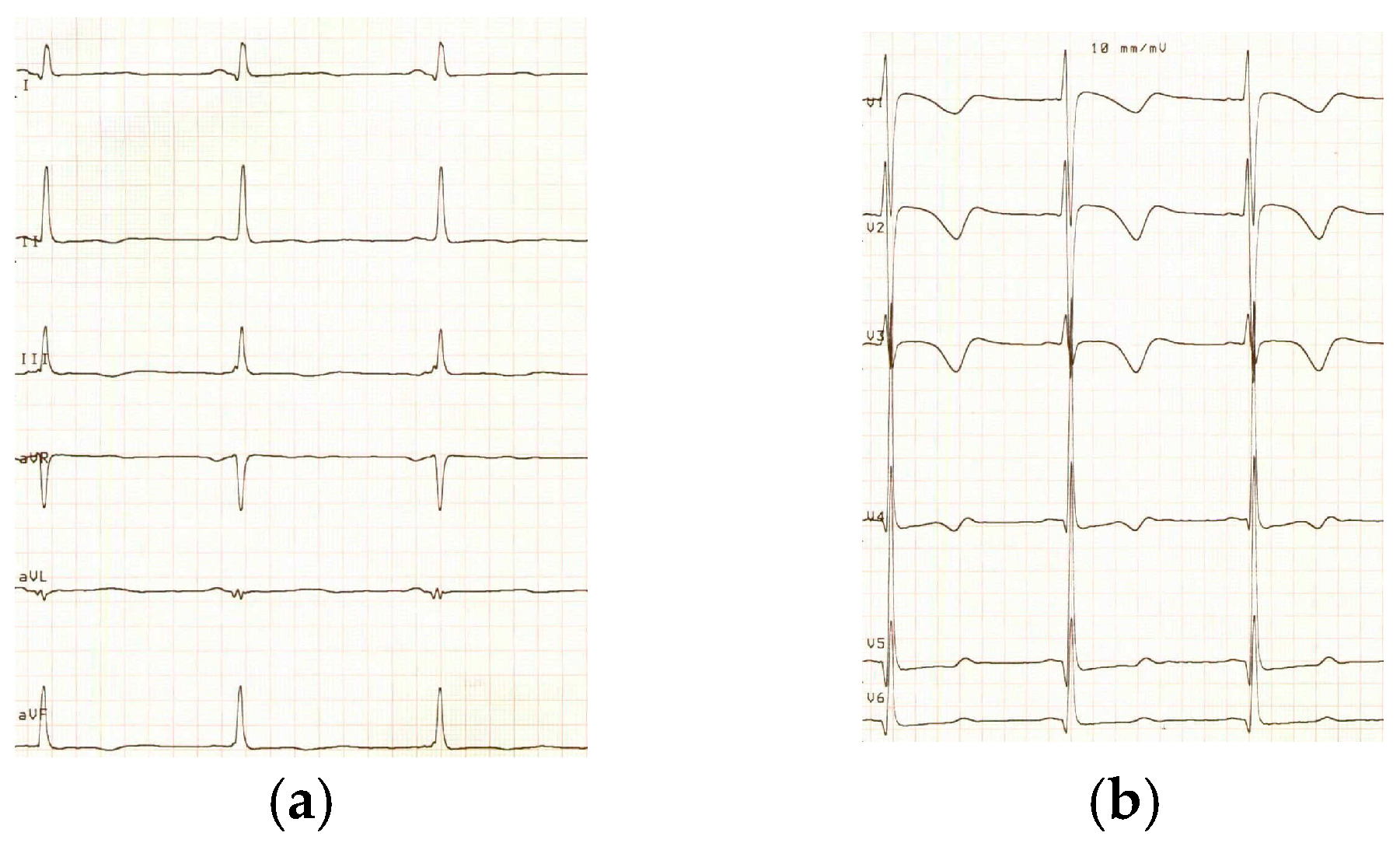
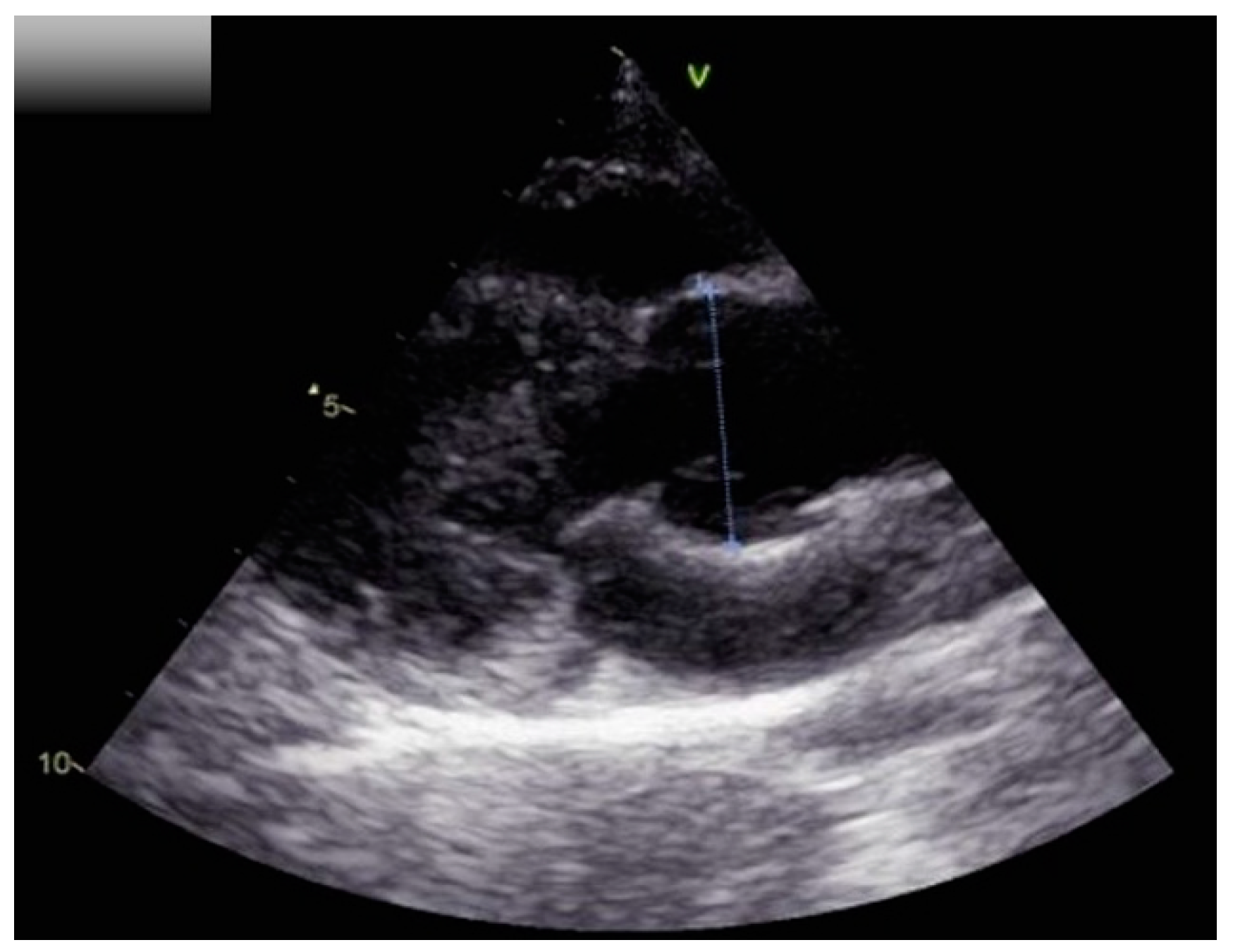
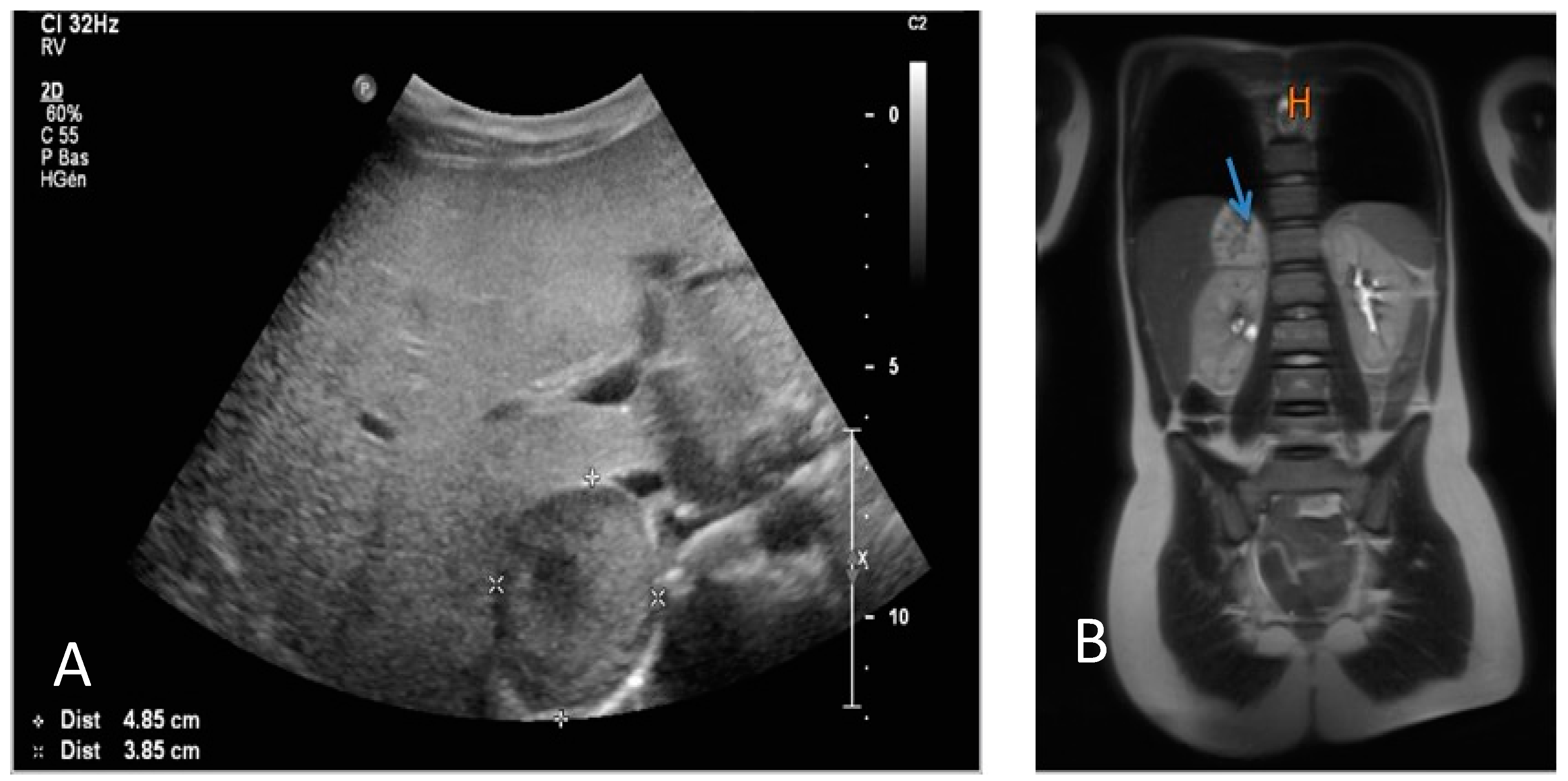
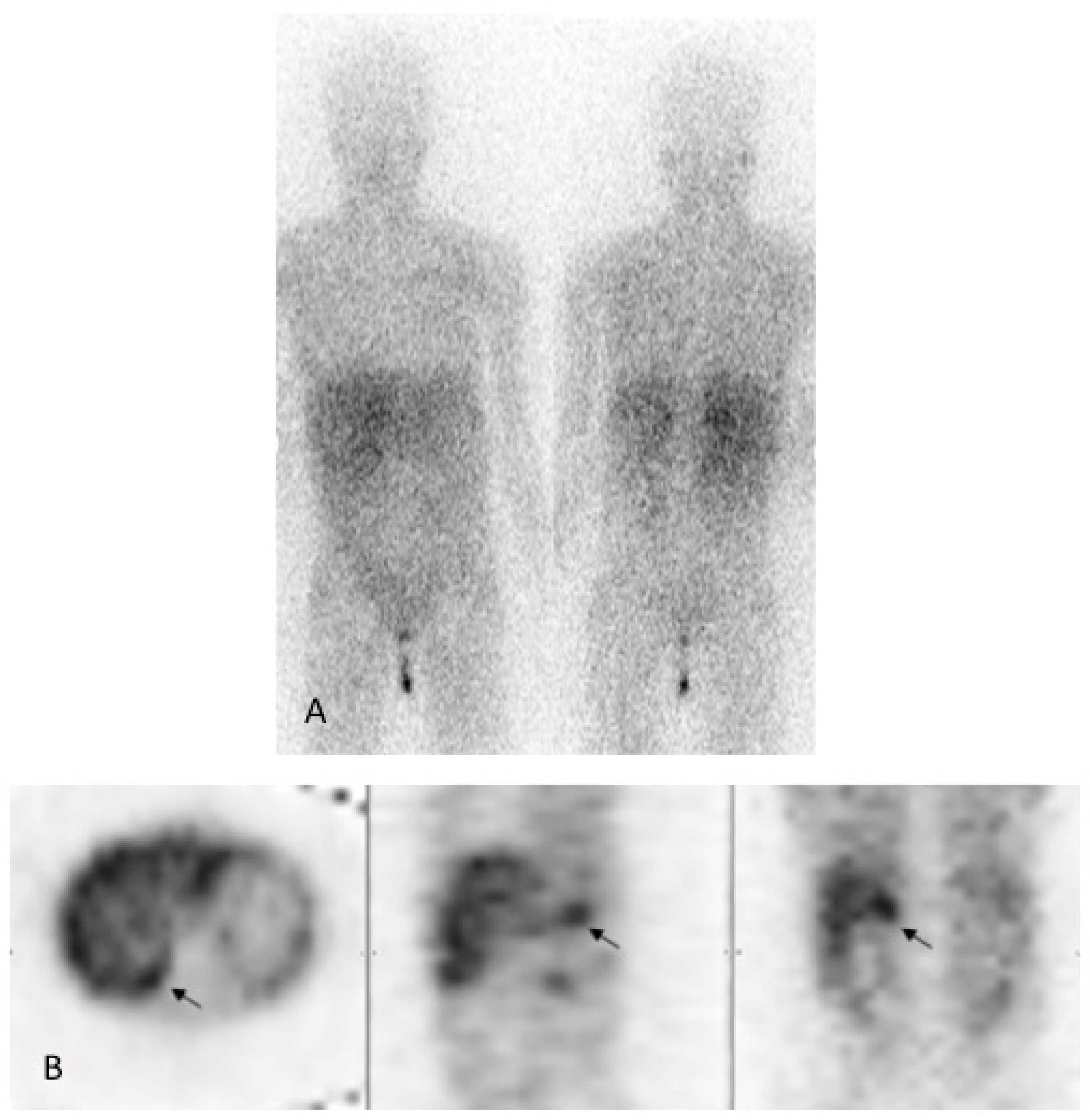
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Armstrong, R.; Sridhar, M.; Greenhalgh, K.L.; Howell, L.; Jones, C.; Landes, C.; McPartland, J.L.; Moores, C.; Losty, P.D.; Didi, M. Phaeochromocytoma in children. Arch. Dis. Child. 2008, 93, 899–904. [Google Scholar] [CrossRef]
- Sarathi, V. Characteristics of pediatric pheochromocytoma/paraganglioma. Indian J. Endocrinol. Metab. 2017, 21, 470. [Google Scholar] [CrossRef]
- Fishbein, L.; Nathanson, K.L. Pheochromocytoma and Paraganglioma: Understanding the Complexities of the Genetic Background. Cancer Genet. 2012, 205, 1–11. [Google Scholar] [CrossRef]
- Lenders, J.W.; Kerstens, M.N.; Laurence, A.M.A.R.; Prejbisz, A.; Robledo, M.; Taieb, D.; Pacak, K.; Crona, J.; Zelinka, T.; Mannelli, M.; et al. Genetics, diagnosis, management and future directions of research of phaeochromocytoma and paraganglioma: A position statement and consensus of the Working Group on Endocrine Hypertension of the European Society of Hypertension. J. Hypertens 2020, 38, 1443–1456. [Google Scholar] [CrossRef]
- Kinuani, R.; Bruyère, P.J.; Schoysman, L.; Kempeneers, C.; Daron, B.; Seghaye, M.C. Total absence of the pericardium associated with hypogammaglobulinemia and bronchiectasis in a girl. Pediatr. Rep. 2019, 11, 5–7. [Google Scholar] [CrossRef]
- Davignon, A.; Rautaharju, P.; Boiselle, E.; Soumis, F.; Mégélas, M.; Choquette, A. Normal ECG standards for infants and children. Ped. Cardiol. 1980, 1, 123–131. [Google Scholar] [CrossRef]
- Colan, S.D. Echocardiography in Pediatric and Congenital Heart Disease from Fetus to Adult, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- Ueda, T.; Oka, N.; Matsumoto, A.; Miyazaki, H.; Ohmura, H.; Kikuchi, T.; Nakayama, M.; Kato, S.; Imaizumi, T. Pheochromocytoma presenting as recurrent hypotension and syncope. Intern. Med. 2005, 44, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Molaei, A.; Akbarzadeh-Bairami, V.; Sadat-Ebrahimi, S.R. A case of pheochromocytoma presenting with cardiac manifestation: Case report. BMC Pediatr. 2020, 20, 299. [Google Scholar] [CrossRef] [PubMed]
- Uçaktürk, S.A.; Mengen, E.; Azak, E.; Çetin, İ.İ.; Kocaay, P.; Şenel, E. Catecholamine-induced myocarditis in a child with pheochromocytoma. J. Clin. Res. Pediatr. Endocrinol. 2020, 12, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Y-Hassan, S.; Falhammar, H. Cardiovascular Manifestations and Complications of Pheochromocytomas and Paragangliomas. J. Clin. Med. 2020, 9, 2435. [Google Scholar] [CrossRef]
- Luca, F.; Holl, N.; Vinzio, S.; Grunenberger, F.; Suna, C.; Taquet, M.C.; Goichot, B.; Schlienger, J.L. Manifestations cardiaques des phéochromocytomes. Ann. Endocrinol. 2009, 70, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.R.U.; Brofferio, A.; Viana, B.; Pacak, K. Catecholamine-induced cardiomyopathy in pheochromocytoma: How to manage a rare complication in a rare disease? Horm. Metab. Res. 2019, 51, 458–469. [Google Scholar] [CrossRef]
- Luiz, H.V.; Tanchee, M.J.; Pavlatou, M.G.; Yu, R.; Nambuba, J.; Wolf, K.; Prodanov, T.; Wesley, R.; Adams, K.; Fojo, T.; et al. Are patients with hormonally functional phaeochromocytoma and paraganglioma initially receiving a proper adrenoceptor blockade? A retrospective cohort study. Clin. Endocrinol. 2016, 85, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Butt, K.; Ali, S.; Sattar, Z.; Rahman, A.U.; Burt, J.R. Funny lumps, flaming pheo, and a broken heart: A rare case of pheochromocytoma. Cureus 2018, 10, e3646. [Google Scholar] [CrossRef]
- Ozyuncu, N.; Akturk, S.; Tan Kurklu, T.S.; Erol, C. Acute coronary syndrome-like presentation with prolonged QT interval: An unusual case of pheochromocytoma. BMJ Case Rep. 2016, 1, bcr2016216142. [Google Scholar] [CrossRef] [PubMed]
- Marzuillo, P.; Benettoni, A.; Germani, C.; Ferrara, G.; D’Agata, B.; Barbi, E. Acquired long QT syndrome. Pediatr. Emerg. Care 2014, 30, 257–261. [Google Scholar] [CrossRef]
- Oruganti, S.S.; Gambeer Rao, M.; Pisapati, V.L.N.M. Adrenal and extra-adrenal pheochromocytomas presenting as life-threatening ventricular arrhythmias: Report of three cases. Indian Heart J. 2016, 68, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Cecilia, G.M.; Emilien, D.; Laurent, H. Acquired long QT interval complicated with torsades de pointes as presentation of a pheochromocytoma in a paediatric patient: A case report. Cardiol. Young 2015, 25, 391–393. [Google Scholar] [CrossRef] [PubMed]
- Toutai, C.; Berrajaa, M.; Aissaoui, H.; Elouafi, N.; Jabi, R.; Bouziane, M.; Latrech, H.; Housni, B.; Ismaili, N. Rare association of aortoarteritis and pheochromocytoma: A case report. Int. J. Surg. Case Rep. 2020, 77, 91–95. [Google Scholar] [CrossRef]
- Lin, A.T.; Min, Y.; Clements, P.J.; Morris, R.; Shayestehfar, B.; Mankin, L.; Wong, A.L. New onset systemic lupus erythematosus with pheochromocytoma. J. Rheumatol. 2002, 29, 1334–1337. [Google Scholar]
- Kota, S.K.; Kota, S.K.; Meher, L.K.; Jammula, S.; Panda, S.; Modi, K.D. Coexistence of pheochromocytoma with uncommon vascular lesions. Indian J. Endocrinol. Metab. 2012, 16, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.Y.; Kaelin, W.G. Role of VHL gene mutation in human cancer. J. Clin. Oncol. 2004, 22, 4991–5004. [Google Scholar] [CrossRef] [PubMed]
- Jhawar, S.; Arakawa, Y.; Kumar, S.; Varghese, D.; Kim, Y.S.; Roper, N.; Elloumi, F.; Pommier, Y.; Pacak, K.; Del Rivero, J. New insights on the genetics of pheochromocytoma and paraganglioma and its clinical implications. Cancers 2022, 14, 594. [Google Scholar] [CrossRef] [PubMed]
- Burnichon, N.; Vescovo, L.; Amar, L.; Libe, R.; de Reynies, A.; Venisse, A.; Jouanno, E.; Laurendeau, I.; Parfait, B.; Bertherat, J.; et al. Integrative genomic analysis reveals somatic mutations in pheochromocytoma and paraganglioma. Hum. Mol. Genet. 2011, 20, 3974–3985. [Google Scholar] [CrossRef]
- Kumar, R.M.; Violette, P.D.; Tran, C.; Tomiak, E.; Izard, J.; Bathini, V.; Rowe, N.E. Canadian Urological Association best practice report: Long-term surveillance following resection of pheochromocytoma. Can. Urol. Assoc. J. 2019, 13, 372–376. [Google Scholar] [CrossRef]
- Amar, L.; Lussey-Lepoutre, C.; Lenders, J.W.; Djadi-Prat, J.; Plouin, P.F.; Steichen, O. Recurrence or new tumors after complete resection of pheochromocytomas and paragangliomas: A systematic review and meta-analysis. Eur. J Endocrinol. 2016, 175, R135–R145. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pre-Operative | 2 Weeks Post-Operative | |
|---|---|---|
| SV2 + RV5 (percentile (P)) | 7.8 mvolts (>P98) | 6.1 mvolts (P95) |
| QTc duration | 470 ms | 304 ms |
| Aortic root size (Z-score) | 29 mm (2.5) | 25 mm (0.7) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Angelo, L.; Parent, A.-S.; Derwael, C.; Hustinx, R.; Seghaye, M.-C. Unusual Cardiac Manifestations of a Pheochromocytoma in a Girl. Pediatr. Rep. 2023, 15, 237-244. https://doi.org/10.3390/pediatric15010019
D’Angelo L, Parent A-S, Derwael C, Hustinx R, Seghaye M-C. Unusual Cardiac Manifestations of a Pheochromocytoma in a Girl. Pediatric Reports. 2023; 15(1):237-244. https://doi.org/10.3390/pediatric15010019
Chicago/Turabian StyleD’Angelo, Lisa, Anne-Simone Parent, Céline Derwael, Roland Hustinx, and Marie-Christine Seghaye. 2023. "Unusual Cardiac Manifestations of a Pheochromocytoma in a Girl" Pediatric Reports 15, no. 1: 237-244. https://doi.org/10.3390/pediatric15010019
APA StyleD’Angelo, L., Parent, A.-S., Derwael, C., Hustinx, R., & Seghaye, M.-C. (2023). Unusual Cardiac Manifestations of a Pheochromocytoma in a Girl. Pediatric Reports, 15(1), 237-244. https://doi.org/10.3390/pediatric15010019