


CAKUT: A Pediatric and Evolutionary Perspective on the Leading Cause of CKD in Childhood

Abstract
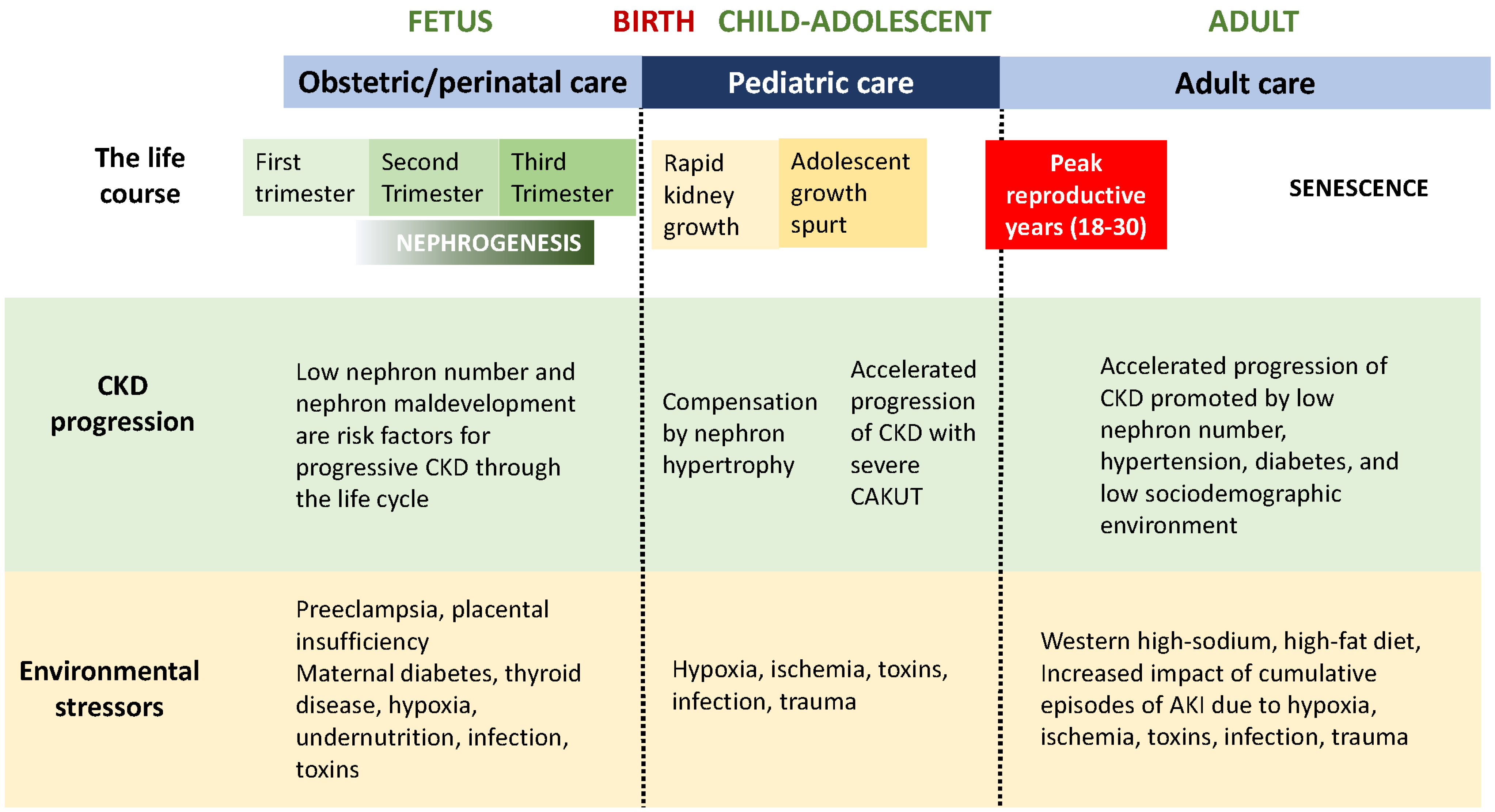
1. Chronic Kidney Disease over the Life Span
2. Nephrogenesis: Determination of Nephron Number
3. CAKUT and Pediatric CKD
4. Genetics of CAKUT
5. Animal Models of Congenital Urinary Tract Obstruction
6. Obstetrics and Fetal Management of CAKUT
7. Postnatal Management by a Pediatrician, Pediatric Nephrologist, and Urologist
8. Transfer to Adult Care: Internist, Nephrologist, and Urologist
9. Evolution of the Kidney: An Explanation for the Epidemiology of Progressive CKD
10. Answering the Questions of the Family and Child with CAKUT
11. The Future: Advances in the Diagnosis and Management of CAKUT
Funding
Conflicts of Interest
References
- Xie, Y.; Bowe, B.; Mokdad, A.H.; Xian, H.; Yan, Y.; Li, T.; Maddukuri, G.; Tsai, C.Y.; Floyd, T.; Al-Aly, Z. Analysis of the Global Burden of Disease study highlights the global, regional, and national trends of chronic kidney disease epidemiology from 1990 to 2016. Kidney Int. 2018, 94, 567–581. [Google Scholar] [CrossRef]
- Becherucci, F.; Roperto, R.M.; Materassi, M.; Romagnani, P. Chronic kidney disease in children. Clin. Kidney J. 2016, 9, 583–591. [Google Scholar] [CrossRef]
- Barker, D.J.P. The origins of the developmental origins theory. J. Intern. Med. 2007, 261, 412–417. [Google Scholar] [CrossRef]
- Eriksson, J.G.; Salonen, M.K.; Kajantie, E.; Osmond, C. Prenatal Growth and CKD in Older Adults: Longitudinal Findings From the Helsinki Birth Cohort Study, 1924–1944. Am. J. Kidney Dis. 2018, 71, 20–26. [Google Scholar] [CrossRef]
- Calderon-Margalit, R.; Golan, E.; Twig, G.; Leiba, A.; Tzur, D.; Afek, A.; Skorecki, K.; Vivante, A. History of Childhood Kidney Disease and Risk of Adult End-Stage Renal Disease. N. Engl. J. Med. 2018, 378, 428–438. [Google Scholar] [CrossRef]
- Murugapoopathy, V.; Gupta, I.R. A Primer on Congenital Anomalies of the Kidneys and Urinary Tracts (CAKUT). Clin. J. Am. Soc. Nephrol. CJASN 2020, 15, 723–731. [Google Scholar] [CrossRef]
- Mañalich, R.; Reyes, L.; Herrera, M.; Melendi, C.; Fundora, I. Relationship between weight at birth and the number and size of renal glomeruli in humans: A histomorphometric study. Kidney Int. 2000, 58, 770–773. [Google Scholar] [CrossRef]
- Crump, C.; Sundquist, J.; Winkleby, M.A.; Sundquist, K. Preterm birth and risk of chronic kidney disease from childhood into mid-adulthood: National cohort study. BMJ 2019, 365, l1346. [Google Scholar] [CrossRef]
- Wardlaw, T.M. Low Birthweight: Country, Regional and Global Estimates; UNICEF: New York, NY, USA, 2004. [Google Scholar]
- Nicolaou, N.; Renkema, K.Y.; Bongers, E.M.; Giles, R.H.; Knoers, N.V. Genetic, environmental, and epigenetic factors involved in CAKUT. Nat. Rev. Nephrol. 2015, 11, 720–731. [Google Scholar] [CrossRef]
- van der Ven, A.T.; Vivante, A.; Hildebrandt, F. Novel Insights into the Pathogenesis of Monogenic Congenital Anomalies of the Kidney and Urinary Tract. J. Am. Soc. Nephrol. 2018, 29, 36–50. [Google Scholar] [CrossRef]
- Capone, V.P.; Morello, W.; Taroni, F.; Montini, G. Genetics of Congenital Anomalies of the Kidney and Urinary Tract: The Current State of Play. Int. J. Mol. Sci. 2017, 18, 796. [Google Scholar] [CrossRef]
- Vivante, A.; Kohl, S.; Hwang, D.-Y.; Dworschak, G.C.; Hildebrandt, F. Single-gene causes of congenital anomalies of the kidney and urinary tract (CAKUT) in humans. Pediatr. Nephrol. 2014, 29, 695–704. [Google Scholar] [CrossRef]
- Thomas, C.P.; Freese, M.E.; Ounda, A.; Jetton, J.G.; Holida, M.; Noureddine, L.; Smith, R.J. Initial experience from a renal genetics clinic demonstrates a distinct role in patient management. Genet. Med. 2020, 22, 1025–1035. [Google Scholar] [CrossRef]
- Chevalier, R.L. Bioenergetic evolution explains prevalence of low nephron number at birth: Risk factor for CKD. Kidney 2020, 360, 863–879. [Google Scholar] [CrossRef]
- Ekulu, P.M.; Nkoy, A.B.; Adebayo, O.C.; Kazadi, O.K.; Aloni, M.N.; Arcolino, F.O.; Ngiyulu, R.M.; Gini, J.E.; Lepira, F.B.; Van den Heuvel, L.P.; et al. A focus on the association of Apol1 with kidney disease in children. Pediatr. Nephrol. 2021, 36, 777–788. [Google Scholar] [CrossRef]
- Peters, C.A. Urinary tract obstruction in children. J. Urol. 1995, 154, 1874–1883; discussion 1883–1874. [Google Scholar] [CrossRef]
- Thornhill, B.A.; Burt, L.A.; Chen, C.; Forbes, M.S.; Chevalier, R.L. Variable chronic partial ureteral obstruction in the neonatal rat: A new model of ureteropelvic junction obstruction. Kidney Int. 2005, 67, 42–52. [Google Scholar] [CrossRef]
- Thornhill, B.A.; Forbes, M.S.; Marcinko, E.S.; Chevalier, R.L. Glomerulotubular disconnection in neonatal mice after relief of partial ureteral obstruction. Kidney Int. 2007, 72, 1103–1112. [Google Scholar] [CrossRef]
- Sergio, M.; Galarreta, C.I.; Thornhill, B.A.; Forbes, M.S.; Chevalier, R.L. The fate of nephrons in congenital obstructive nephropathy: Adult recovery is limited by nephron number. J. Urol. 2015, 194, 1463–1472. [Google Scholar] [CrossRef]
- Williams, G.; Fletcher, J.T.; Alexander, S.I.; Craig, J.C. Vesicoureteral Reflux. J. Am. Soc. Nephrol. 2008, 19, 847. [Google Scholar] [CrossRef]
- Vemulakonda, V.M. Ureteropelvic junction obstruction: Diagnosis and management. Curr. Opin. Pediatr. 2021, 33, 227–234. [Google Scholar] [CrossRef]
- Hodges, S.J.; Patel, B.; McLorie, G.; Atala, A. Posterior urethral valves. ScientificWorldJournal 2009, 9, 1119–1126. [Google Scholar] [CrossRef]
- Hains, D.S.; Bates, C.M.; Ingraham, S.; Schwaderer, A.L. Management and etiology of the unilateral multicystic dysplastic kidney: A review. Pediatr. Nephrol. 2009, 24, 233–241. [Google Scholar] [CrossRef]
- VanNoy, G.E.; Wojcik, M.H.; Genetti, C.A.; Mullen, T.E.; Agrawal, P.B.; Stein, D.R. Reconsidering Genetic Testing for Neonatal Polycystic Kidney Disease. Kidney Int. Rep. 2020, 5, 1316–1319. [Google Scholar] [CrossRef]
- Thomas, I.T.; Smith, D.W. Oligohydramnios, cause of the nonrenal features of Potter’s syndrome, including pulmonary hypoplasia. J. Pediatr. 1974, 84, 811–815. [Google Scholar] [CrossRef]
- Clark, D.A. Times of first void and first stool in 500 newborns. Pediatrics 1977, 60, 457–459. [Google Scholar] [CrossRef]
- Chevalier, R.L. Congenital urinary tract obstruction: The long view. Adv. Chronic Kidney Dis. 2015, 22, 312–319. [Google Scholar] [CrossRef]
- Yamaçake, K.G.; Nguyen, H.T. Current management of antenatal hydronephrosis. Pediatr. Nephrol. 2013, 28, 237–243. [Google Scholar] [CrossRef]
- Chevalier, R.L.; Campbell, F.; Brenbridge, A.N. Nephrosonography and renal scintigraphy in evaluation of newborn with renomegaly. Urology 1984, 24, 96–103. [Google Scholar] [CrossRef]
- Kumar, J.; Gordillo, R.; Kaskel, F.J.; Druschel, C.M.; Woroniecki, R.P. Increased prevalence of renal and urinary tract anomalies in children with congenital hypothyroidism. J. Pediatr. 2009, 154, 263–266. [Google Scholar] [CrossRef]
- Postolache, L.; Parsa, A.; Simoni, P.; Boitsios, G.; Ismaili, K.; Schurmans, T.; Monier, A.; Casimir, G.; Albert, A.; Parsa, C.F. Widespread kidney anomalies in children with Down syndrome. Pediatr. Nephrol. 2022, 37, 2361–2368. [Google Scholar] [CrossRef]
- Rhone, E.T.; Carmody, J.B.; Swanson, J.R.; Charlton, J.R. Nephrotoxic medication exposure in very low birth weight infants. J. Matern. -Fetal Neonatal Med. 2014, 27, 1485–1490. [Google Scholar] [CrossRef]
- Gjerde, A.; Lillås, B.S.; Marti, H.P.; Reisæter, A.V.; Vikse, B.E. Intrauterine growth restriction, preterm birth and risk of end-stage renal disease during the first 50 years of life. Nephrol. Dial. Transplant. 2020, 35, 1157–1163. [Google Scholar] [CrossRef]
- Ardissino, G.; Vigano, S.; Testa, S.; Dacco, V.; Paglialonga, F.; Leoni, A.; Belingheri, M.; Avolio, L.; Ciofani, A.; Claris-Appiani, A.; et al. No clear evidence of ACEi efficacy on the progression of chronic kidney disease in children with hypodysplastic nephropathy--report from the ItalKid project database. Nephrol. Dial. Transplant. 2007, 22, 2525–2530. [Google Scholar] [CrossRef]
- Wuhl, E.; van Stralen, K.J.; Verrina, E.; Bjerre, A.; Wanner, C.; Heaf, J.G.; Zurriaga, O.; Hoitsma, A.; Niaudet, P.; Palsson, R.; et al. Timing and outcome of renal replacement therapy in patients with congenital malformations of the kidney and urinary tract. Clin. J. Am. Soc. Nephrol. 2013, 8, 67–74. [Google Scholar] [CrossRef]
- Sanna-Cherchi, S.; Ravani, P.; Corbani, V.; Parodi, S.; Haupt, R.; Piaggio, G.; Degli Innocenti, M.L.; Somenzi, D.; Trivelli, A.; Caridi, G.; et al. Congenital anomalies of the kidney and urinary tract (CAKUT): Longitudinal cohort study on renal outcome. Kidney Int. 2009, 76, 528–533. [Google Scholar] [CrossRef]
- Marzuillo, P.; Guarino, S.; Grandone, A.; Di Somma, A.; Diplomatico, M.; Rambaldi, P.F.; Decimo, F.; Miraglia Del Giudice, E.; La Manna, A.; Polito, C. Congenital solitary kidney size at birth could predict reduced eGFR levels later in life. J. Perinatol. Off. J. Calif. Perinat. Assoc. 2019, 39, 129–134. [Google Scholar] [CrossRef]
- McArdle, Z.; Schreuder, M.F.; Moritz, K.M.; Denton, K.M.; Singh, R.R. Physiology and Pathophysiology of Compensatory Adaptations of a Solitary Functioning Kidney. Front. Physiol. 2020, 11, 725. [Google Scholar] [CrossRef]
- Westland, R.; Kurvers, R.A.; van Wijk, J.A.; Schreuder, M.F. Risk factors for renal injury in children with a solitary functioning kidney. Pediatrics 2013, 131, e478–e485. [Google Scholar] [CrossRef]
- Odermatt, A. The Western-style diet: A major risk factor for impaired kidney function and chronic kidney disease. Am. J. Physiol. Renal. Physiol. 2011, 301, F919–F931. [Google Scholar] [CrossRef]
- Chevalier, R.L. Evolution, kidney development, and chronic kidney disease. Semin. Cell Dev. Biol. 2019, 91, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, R.L. Evolutionary Nephrology. Kidney Int. Rep. 2017, 2, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.W. From Fish to Philosopher; Little, Brown: Boston, MA, USA, 1953. [Google Scholar]
- Wallace, D.C. A mitochondrial bioenergetic etiology of disease. J. Clin. Investig. 2013, 123, 1405–1412. [Google Scholar] [CrossRef]
- Luyckx, V.A.; Chevalier, R.L. Impact of early life development on later onset chronic kidney disease and hypertension and the role of evolutionary trade-offs. Exp. Physiol. 2022, 107, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Kumar, S.; Sharma, R.; Gadodia, A.; Roy, K.K.; Sharma, J.B. The role of magnetic resonance imaging in fetal renal anomalies. Int. J. Gynaecol. Obstet. 2010, 111, 209–212. [Google Scholar] [CrossRef]
- Klein, J.; Buffin-Meyer, B.; Boizard, F.; Moussaoui, N.; Lescat, O.; Breuil, B.; Fedou, C.; Feuillet, G.; Casemayou, A.; Neau, E.; et al. Amniotic fluid peptides predict postnatal kidney survival in developmental kidney disease. Kidney Int. 2021, 99, 737–749. [Google Scholar] [CrossRef]
- Hamada, R.; Kikunaga, K.; Kaneko, T.; Okamoto, S.; Tomotsune, M.; Uemura, O.; Kamei, K.; Wada, N.; Matsuyama, T.; Ishikura, K.; et al. Urine alpha 1-microglobulin-to-creatinine ratio and beta 2-microglobulin-to-creatinine ratio for detecting CAKUT with kidney dysfunction in children. Pediatr. Nephrol. 2023, 38, 479–487. [Google Scholar] [CrossRef]
- Bennett, K.M.; Baldelomar, E.J.; Morozov, D.; Chevalier, R.L.; Charlton, J.R. New imaging tools to measure nephron number in vivo: Opportunities for developmental nephrology. J. Dev. Orig. Health Dis. 2020, in press. [Google Scholar] [CrossRef]
- Charlton, J.R.; Xu, Y.; Wu, T.; deRonde, K.A.; Hughes, J.L.; Dutta, S.; Oxley, G.T.; Cwiek, A.; Cathro, H.P.; Charlton, N.P.; et al. Magnetic resonance imaging accurately tracks kidney pathology and heterogeneity in the transition from acute kidney injury to chronic kidney disease. Kidney Int. 2021, 99, 173–185. [Google Scholar] [CrossRef]
- Carmody, J.B.; Charlton, J.R. Short-term gestation, long-term risk: Prematurity and chronic kidney disease. Pediatrics 2013, 131, 1168–1179. [Google Scholar] [CrossRef]
- Naved, B.A.; Bonventre, J.V.; Hubbell, J.A.; Hukriede, N.A.; Humphreys, B.D.; Kesselman, C.; Valerius, M.T.; McMahon, A.P.; Shankland, S.J.; Wertheim, J.A.; et al. Kidney repair and regeneration: Perspectives of the NIDDK (Re)Building a Kidney consortium. Kidney Int. 2022, 101, 845–853. [Google Scholar] [CrossRef] [PubMed]
- Benigni, A.; Morigi, M.; Remuzzi, G. Kidney regeneration. Lancet 2010, 375, 1310–1317. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Abnormal Collecting System (Duplicated, Refluxing, Obstructed) |
|---|
| Abnormal number or position of kidneys (ectopic, supernumerary, horseshoe kidney; renal agenesis) |
| Renal hypoplasia/dysplasia (with or without associated extrarenal anomalies) |
| Multicystic/polycystic kidney disease (unilateral or bilateral) |
| Oligohydramnios: Potter sequence (pulmonary hypoplasia with bilateral renal agenesis, severe posterior urethral valves, bilateral hypoplasia/dysplasia, or cystic kidney disease) |
| Rapid kidney growth (compensatory growth with decreased contralateral kidney function) |
| Upper Tract |
| Ureteropelvic junction obstruction |
| Lower tract |
| Vesicoureteral obstruction, ureterocele |
| Posterior urethral valves, anterior urethral valves, urethral atresia |
| Prenatal |
| Fetal ultrasound: rule out decreased amniotic fluid volume, increased renal echogenicity, hydronephrosis, bladder dilatation |
| Fetal karyotyping: rule out chromosomal anomaly |
| Serial fetal urine sampling (oligohydramnios or bilateral hydronephrosis): rule out critical obstruction and consider prenatal surgery |
| Postnatal |
| Complete physical examination: rule out extrarenal anomalies |
| Postnatal ultrasound: rule out bladder thickening/trabeculation |
Prune-belly syndrome (Eagle-Barrett syndrome)
|
VATER, VACTRL syndrome
|
Bladder exstrophy
|
Myelomeningocele
|
Sacrococcygeal teratoma
|
Congenital hypothyroidism
|
Presence of outer ear abnormalities or single umbilical artery
|
Chromosomal anomalies
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chevalier, R.L. CAKUT: A Pediatric and Evolutionary Perspective on the Leading Cause of CKD in Childhood. Pediatr. Rep. 2023, 15, 143-153. https://doi.org/10.3390/pediatric15010012
Chevalier RL. CAKUT: A Pediatric and Evolutionary Perspective on the Leading Cause of CKD in Childhood. Pediatric Reports. 2023; 15(1):143-153. https://doi.org/10.3390/pediatric15010012
Chicago/Turabian StyleChevalier, Robert L. 2023. "CAKUT: A Pediatric and Evolutionary Perspective on the Leading Cause of CKD in Childhood" Pediatric Reports 15, no. 1: 143-153. https://doi.org/10.3390/pediatric15010012
APA StyleChevalier, R. L. (2023). CAKUT: A Pediatric and Evolutionary Perspective on the Leading Cause of CKD in Childhood. Pediatric Reports, 15(1), 143-153. https://doi.org/10.3390/pediatric15010012