
Alpha-Lipoic Acid and Biotin in Neurodegenerative Diseases: Convergent Mechanistic Insights from Preclinical Models to Clinical Perspectives

 , , , , , and
, , , , , and
Abstract
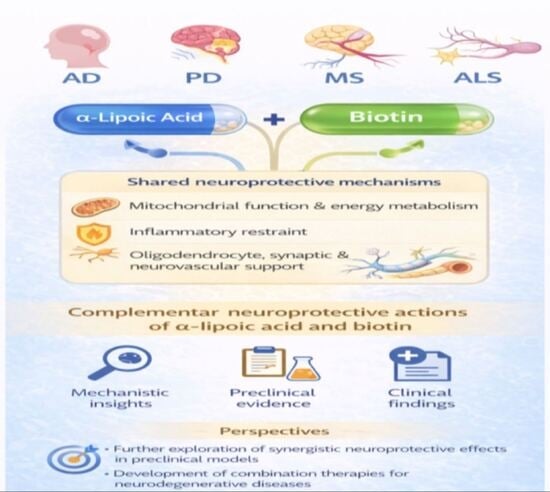
1. Introduction
2. Materials and Methods
3. Results
3.1. Alpha-Lipoic Acid
3.2. Biotin
3.3. Probable Complementary and Potentially Additive Interaction Between Alpha-Lipoic Acid and Biotin: Molecular Rationale for Neuroprotection in Neurodegenerative Diseases
3.4. Preclinical Evidence on Alpha-Lipoic Acid and Biotin in Neurodegenerative Disorders
3.4.1. Alzheimer’s Disease
3.4.2. Parkinson’s Disease
3.4.3. Multiple Sclerosis
3.4.4. Amyotrophic Lateral Sclerosis
3.4.5. Huntington’s Disease
3.5. Clinical Evidence and Translational Perspectives
3.5.1. Alzheimer’s Disease
3.5.2. Parkinson’s Disease
3.5.3. Multiple Sclerosis
3.5.4. Amyotrophic Lateral Sclerosis
3.5.5. Huntington’s Disease
3.6. Safety, Pharmacokinetics, and Limitations Regarding the Use of Alpha-Lipoic Acid and Biotin
3.6.1. Alpha-Lipoic Acid
3.6.2. Biotin
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gadhave, D.G.; Sugandhi, V.V.; Jha, S.K.; Nangare, S.N.; Gupta, G.; Singh, S.K.; Dua, K.; Cho, H.; Hansbro, P.M.; Paudel, K.R. Neurodegenerative Disorders: Mechanisms of Degeneration and Therapeutic Approaches with Their Clinical Relevance. Ageing Res. Rev. 2024, 99, 102357. [Google Scholar] [CrossRef]
- Lamptey, R.N.L.; Chaulagain, B.; Trivedi, R.; Gothwal, A.; Layek, B.; Singh, J. A Review of the Common Neurodegenerative Disorders: Current Therapeutic Approaches and the Potential Role of Nanotherapeutics. Int. J. Mol. Sci. 2022, 23, 1851. [Google Scholar] [CrossRef]
- Kelser, B.M.; Teichner, E.M.; Subtirelu, R.C.; Hoss, K.N. A Review of Proposed Mechanisms for Neurodegenerative Disease. Front. Aging Neurosci. 2024, 16, 1370580. [Google Scholar] [CrossRef]
- Wilson, D.M., III; Cookson, M.R.; Van Den Bosch, L.; Zetterberg, H.; Holtzman, D.M.; Dewachter, I. Hallmarks of Neurodegenerative Diseases. Cell 2023, 186, 693–714. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, X.W.; Zhang, Y.; Li, X. Advances in Research on Mitochondrial Dysfunction in Neurodegenerative Diseases. J. Neurol. 2025, 272, 364. [Google Scholar] [CrossRef]
- Ristori, S.; Bertoni, G.; Bientinesi, E.; Monti, D. The Role of Nutraceuticals and Functional Foods in Mitigating Cellular Senescence and Its Related Aspects: A Key Strategy for Delaying or Preventing Aging and Neurodegenerative Disorders. Nutrients 2025, 17, 1837. [Google Scholar] [CrossRef]
- Riveron-Negrete, L.; Fernández-Mejía, C. Pharmacological Effects of Biotin in Animals. Mini-Rev. Med. Chem. 2017, 17, 529–540. [Google Scholar] [CrossRef]
- Aguilera-Méndez, A.; Boone-Villa, D.; Nieto-Aguilar, R.; Villafaña-Rauda, S.; Saavedra-Molina, A.; Sobrevilla, J.V. Role of Vitamins in the Metabolic Syndrome and Cardiovascular Disease. Pflug. Arch. Eur. J. Physiol. 2022, 474, 117–140. [Google Scholar] [CrossRef]
- Karachaliou, C.E.; Livaniou, E. Biotin Homeostasis and Human Disorders: Recent Findings and Perspectives. Int. J. Mol. Sci. 2024, 25, 6578. [Google Scholar] [CrossRef]
- Manavi, M.A.; Nourhashemi, M.; Emami, S.; Fathian Nasab, M.H.; Dehnavi, F.; Küçükkılınç, T.T.; Foroumadi, A.; Sharifzadeh, M.; Khoobi, M. Lipoic Acid Scaffold Applications in the Design of Multitarget-Directed Ligands Against Alzheimer’s Disease. Bioorg. Chem. 2025, 157, 108241. [Google Scholar] [CrossRef]
- Superti, F.; Russo, R. Alpha-Lipoic Acid: Biological Mechanisms and Health Benefits. Antioxidants 2024, 13, 1228. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, S.; He, Y.; Pang, P.; Shan, H. Advances in α-Lipoic Acid for Disease Prevention: Mechanisms and Therapeutic Insights. Molecules 2025, 30, 1972. [Google Scholar] [CrossRef]
- Kodentsova, V.M.; Risnik, D.V.; Kryukova, E.V.; Dariy, S.G. Functional Ingredients for Specialized Foods: Issues to Be Addressed. Med. Alph. 2023, 8, 8–13. [Google Scholar] [CrossRef]
- Xie, H.; Yang, X.; Cao, Y.; Long, X.; Shang, H.; Jia, Z. Role of Lipoic Acid in Multiple Sclerosis. CNS Neurosci. Ther. 2022, 28, 319–331. [Google Scholar] [CrossRef]
- Molz, P.; Schröder, N. Potential Therapeutic Effects of Lipoic Acid on Memory Deficits Related to Aging and Neurodegeneration. Front. Pharmacol. 2017, 8, 849. [Google Scholar] [CrossRef]
- Kyung, S.; Lim, J.W.; Kim, H. α-Lipoic Acid Inhibits IL-8 Expression by Activating Nrf2 Signaling in Helicobacter pylori-Infected Gastric Epithelial Cells. Nutrients 2019, 11, 2524. [Google Scholar] [CrossRef]
- Wood, S.H.; van Dam, S.; Craig, T.; Tacutu, R.; O’Toole, A.; Merry, B.J.; de Magalhães, J.P. Transcriptome Analysis in Calorie-Restricted Rats Implicates Epigenetic and Post-Translational Mechanisms in Neuroprotection and Aging. Genome Biol. 2015, 16, 285. [Google Scholar] [CrossRef]
- Kim, S.M.; Ha, J.S.; Han, A.R.; Cho, S.W.; Yang, S.J. Effects of α-Lipoic Acid on LPS-Induced Neuroinflammation and NLRP3 Inflammasome Activation through the Regulation of BV-2 Microglial Cell Activation. BMB Rep. 2019, 52, 613–618. [Google Scholar] [CrossRef]
- Lin, C.; He, C.; Li, L.; Liu, Y.; Tang, L.; Ni, Z.; Zhang, N.; Lai, T.; Chen, X.; Wang, X. Alpha-Lipoic Acid (ALA) Ameliorates Early Brain Injury After Subarachnoid Hemorrhage via Inhibiting STING-NLRP3 Inflammatory Signaling in SD Rats. Neuroreport 2024, 35, 250–257. [Google Scholar] [CrossRef]
- Salinthone, S.; Yadav, V.; Schillace, R.V.; Bourdette, D.N.; Carr, D.W. Lipoic Acid Attenuates Inflammation via cAMP and Protein Kinase A Signaling. PLoS ONE 2010, 5, e13058. [Google Scholar] [CrossRef]
- Gomes, B.A.Q.; Santos, S.M.D.; Gato, L.D.S.; Espíndola, K.M.M.; Silva, R.K.M.D.; Davis, K.; Navegantes-Lima, K.C.; Burbano, R.M.R.; Romao, P.R.T.; Coleman, M.D.; et al. Alpha-Lipoic Acid Reduces Neuroinflammation and Oxidative Stress Induced by Dapsone in an Animal Model. Nutrients 2025, 17, 791. [Google Scholar] [CrossRef]
- Jiang, T.; Yin, F.; Yao, J.; Brinton, R.D.; Cadenas, E. Lipoic Acid Restores Age-Associated Impairment of Brain Energy Metabolism through the Modulation of Akt/JNK Signaling and PGC1α Transcriptional Pathway. Aging Cell 2013, 12, 1021–1031. [Google Scholar] [CrossRef]
- Fu, B.; Zhang, J.; Zhang, X.; Zhang, C.; Li, Y.; Zhang, Y.; He, T.; Li, P.; Zhu, X.; Zhao, Y.; et al. Alpha-Lipoic Acid Upregulates SIRT1-Dependent PGC-1α Expression and Protects Mouse Brain against Focal Ischemia. Neuroscience 2014, 281, 251–257. [Google Scholar] [CrossRef]
- Zhang, J.; Jiang, Y.; Dong, X.; Meng, Z.; Ji, L.; Kang, Y.; Liu, M.; Zhou, W.; Song, W. Alpha-Lipoic Acid Alleviates Cognitive Deficits in Transgenic APP23/PS45 Mice through a Mitophagy-Mediated Increase in ADAM10 α-Secretase Cleavage of APP. Alzheimer’s Res. Ther. 2024, 16, 160. [Google Scholar] [CrossRef]
- Molinari, C.; Morsanuto, V.; Ghirlanda, S.; Ruga, S.; Notte, F.; Gaetano, L.; Uberti, F. Role of Combined Lipoic Acid and Vitamin D3 on Astrocytes as a Way to Prevent Brain Ageing by Induced Oxidative Stress and Iron Accumulation. Oxidative Med. Cell. Longev. 2019, 2019, 2843121. [Google Scholar] [CrossRef]
- Sun, L.Q.; Chen, Y.Y.; Wang, X.; Li, X.J.; Xue, B.; Qu, L.; Zhang, T.T.; Mu, Y.M.; Lu, J.M. The Protective Effect of Alpha Lipoic Acid on Schwann Cells Exposed to Constant or Intermittent High Glucose. Biochem. Pharmacol. 2012, 84, 961–973. [Google Scholar] [CrossRef]
- Fahmy, M.I.; Khalaf, S.S.; Elrayess, R.A. The Neuroprotective Effects of Alpha Lipoic Acid in Rotenone-Induced Parkinson’s Disease in Mice via Activating PI3K/AKT Pathway and Antagonizing Related Inflammatory Cascades. Eur. J. Pharmacol. 2024, 980, 176878. [Google Scholar] [CrossRef]
- Kamarudin, M.N.; Mohd Raflee, N.A.; Hussein, S.S.; Lo, J.Y.; Supriady, H.; Abdul Kadir, H. (R)-(+)-α-Lipoic Acid Protected NG108-15 Cells against H2O2-Induced Cell Death through PI3K-Akt/GSK-3β Pathway and Suppression of NF-κB-Cytokines. Drug Des. Dev. Ther. 2014, 8, 1765–1780. [Google Scholar] [CrossRef][Green Version]
- Capece, U.; Moffa, S.; Improta, I.; Di Giuseppe, G.; Nista, E.C.; Cefalo, C.M.A.; Cinti, F.; Pontecorvi, A.; Gasbarrini, A.; Giaccari, A.; et al. Alpha-Lipoic Acid and Glucose Metabolism: A Comprehensive Update on Biochemical and Therapeutic Features. Nutrients 2022, 15, 18. [Google Scholar] [CrossRef]
- Shay, K.P.; Moreau, R.F.; Smith, E.J.; Smith, A.R.; Hagen, T.M. Alpha-Lipoic Acid as a Dietary Supplement: Molecular Mechanisms and Therapeutic Potential. Biochim. Biophys. Acta 2009, 1790, 1149–1160. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, D.; Zhang, Z.; Tian, J.; An, J.; Zhang, W.; Ben, Y. Alpha-Lipoic Acid Activates AMPK to Protect against Oxidative Stress and Apoptosis in Rats with Diabetic Peripheral Neuropathy. Hormones 2023, 22, 95–105. [Google Scholar] [CrossRef] [PubMed]
- McCarty, M.F.; DiNicolantonio, J.J. Neuroprotective Potential of High-Dose Biotin. Med. Hypotheses 2017, 109, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Canda, E.; Kalkan Uçar, S.; Çoker, M. Biotinidase Deficiency: Prevalence, Impact and Management Strategies. Pediatr. Health Med. Ther. 2020, 11, 127–133. [Google Scholar]
- Sedel, F.; Bernard, D.; Mock, D.M.; Tourbah, A. Targeting Demyelination and Virtual Hypoxia with High-Dose Biotin as a Treatment for Progressive Multiple Sclerosis. Neuropharmacology 2016, 110, 644–653. [Google Scholar] [CrossRef]
- Tourbah, A.; Lebrun-Frenay, C.; Edan, G.; Clanet, M.; Papeix, C.; Vukusic, S.; De Sèze, J.; Debouverie, M.; Gout, O.; Clavelou, P.; et al. MD1003 (High-Dose Biotin) for the Treatment of Progressive Multiple Sclerosis: A Randomised, Double-Blind, Placebo-Controlled Study. Mult. Scler. 2016, 22, 1719–1731. [Google Scholar] [CrossRef]
- Wal, A.; Sasmal, A.; Singh, R.; Yadav, P.; Singh, Y.; Garg, V.; Wal, P. Regulatory Role, Mechanism, and Metabolic Profile of Biotin in Gene Expression. Curr. Pharmacogenomics Pers. Med. 2023, 20, e120723218676. [Google Scholar] [CrossRef]
- Hassan, Y.I.; Zempleni, J. Epigenetic Regulation of Chromatin Structure and Gene Function by Biotin. J. Nutr. 2006, 136, 1763–1765. [Google Scholar] [CrossRef]
- Aguilera-Méndez, A.; Figueroa-Fierros, I.; Ruiz-Pérez, X.; Godínez-Hernández, D.; Saavedra-Molina, A.; Rios-Chavez, P.; Villafaña, S.; Boone-Villa, D.; Ortega-Cuellar, D.; Gauthereau-Torres, M.Y.; et al. The Beneficial Effects of Prenatal Biotin Supplementation in a Rat Model of Intrauterine Caloric Restriction to Prevent Cardiometabolic Risk in Adult Female Offspring. Int. J. Mol. Sci. 2024, 25, 9052. [Google Scholar] [CrossRef]
- Vilches-Flores, A.; Tovar, A.R.; Marin-Hernandez, A.; Rojas-Ochoa, A.; Fernandez-Mejia, C. Biotin Increases Glucokinase Expression via Soluble Guanylate Cyclase/Protein Kinase G, Adenosine Triphosphate Production and Autocrine Action of Insulin in Pancreatic Rat Islets. J. Nutr. Biochem. 2010, 21, 606–612. [Google Scholar] [CrossRef]
- Aguilera-Méndez, A.; Espino-García, R.; Toledo-López, Z.J.; Hernández-Gallegos, Z.; Villafaña-Rauda, S.; Nieto-Aguilar, R.; Serrato-Ochoa, D.; Manuel-Jacobo, G.C. Biotin Improves Relaxation of Rat Aortic Rings in Combination with Antihypertensive Drugs. PharmaNutrition 2019, 8, 100147. [Google Scholar] [CrossRef]
- Park, M.K.; Yang, H.W.; Woo, S.Y.; Kim, D.Y.; Son, D.S.; Choi, B.Y.; Suh, S.W. Modulation of Second Messenger Signaling in the Brain through PDE4 and PDE5 Inhibition: Therapeutic Implications for Neurological Disorders. Cells 2025, 14, 86. [Google Scholar] [CrossRef]
- Borovac, J.; Rai, J.; Valencia, M.; Li, H.; Georgiou, J.; Collingridge, G.L.; Takao, K.; Okamoto, K. Optogenetic Elevation of Postsynaptic cGMP in the Hippocampal Dentate Gyrus Enhances LTP and Modifies Mouse Behaviors. Front. Mol. Neurosci. 2024, 17, 1479360. [Google Scholar] [CrossRef]
- Tropea, M.R.; Gulisano, W.; Vacanti, V.; Arancio, O.; Puzzo, D.; Palmeri, A. Nitric Oxide/cGMP/CREB Pathway and Amyloid-Beta Crosstalk: From Physiology to Alzheimer’s Disease. Free Radic. Biol. Med. 2022, 193, 657–668. [Google Scholar] [CrossRef]
- Puzzo, D.; Staniszewski, A.; Deng, S.X.; Privitera, L.; Leznik, E.; Liu, S.; Zhang, H.; Feng, Y.; Palmeri, A.; Landry, D.W.; et al. Phosphodiesterase 5 Inhibition Improves Synaptic Function, Memory, and Amyloid-Beta Load in an Alzheimer’s Disease Mouse Model. J. Neurosci. 2009, 29, 8075–8086. [Google Scholar] [CrossRef]
- Sghaier, R.; Zarrouk, A.; Nury, T.; Badreddine, I.; O’Brien, N.; Mackrill, J.J.; Vejux, A.; Samadi, M.; Nasser, B.; Caccia, C.; et al. Biotin Attenuation of Oxidative Stress, Mitochondrial Dysfunction, Lipid Metabolism Alteration and 7β-Hydroxycholesterol-Induced Cell Death in 158N Murine Oligodendrocytes. Free Radic. Res. 2019, 53, 535–561. [Google Scholar] [CrossRef]
- Aguilera-Méndez, A.; Fernández-Mejía, C. The Hypotriglyceridemic Effect of Biotin Supplementation Involves Increased Levels of cGMP and AMPK Activation. BioFactors 2012, 38, 387–394. [Google Scholar] [CrossRef]
- Moreno-Méndez, E.; Hernández-Vázquez, A.; Fernández-Mejía, C. Effect of Biotin Supplementation on Fatty Acid Metabolic Pathways in 3T3-L1 Adipocytes. BioFactors 2019, 45, 259–270. [Google Scholar] [CrossRef]
- Fourcade, S.; Goicoechea, L.; Parameswaran, J.; Schlüter, A.; Launay, N.; Ruiz, M.; Seyer, A.; Colsch, B.; Calingasan, N.Y.; Ferrer, I.; et al. High-Dose Biotin Restores Redox Balance, Energy and Lipid Homeostasis, and Axonal Health in a Model of Adrenoleukodystrophy. Brain Pathol. 2020, 30, 945–963. [Google Scholar] [CrossRef]
- Lohr, K.M.; Frost, B.; Scherzer, C.; Feany, M.B. Biotin Rescues Mitochondrial Dysfunction and Neurotoxicity in a Tauopathy Model. Proc. Natl. Acad. Sci. USA 2020, 117, 33608–33618. [Google Scholar] [CrossRef]
- Cui, Q.L.; Lin, Y.H.; Xu, Y.K.T.; Fernandes, M.G.F.; Rao, V.T.S.; Kennedy, T.E.; Antel, J. Effects of Biotin on Survival, Ensheathment, and ATP Production by Oligodendrocyte Lineage Cells In Vitro. PLoS ONE 2020, 15, e0233859. [Google Scholar] [CrossRef]
- Shen, W.; Hao, J.; Tian, C.; Ren, J.; Yang, L.; Li, X.; Luo, C.; Cotma, C.W.; Liu, J. A Combination of Nutriments Improves Mitochondrial Biogenesis and Function in Skeletal Muscle of Type 2 Diabetic Goto-Kakizaki Rats. PLoS ONE 2008, 3, e2328. [Google Scholar] [CrossRef]
- Zempleni, J.; Helm, R.M.; Mock, D.M. In Vivo Biotin Supplementation at a Pharmacologic Dose Decreases Proliferation Rates of Human Peripheral Blood Mononuclear Cells and Cytokine Release. J. Nutr. 2001, 131, 1479–1484. [Google Scholar] [CrossRef]
- Wiedmann, S.; Eudy, J.D.; Zempleni, J. Biotin Supplementation Increases Expression of Genes Encoding Interferon-Gamma, Interleukin-1Beta, and 3-Methylcrotonyl-CoA Carboxylase, and Decreases Expression of the Gene Encoding Interleukin-4 in Human Peripheral Blood Mononuclear Cells. J. Nutr. 2003, 133, 716–719. [Google Scholar] [CrossRef]
- Sahin, K.; Orhan, C.; Karatoprak, S.; Tuzcu, M.; Deeh, P.B.D.; Ozercan, I.H.; Sahin, N.; Bozoglan, M.Y.; Sylla, S.; Ojalvo, S.P.; et al. Therapeutic Effects of a Novel Form of Biotin on Propionic Acid-Induced Autistic Features in Rats. Nutrients 2022, 14, 1280. [Google Scholar] [CrossRef]
- Helmy, S.A.; Samaha, M.M.; Abd El Salam, A.S.G.; Abd Elrazik, N.A.; El-Sayed, S.M. Biotin and sulfasalazine combination therapy alleviates acetic acid-induced ulcerative colitis in rats through modulation of oxidative stress and inflammatory signaling pathways. Sci. Rep. 2025, 15, 9932. [Google Scholar] [CrossRef]
- Kuroishi, T.; Endo, Y.; Muramoto, K.; Sugawara, S. Biotin deficiency up-regulates TNF-α production in murine macrophages. J. Leukoc. Biol. 2008, 83, 912–920. [Google Scholar] [CrossRef]
- Skupsky, J.; Sabui, S.; Hwang, M.; Nakasaki, M.; Cahalan, M.D.; Said, H.M. Biotin supplementation ameliorates murine colitis by preventing NF-κB activation. Cell. Mol. Gastroenterol. Hepatol. 2020, 9, 557–567. [Google Scholar] [CrossRef]
- Kuroishi, T. Regulation of immunological and inflammatory functions by biotin. Can. J. Physiol. Pharmacol. 2015, 93, 1091–1096. [Google Scholar] [CrossRef]
- Sakurai-Yageta, M.; Suzuki, Y. Molecular Mechanisms of Biotin in Modulating Inflammatory Diseases. Nutrients 2024, 16, 2444. [Google Scholar] [CrossRef]
- Moretti, R.; Peinkhofer, C. B Vitamins and Fatty Acids: What Do They Share with Small Vessel Disease-Related Dementia? Int. J. Mol. Sci. 2019, 20, 5797. [Google Scholar] [CrossRef]
- Agrawal, S.; Agrawal, A.; Said, H.M. Biotin Deficiency Enhances the Inflammatory Response of Human Dendritic Cells. Am. J. Physiol. Cell Physiol. 2016, 311, C386–C391. [Google Scholar] [CrossRef]
- Aguilera-Mendez, A.; Hernández-Equihua, M.G.; Rueda-Rocha, A.C.; Guajardo-López, C.; Nieto-Aguilar, R.; Serrato-Ochoa, D.; Ruíz Herrera, L.F.; Guzmán-Nateras, J.A. Protective Effect of Supplementation with Biotin against High-Fructose-Induced Metabolic Syndrome in Rats. Nutr. Res. 2018, 57, 86–96. [Google Scholar] [CrossRef]
- Shahid, A.; Nasir, K.; Bhatia, M. Therapeutic Potential of Alpha-Lipoic Acid: Unraveling Its Role in Oxidative Stress and Inflammatory Conditions. Curr. Issues Mol. Biol. 2025, 47, 322. [Google Scholar] [CrossRef]
- dos Santos, P.S.; Feitosa, C.M.; Saldanha, G.B.; da Rocha Tomé, A.; Feng, D.; de Freitas, R.M. Lipoic Acid Inhibits Caspase-Dependent and -Independent Cell Death Pathways and Is Neuroprotective against Hippocampal Damage after Pilocarpine-Induced Seizures. Pharmacol. Biochem. Behav. 2011, 97, 531–536. [Google Scholar] [CrossRef]
- Wei, W.; Wang, H.; Wu, Y.; Ding, K.; Li, T.; Cong, Z.; Xu, J.; Zhou, M.; Huang, L.; Ding, H.; et al. Alpha Lipoic Acid Inhibits Neural Apoptosis via a Mitochondrial Pathway in Rats following Traumatic Brain Injury. Neurochem. Int. 2015, 87, 85–91. [Google Scholar] [CrossRef]
- Vasudevan, D.; Naik, M.M.; Mukaddam, Q.I. Efficacy and Safety of Methylcobalamin, Alpha Lipoic Acid and Pregabalin Combination versus Pregabalin Monotherapy in Improving Pain and Nerve Conduction Velocity in Type 2 Diabetes-Associated Peripheral Neuropathy: Results of a Pilot Study. Ann. Indian Acad. Neurol. 2014, 17, 19–24. [Google Scholar] [CrossRef]
- Maladkar, M.; Tekchandani, C.; Dave, U. Post-Marketing Surveillance of Fixed Dose Combination of Methylcobalamin, Alpha Lipoic Acid, Folic Acid, Biotin, Benfotiamine, and Vitamin B6 (Nutripathy) for the Management of Peripheral Neuropathy. J. Diabetes Mellit. 2014, 4, 124–132. [Google Scholar] [CrossRef]
- Holmquist, L.; Stuchbury, G.; Berbaum, K.; Muscat, S.; Young, S.; Hager, K.; Engel, J.; Münch, G. Lipoic Acid as a Novel Treatment for Alzheimer’s Disease and Related Dementias. Pharmacol. Ther. 2007, 113, 154–164. [Google Scholar] [CrossRef]
- Seifar, F.; Khalili, M.; Khaledyan, H.; Amiri Moghadam, S.; Izadi, A.; Azimi, A.; Shakouri, S.K. α-Lipoic Acid, Functional Fatty Acid, as a Novel Therapeutic Alternative for Central Nervous System Diseases: A Review. Nutr. Neurosci. 2019, 22, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Dieter, F.; Esselun, C.; Eckert, G.P. Redox Active α-Lipoic Acid Differentially Improves Mitochondrial Dysfunction in a Cellular Model of Alzheimer and Its Control Cells. Int. J. Mol. Sci. 2022, 23, 9186. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.F.; Bussiere, J.R.; Hammond, R.S.; Montine, T.J.; Henson, E.; Jones, R.E.; Stackman, R.W., Jr. Chronic Dietary Alpha-Lipoic Acid Reduces Deficits in Hippocampal Memory of Aged Tg2576 Mice. Neurobiol. Aging 2007, 28, 213–225. [Google Scholar] [CrossRef]
- Sancheti, H.; Kanamori, K.; Patil, I.; Díaz Brinton, R.; Ross, B.D.; Cadenas, E. Reversal of Metabolic Deficits by Lipoic Acid in a Triple Transgenic Mouse Model of Alzheimer’s Disease: A 13C NMR Study. J. Cereb. Blood Flow Metab. 2014, 34, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H.; Yan, X.Z.; Xu, S.F.; Pang, Z.Q.; Li, L.B.; Yang, Y.; Fan, Y.G.; Wang, Z.; Yu, X.; Guo, C.; et al. α-Lipoic Acid Maintains Brain Glucose Metabolism via BDNF/TrkB/HIF-1α Signaling Pathway in P301S Mice. Front. Aging Neurosci. 2020, 12, 262. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.Y.; Xu, J.H.; Chang, Y.W.; Lo, Y.M.; Wu, J.S.; Huang, W.C.; Shen, S.C. Effects of α-Lipoic Acid on Phagocytosis of Oligomeric Beta-Amyloid1–42 in BV-2 Mouse Microglial Cells. Front. Aging Neurosci. 2022, 13, 788723. [Google Scholar] [CrossRef]
- Almasi, S.; Jafarzadeh Shirazi, M.R.; Rezvani, M.R.; Ramezani, M.; Salehi, I.; Pegah, A.; Komaki, A. The Protective Effect of Biotin Supplementation and Swimming Training on Cognitive Impairment and Mental Symptoms in a Rat Model of Alzheimer’s Disease: A Behavioral, Biochemical, and Histological Study. Heliyon 2024, 10, e32299. [Google Scholar] [CrossRef]
- Andreeva-Gateva, P.; Traikov, L.; Sabit, Z.; Bakalov, D.; Tafradjiiska-Hadjiolova, R. Antioxidant Effect of Alpha-Lipoic Acid in 6-Hydroxydopamine Unilateral Intrastriatal Injected Rats. Antioxidants 2020, 9, 122. [Google Scholar] [CrossRef]
- Zhang, J.; Gao, Y.; Zhang, L.; Zhang, C.; Zhao, Y.; Zhang, Y.; Li, S.; Chang, C.; Zhang, X.; Yang, G. Alpha-Lipoic Acid Attenuates MPTP/MPP+-Induced Neurotoxicity: Roles of SIRT1-Dependent PGC-1α Signaling Pathways. Neurotox. Res. 2022, 40, 410–419. [Google Scholar] [CrossRef]
- Tai, S.; Zheng, Q.; Zhai, S.; Cai, T.; Xu, L.; Yang, L.; Jiao, L.; Zhang, C. Alpha-Lipoic Acid Mediates Clearance of Iron Accumulation by Regulating Iron Metabolism in a Parkinson’s Disease Model Induced by 6-OHDA. Front. Neurosci. 2020, 14, 612. [Google Scholar] [CrossRef]
- Zheng, Q.; Ma, P.; Yang, P.; Zhai, S.; He, M.; Zhang, X.; Tu, Q.; Jiao, L.; Ye, L.; Feng, Z.; et al. Alpha Lipoic Acid Ameliorates Motor Deficits by Inhibiting Ferroptosis in Parkinson’s Disease. Neurosci. Lett. 2023, 810, 137346. [Google Scholar] [CrossRef] [PubMed]
- de Araújo, D.P.; De Sousa, C.N.; Araújo, P.V.; Menezes, C.E.; Sousa Rodrigues, F.T.; Escudeiro, S.S.; Lima, N.B.; Patrocínio, M.C.; Aguiar, L.M.; Viana, G.S.; et al. Behavioral and Neurochemical Effects of Alpha-Lipoic Acid in the Model of Parkinson’s Disease Induced by Unilateral Stereotaxic Injection of 6-OHDA in Rat. Evid.-Based Complement. Altern. Med. 2013, 2013, 571378. [Google Scholar] [CrossRef]
- Zhang, S.F.; Xie, C.L.; Lin, J.Y.; Wang, M.H.; Wang, X.J.; Liu, Z.G. Lipoic Acid Alleviates L-DOPA-Induced Dyskinesia in 6-OHDA Parkinsonian Rats via Anti-Oxidative Stress. Mol. Med. Rep. 2018, 17, 1118–1124. [Google Scholar] [CrossRef] [PubMed]
- Kulikova, O.; Troshev, D.; Berezhnoy, D.; Stvolinsky, S.; Timoshina, Y.; Abaimov, D.; Muzychuk, O.; Latanov, A.; Fedorova, T. Neuroprotective Efficacy of a Nanomicellar Complex of Carnosine and Lipoic Acid in a Rat Model of Rotenone-Induced Parkinson’s Disease. Antioxidants 2023, 12, 1215. [Google Scholar] [CrossRef]
- Lai, Y.; Reina-Gonzalez, P.; Maor, G.; Miller, G.W.; Sarkar, S. Biotin Mitigates the Development of Manganese-Induced, Parkinson’s Disease-Related Neurotoxicity in Drosophila and Human Neurons. Sci. Signal. 2025, 18, eadn9868. [Google Scholar] [CrossRef]
- Dovonou, A.; Bolduc, C.; Soto Linan, V.; Gora, C.; Peralta, M.R., III; Lévesque, M. Animal Models of Parkinson’s Disease: Bridging the Gap between Disease Hallmarks and Research Questions. Transl. Neurodegener. 2023, 12, 36. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.E.; Moes, N.; Zwickey, H.; Cunningham, C.L.; Gregory, W.L.; Oken, B. Treatment of Experimental Autoimmune Encephalomyelitis with Alpha Lipoic Acid and Associative Conditioning. Brain Behav. Immun. 2008, 22, 538–543. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, B.; Gong, S.; Wu, Q.; Gao, J. Neuroprotective Effects of α-Lipoic Acid on Long-Term Experimental Autoimmune Encephalomyelitis. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 6517–6528. [Google Scholar] [CrossRef]
- Chaudhary, P.; Marracci, G.; Galipeau, D.; Pocius, E.; Morris, B.; Bourdette, D. Lipoic Acid Reduces Inflammation in a Mouse Focal Cortical Experimental Autoimmune Encephalomyelitis Model. J. Neuroimmunol. 2015, 289, 68–74. [Google Scholar] [CrossRef][Green Version]
- Levy, M.J.F.; Garcia-Diaz, B.; Sedel, F.; Baron-Van Evercooren, A.; Mozafari, S. High-Dose Pharmaceutical Grade Biotin (MD1003) Accelerates Differentiation of Murine and Grafted Human Oligodendrocyte Progenitor Cells In Vivo. Int. J. Mol. Sci. 2022, 23, 15733. [Google Scholar] [CrossRef]
- Yulug, B.; Kilic, E.; Oğuz, T.; Orhan, C.; Er, B.; Tuzcu, M.; Ozercan, I.H.; Sahin, N.; Canpolat, S.; Komorowski, J.; et al. Dose-Dependent Effect of a New Biotin Compound in Hippocampal Remyelination in Rats. Mol. Neurobiol. 2025, 62, 6503–6520. [Google Scholar] [CrossRef]
- Andreassen, O.A.; Dedeoglu, A.; Friedlich, A.; Ferrante, K.L.; Hughes, D.; Szabo, C.; Beal, M.F. Effects of an Inhibitor of Poly(ADP-Ribose) Polymerase, Desmethylselegiline, Trientine, and Lipoic Acid in Transgenic ALS Mice. Exp. Neurol. 2001, 168, 419–424. [Google Scholar] [CrossRef]
- Wang, T.; Cheng, J.; Wang, S.; Wang, X.; Jiang, H.; Yang, Y.; Wang, Y.; Zhang, C.; Liang, W.; Feng, H. α-Lipoic Acid Attenuates Oxidative Stress and Neurotoxicity via the ERK/Akt-Dependent Pathway in the Mutant hSOD1-Related Drosophila Model and the NSC34 Cell Line of Amyotrophic Lateral Sclerosis. Brain Res. Bull. 2018, 140, 299–310. [Google Scholar] [CrossRef]
- Giacobbe, A.; Hiana, J.; Wang, O.; Benatar, M.; Wicks, P.; Mascias Cadavid, J.; ALSUntangled Investigators. ALSUntangled #79: Alpha-Lipoic Acid. Amyotroph. Lateral Scler. Front. Degener. 2025, 27, 233–237. [Google Scholar] [CrossRef]
- World Intellectual Property Organization. Biotin for Treating Amyotrophic Lateral Sclerosis. WO2016151132A1, 25 March 2016. Available online: https://patents.google.com/patent/WO2016151132A1 (accessed on 18 March 2026).
- Andreassen, O.A.; Ferrante, R.J.; Dedeoglu, A.; Beal, M.F. Lipoic Acid Improves Survival in Transgenic Mouse Models of Huntington’s Disease. NeuroReport 2001, 12, 3371–3373. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, A.; Kanwal, A.; Banerjee, S.K.; Sandhir, R. Mitochondrial Modulators in Experimental Huntington’s Disease: Reversal of Mitochondrial Dysfunctions and Cognitive Deficits. Neurobiol. Aging 2015, 36, 2186–2200. [Google Scholar] [CrossRef] [PubMed]
- Bono-Yagüe, J.; Gómez-Escribano, A.P.; Millán, J.M.; Vázquez-Manrique, R.P. Reactive Species in Huntington Disease: Are They Really the Radicals You Want to Catch? Antioxidants 2020, 9, 577. [Google Scholar] [CrossRef]
- Lim, R.G.; Al-Dalahmah, O.; Wu, J.; Gold, M.P.; Reidling, J.C.; Tang, G.; Adam, M.; Dansu, D.K.; Park, H.J.; Casaccia, P.; et al. Huntington Disease Oligodendrocyte Maturation Deficits Revealed by Single-Nucleus RNAseq Are Rescued by Thiamine–Biotin Supplementation. Nat. Commun. 2022, 13, 7791. [Google Scholar] [CrossRef] [PubMed]
- Hager, K.; Kenklies, M.; McAfoose, J.; Engel, J.; Münch, G. Alpha-Lipoic Acid as a New Treatment Option for Alzheimer’s Disease—A 48 Months Follow-Up Analysis. In Neuropsychiatric Disorders: An Integrative Approach; Journal of Neural Transmission. Supplementa; Springer: New York, NY, USA, 2007; Volume 72, pp. 189–193. [Google Scholar] [CrossRef]
- Shinto, L.; Quinn, J.; Montine, T.; Dodge, H.H.; Woodward, W.; Baldauf-Wagner, S.; Waichunas, D.; Bumgarner, L.; Bourdette, D.; Silbert, L.; et al. A Randomized Placebo-Controlled Pilot Trial of Omega-3 Fatty Acids and Alpha Lipoic Acid in Alzheimer’s Disease. J. Alzheimer’s Dis. 2014, 38, 111–120. [Google Scholar] [CrossRef]
- Fava, A.; Pirritano, D.; Plastino, M.; Cristiano, D.; Puccio, G.; Colica, C.; Ermio, C.; De Bartolo, M.; Mauro, G.; Bosco, D. The Effect of Lipoic Acid Therapy on Cognitive Functioning in Patients with Alzheimer’s Disease. J. Neurodegener. Dis. 2013, 2013, 454253. [Google Scholar] [CrossRef]
- Hager, K.; Marahrens, A.; Kenklies, M.; Riederer, P.; Münch, G. Alpha-Lipoic Acid as a New Treatment Option for Alzheimer Type Dementia. Arch. Gerontol. Geriatr. 2001, 32, 275–282. [Google Scholar] [CrossRef]
- Galasko, D.R.; Peskind, E.; Clark, C.M.; Quinn, J.F.; Ringman, J.M.; Jicha, G.A.; Cotman, C.; Cottrell, B.; Montine, T.J.; Thomas, R.G.; et al. Alzheimer’s Disease Cooperative Study. Antioxidants for Alzheimer Disease: A Randomized Clinical Trial with Cerebrospinal Fluid Biomarker Measures. Arch. Neurol. 2012, 69, 836–841. [Google Scholar] [CrossRef]
- Kong, Y.; Zhong, J.; Wang, T.; Zhang, D. Association between Dietary Biotin Intake and Dementia Risk, Including Alzheimer’s Disease: A Prospective Study of 122,959 UK Biobank Participants. Mol. Nutr. Food Res. 2025, 69, e70252. [Google Scholar] [CrossRef]
- Cooper, J.L. P3-400: Biotin Deficiency and Abnormal Pantothenic Acid Levels in Dementia. Alzheimer’s Dement. 2008, 4, T638–T639. [Google Scholar] [CrossRef]
- Rabin, M.L.; Stevens-Haas, C.; Havrilla, E.; Rosenstein, A.; Toffey, B.; Devi, T.; Earnhardt, M.C.; Kurlan, R. Complementary Therapies for Parkinson’s Disease: What’s Promoted, Rationale, Potential Risks and Benefits. Mov. Disord. Clin. Pract. 2015, 2, 205–212. [Google Scholar] [CrossRef]
- Przewodowska, D.; Marzec, W.; Madetko, N. Novel Therapies for Parkinsonian Syndromes—Recent Progress and Future Perspectives. Front. Mol. Neurosci. 2021, 14, 720220. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. Alpha-Lipoic Acid/L-Acetyl Carnitine for Progressive Supranuclear Palsy (NCT01537549). 2017. Available online: https://clinicaltrials.gov/ct2/show/study/NCT01537549 (accessed on 18 March 2026).
- Nishiwaki, H.; Ueyama, J.; Ito, M.; Hamaguchi, T.; Takimoto, K.; Maeda, T.; Kashihara, K.; Tsuboi, Y.; Mori, H.; Kurokawa, K.; et al. Meta-Analysis of Shotgun Sequencing of Gut Microbiota in Parkinson’s Disease. NPJ Park. Dis. 2024, 10, 106. [Google Scholar] [CrossRef]
- Rodrigues, P.; Viero, F.T.; Trevisan, G. The Impact of α-Lipoic Acid Treatment on Multiple Sclerosis Disability: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Sclerosis 2025, 3, 4. [Google Scholar] [CrossRef]
- Yadav, V.; Marracci, G.; Lovera, J.; Woodward, W.; Bogardus, K.; Marquardt, W.; Shinto, L.; Morris, C.; Bourdette, D. Lipoic Acid in Multiple Sclerosis: A Pilot Study. Mult. Scler. 2005, 11, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Loy, B.D.; Fling, B.W.; Horak, F.B.; Bourdette, D.N.; Spain, R.I. Effects of Lipoic Acid on Walking Performance, Gait, and Balance in Secondary Progressive Multiple Sclerosis. Complement. Ther. Med. 2018, 41, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, S.E.; Yadav, V.; Kerns, A.R.; Tsang, C.; Markwardt, S.; Kim, E.; Spain, R.; Bourdette, D.; Salinthone, S. Lipoic Acid Stimulates cAMP Production in Healthy Control and Secondary Progressive MS Subjects. Mol. Neurobiol. 2018, 55, 6037–6049. [Google Scholar] [CrossRef]
- Sedel, F.; Papeix, C.; Bellanger, A.; Touitou, V.; Lebrun-Frenay, C.; Galanaud, D.; Gout, O.; Lyon-Caen, O.; Tourbah, A.; Fontaine, B. High Doses of Biotin in Chronic Progressive Multiple Sclerosis: A Pilot Study. Mult. Scler. Relat. Disord. 2015, 4, 159–169. [Google Scholar] [CrossRef]
- Birnbaum, G.; Stulc, J. High-Dose Biotin as Treatment for Progressive Multiple Sclerosis. Mult. Scler. Relat. Disord. 2017, 18, 141–143. [Google Scholar] [CrossRef]
- Tourbah, A.; Gout, O.; Vighetto, A.; Deburghgraeve, V.; Pelletier, J.; Papeix, C.; Lebrun-Frenay, C.; Labauge, P.; Brassat, D.; Toosy, A.; et al. MD1003 (High-Dose Pharmaceutical-Grade Biotin) for the Treatment of Chronic Visual Loss Related to Optic Neuritis in Multiple Sclerosis: A Randomized, Double-Blind, Placebo-Controlled Study. CNS Drugs 2018, 32, 661–672. [Google Scholar] [CrossRef]
- Cree, B.A.C.; Cutter, G.; Wolinsky, J.S.; Freedman, M.S.; Comi, G.; Giovannoni, G.; Hartung, H.P.; Arnold, D.; Kuhle, J.; Block, V.; et al. Safety and Efficacy of MD1003 (High-Dose Biotin) in Patients with Progressive Multiple Sclerosis (SPI2): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Neurol. 2020, 19, 988–997. [Google Scholar] [CrossRef]
- Espiritu, A.I.; Remalante-Rayco, P.P.M. High-Dose Biotin for Multiple Sclerosis: A Systematic Review and Meta-Analyses of Randomized Controlled Trials. Mult. Scler. Relat. Disord. 2021, 55, 103159. [Google Scholar] [CrossRef] [PubMed]
- Créange, A.; Hutin, E.; Sedel, F.; Le Vigouroux, L.; Lefaucheur, J.P. High-Dose Pharmaceutical-Grade Biotin in Patients with Demyelinating Neuropathies: A Phase 2b Open-Label, Uncontrolled, Pilot Study. BMC Neurol. 2023, 23, 389. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. Explore Neuroprotective Effect of Lipoic Acid in Amyotrophic Lateral Sclerosis (NCT04518540). 2020. Available online: https://clinicaltrials.gov/study/NCT04518540 (accessed on 18 March 2026).
- Juntas-Morales, R.; Pageot, N.; Bendarraz, A.; Alphandéry, S.; Sedel, F.; Seigle, S.; Camu, W. High-Dose Pharmaceutical Grade Biotin (MD1003) in Amyotrophic Lateral Sclerosis: A Pilot Study. EClinicalMedicine 2020, 19, 100254. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. High Doses of Pharmaceutical-Grade Biotin (MD1003) in Amyotrophic Lateral Sclerosis (NCT03427086). 2018. Available online: https://clinicaltrials.gov/study/NCT03427086 (accessed on 18 March 2026).
- Vaddadi, K.S.; Soosai, E.; Chiu, E.; Dingjan, P. A Randomised, Placebo-Controlled, Double-Blind Study of Treatment of Huntington’s Disease with Unsaturated Fatty Acids. NeuroReport 2002, 13, 29–33. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. Trial of the Combined Use of Thiamine and Biotin in Patients with Huntington’s Disease (HUNTIAM) (NCT04478734). 2020. Available online: https://clinicaltrials.gov/study/NCT04478734 (accessed on 18 March 2026).
- Spain, R.; Powers, K.; Murchison, C.; Heriza, E.; Winges, K.; Yadav, V.; Cameron, M.; Kim, E.; Horak, F.; Simon, J.; et al. Lipoic Acid in Secondary Progressive MS: A Randomized Controlled Pilot Trial. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e374. [Google Scholar] [CrossRef]
- Teichert, J.; Hermann, R.; Ruus, P.; Preiss, R. Plasma Kinetics, Metabolism, and Urinary Excretion of Alpha-Lipoic Acid following Oral Administration in Healthy Volunteers. J. Clin. Pharmacol. 2003, 43, 1256–1267. [Google Scholar] [CrossRef] [PubMed]
- Hermann, R.; Mungo, J.; Cnota, P.J.; Ziegler, D. Enantiomer-Selective Pharmacokinetics, Oral Bioavailability, and Sex Effects of Various Alpha-Lipoic Acid Dosage Forms. Clin. Pharmacol. Adv. Appl. 2014, 6, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Chng, H.T.; New, L.S.; Neo, A.H.; Goh, C.W.; Browne, E.R.; Chan, E.C. Distribution Study of Orally Administered Lipoic Acid in Rat Brain Tissues. Brain Res. 2009, 1251, 80–86. [Google Scholar] [CrossRef]
- Ziegler, D.; Low, P.A.; Litchy, W.J.; Boulton, A.J.; Vinik, A.I.; Freeman, R.; Samigullin, R.; Tritschler, H.; Munzel, U.; Maus, J.; et al. Efficacy and Safety of Antioxidant Treatment with α-Lipoic Acid over 4 Years in Diabetic Polyneuropathy: The NATHAN 1 Trial. Diabetes Care 2011, 34, 2054–2060. [Google Scholar] [CrossRef]
- Ziegler, D.; Ametov, A.; Barinov, A.; Dyck, P.J.; Gurieva, I.; Low, P.A.; Munzel, U.; Yakhno, N.; Raz, I.; Novosadova, M.; et al. Oral Treatment with Alpha-Lipoic Acid Improves Symptomatic Diabetic Polyneuropathy: The SYDNEY 2 Trial. Diabetes Care 2006, 29, 2365–2370. [Google Scholar] [CrossRef] [PubMed]
- National Institute of Diabetes and Digestive and Kidney Diseases. LiverTox: Clinical and Research Information on Drug-Induced Liver Injury—Alpha-Lipoic Acid. 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK591554/ (accessed on 18 March 2026).
- EFSA Panel on Nutrition, Novel Foods and Food Allergens (NDA); Turck, D.; Castenmiller, J.; de Henauw, S.; Hirsch-Ernst, K.I.; Kearney, J.; Knutsen, H.K.; Mangelsdorf, I.; McArdle, H.J.; Naska, A.; et al. Scientific Opinion on the Relationship between Intake of Alpha-Lipoic Acid (Thioctic Acid) and the Risk of Insulin Autoimmune Syndrome. EFSA J. 2021, 19, e06577. [Google Scholar] [CrossRef]
- Uchida, Y.; Ito, K.; Ohtsuki, S.; Kubo, Y.; Suzuki, T.; Terasaki, T. Major Involvement of Na(+)-Dependent Multivitamin Transporter (SLC5A6/SMVT) in Uptake of Biotin and Pantothenic Acid by Human Brain Capillary Endothelial Cells. J. Neurochem. 2015, 134, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Reidling, J.C.; Nabokina, S.M.; Said, H.M. Molecular Mechanisms Involved in the Adaptive Regulation of Human Intestinal Biotin Uptake: A Study of the hSMVT System. Am. J. Physiol.-Gastrointest. Liver Physiol. 2007, 292, G275–G281. [Google Scholar] [CrossRef]
- Li, D.; Ferguson, A.; Cervinski, M.A.; Lynch, K.L.; Kyle, P.B. AACC Guidance Document on Biotin Interference in Laboratory Tests. J. Appl. Lab. Med. 2020, 5, 575–587. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Biotin Interference with Troponin Lab Tests: Assays Subject to Biotin Interference. 2022. Available online: https://www.fda.gov/medical-devices/in-vitro-diagnostics/biotin-interference-troponin-lab-tests-assays-subject-biotin-interference (accessed on 18 March 2026).
- Bongarzone, S.; Sementa, T.; Dunn, J.; Bordoloi, J.; Sunassee, K.; Blower, P.J.; Gee, A. Imaging Biotin Trafficking In Vivo with Positron Emission Tomography. J. Med. Chem. 2020, 63, 8265–8275. [Google Scholar] [CrossRef]
- Office of Dietary Supplements, National Institutes of Health. Biotin—Health Professional Fact Sheet. 2022. Available online: https://ods.od.nih.gov/factsheets/Biotin-HealthProfessional (accessed on 18 March 2026).
- Riverón-Negrete, L.; Sicilia-Argumedo, G.; Álvarez-Delgado, C.; Coballase-Urrutia, E.; Alcántar-Fernández, J.; Fernandez-Mejia, C. Dietary Biotin Supplementation Modifies Hepatic Morphology without Changes in Liver Toxicity Markers. BioMed Res. Int. 2016, 2016, 7276463. [Google Scholar] [CrossRef] [PubMed]
- Ronquillo-Sánchez, M.D.; Camacho-Carranza, R.; Fernandez-Mejia, C.; Hernández-Ojeda, S.; Elinos-Baez, M.; Espinosa-Aguirre, J.J. Modulation of the Rat Hepatic Cytochrome P4501A Subfamily Using Biotin Supplementation. BioMed Res. Int. 2013, 2013, 627907. [Google Scholar] [CrossRef][Green Version]
- Lodewyk, K.; Courtney, D.B.; Bagnell, A.; Newton, A.S. Adverse Event Monitoring, Assessment, and Reporting in Nutraceutical and Phytoceutical Trials for Pediatric Neuropsychiatric Conditions: A Systematic Review. J. Psychopharmacol. 2025, 39, 1232–1244. [Google Scholar] [CrossRef]
- Ashrafpour, S.; Ashrafpour, M. The Double-Edged Sword of Nutraceuticals: Comprehensive Review of Protective Agents and Their Hidden Risks. Front. Nutr. 2025, 12, 1524627. [Google Scholar] [CrossRef] [PubMed]
- Timalsina, D.R.; Abichandani, L.; Ambad, R. A Review Article on Oxidative Stress Markers F2-Isoprostanes and Presenilin-1 in Alzheimer’s Disease. J. Pharm. Bioallied Sci. 2025, 17, S109–S112. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Kang, Y.; Gallagher, D.; Herrmann, N.; Survilla, K.; Vieira, D.; Mah, E.; Graham, S.J.; Kiss, A.; Black, S.E.; et al. MRS Demonstrates Elevated Brain Glutathione in Vascular Mild Cognitive Impairment Compared to Cognitively Normal Coronary Artery Disease Controls. Alzheimer’s Dement. 2025, 21, e70230. [Google Scholar] [CrossRef] [PubMed]
- Puliyakkara, A.; Shirlal, A.; Pendem, S.; Priyanka; Kadavigere, R.; Marike, T.S. Myelin Water Imaging as a Quantitative Diagnostic Tool for Neurodegenerative Diseases: A Systematic Review. Diagnosis 2025, 13, 9–19. [Google Scholar] [CrossRef]
- Banach, M.; Katsiki, N.; Latkovskis, G.; Gaita, D.; Escobar, C.; Pella, D.; Penson, P.E.; Fogacci, F.; Reiner, Z.; Cicero, A.F.G. 2024 Update on Postmarketing Nutrivigilance Safety Profile: A Line of Dietary Food Supplements Containing Red Yeast Rice for Dyslipidemia. Arch. Med. Sci. 2024, 21, 729–737. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Compound/Model | Design/Dose/Duration | Primary Outcome/Mechanism | Ref. |
|---|---|---|---|
| Alpha-Lipoic Acid (ALA) | |||
| Preclinical in vivo studies (Neurodegenerative models) | |||
| In vitro human immune cells (T cell-enriched PBMCs, NK cells) + ex vivo PBMCs from multiple sclerosis (MS) subjects | In vitro: ALA 10–100 µg/mL, short pretreatment (1–5 min), immune stimulation 6–24 h. In vivo: (MS): Oral ALA 1200 mg, single dose; PBMCs analyzed at 4 h | ↓ IL-6 and IL-17 secretion in T cell-enriched PBMCs; ↓ T-cell activation (IL-2) and proliferation; ↑ cAMP → ↑ PKA signaling (PKA inhibition blocks these effects) | [20] |
| Dapsone-treated mouse model (prefrontal cortex and hippocampus) | ALA administered at 25 mg/kg → co-treatment with DDS over chronic exposure in mice | ↓ Microglial and astrocytic activation; ↓ pro-inflammatory cytokines (IL-1β, IL-17, IL-4); ↑ BDNF; ↑ antioxidant capacity (TEAC, GSH, SOD, CAT) → ↓ oxidative damage and iron accumulation in PFC/hippocampus | [21] |
| Fischer 344 rats (young, middle, old)—brain aging model | In vivo; Fischer 344 rats of 6, 12, 24 months; ALA given (dose ~100 mg/kg diet or similar) over aging period (weeks/months) | ↑ Brain glucose uptake (µPET); restoration of Akt–JNK signaling balance; ↑ PGC-1α-mediated mitochondrial biogenesis → improved mitochondrial bioenergetics and energy metabolism | [22] |
| APP23/PS45 transgenic mice (Alzheimer’s model) | In vivo; male APP23/PS45 mice, ALA 5 mg/kg/day orally for 4 months starting at 2 months of age | ↑ Mitophagy (BNIP3L-mediated) → ↑ ADAM10 α-secretase cleavage of APP → ↓ amyloid-β plaques; ↑ cognitive performance in behavioral tests | [23] |
| Rotenone—induced Parkinson’s disease in mice | In vivo; male mice (n ≈ 40) divided into four groups: control; ALA 100 mg/kg/day i.p.; rotenone 1.5 mg/kg i.p. every 2 days; rotenone + ALA; duration: 21 days | Activation of the PI3K–Akt pathway; ↓ caspase-3 activation; ↓ IL-1β, TNF-α, and NF-κB; ↑ antioxidant markers (GSH, SOD); ↓ MDA → improved behavioral and motor outcomes | [27] |
| Aged Tg2576 transgenic mice (Alzheimer’s model) | In vivo; 10-month-old Tg2576 and wild-type mice; diet containing 0.1% ALA (~100 mg/kg/day) for 6 months | ↑ Hippocampal-dependent learning and memory (Morris water maze, contextual fear paradigm); ↓ nitrotyrosine (nitrative stress marker); no change in Aβ levels → neuroprotection via reduction of nitrative and oxidative stress | [71] |
| 3 × Tg-AD mice (Alzheimer’s model) | In vivo; Tg-AD mice; ALA 0.23% (≈60 mg/kg/day) in diet for 4 weeks | Restoration of cerebral glucose oxidation and TCA cycle flux (↑ Glu, Gln, Asp labeling); reversal of age-associated hypometabolism → improved mitochondrial bioenergetics via modulation of Akt/JNK/PGC-1α pathways | [72] |
| P301S transgenic mice (tauopathy/Alzheimer’s model) | In vivo; ALA 3 mg/kg and 10 mg/kg administered chronically in P301S mice (duration ~8–12 weeks) | ↑ GLUT3/GLUT4; ↑ HK activity; ↑ BDNF/TrkB → ↑ HIF-1α → restored brain glucose metabolism → neuroprotection | [73] |
| Rat unilateral intrastriatal 6-OHDA model (Parkinsonism-like oxidative damage) | ALA 35 mg/kg, i.p., once daily for 14 days after 6-OHDA lesion (unilateral intrastriatal injection) | ↓ TBARS; ↑ antioxidant defense (↑ GPx activity); ↓ motor asymmetry (trend): apomorphine-induced rotations ↓ at day 14 vs. lesion-only group, although not reaching statistical significance | [75] |
| Parkinson’s disease models: C57BL/6 mice (MPTP-induced) + SH-SY5Y neuroblastoma cells (MPP+-induced) | In vivo: ALA 50 mg/kg/day i.p., administered for 14 days; MPTP 30 mg/kg/day i.p., for 5 consecutive days; EX527 (SIRT1 inhibitor) 10 mg/kg/day i.p. (mechanistic validation). In vitro: ALA 200 µM, pretreatment 3 h before MPP+; MPP+ 1 mM, exposure 24 h; EX527 10 µM | ↑ Motor performance (rotarod, pole test); ↑ Dopaminergic neuron survival (↑ TH+ neurons); ↑ Cell viability (SH-SY5Y); ↓ ROS production; ↑ SOD activity; ↓ MDA levels; ↑ SIRT1 and ↑ PGC-1α signaling; ↑ Mitochondrial function and oxidative stress resistance; Effects reversed by SIRT1 inhibition (EX527) → confirmation of a SIRT1-dependent PGC-1α mechanism | [76] |
| 6-OHDA-induced PD model in Sprague—Dawley rats (unilateral intrastriatal lesion; substantia nigra endpoints). In vitro: PC12 cells exposed to 6-OHDA | In vivo: Lesion: 6-OHDA 20 μg total injected into right striatum (two sites; 10 μg/site). Treatment: ALA 100 mg/kg i.p., once daily for 14 days, initiated 4 weeks after 6-OHDA lesion (after behavioral screening). In vitro (PC12): ALA pretreatment 1 h (commonly 10 μM in key assays) → then 6-OHDA 200 μM for 24 h | ↓ Motor deficits; Dopaminergic integrity preserved (prevention of TH loss); ↓ ROS (PC12); ↑ SOD and GSH in substantia nigra; ↓ iron accumulation; Iron metabolism signaling normalized: ↑ 6-OHDA-induced IRP2 and ↑ DMT1 are antagonized by ALA (rats SN + PC12) → ↑ iron homeostasis | [77] |
| Parkinson’s disease models (in vivo and in vitro; ferroptosis-associated neurodegeneration) | Dose: Not reported in abstract. Duration: Not reported in abstract | ↓ Motor deficits (behavioral performance improved with ALA); ↓ Ferroptosis; ↑ GPX4; ↑ xCT (SLC7A11); ↓ Iron overload; ↑ FPN; ↑ FTH1; ↓ DMT1; ↓ Oxidative damage; ↓ ROS; ↓ Lipid peroxidation; ↑ SIRT1–NRF2 signaling, leading to enhanced antioxidant defense and suppression of ferroptotic cell death. | [78] |
| Model of Parkinson’s disease induced by (6-OHDA) | Male rats; ALA intraperitoneally (100 mg/kg or 200 mg/kg) daily for 15 days following lesion induction | Improved motor behavior (↓ apomorphine-induced rotations, ↑ locomotor activity, ↑ contralateral paw use); ↓ TBARS and nitrite levels; restoration of catalase activity | [79] |
| Model of Parkinson’s disease-induced dyskinesia | Male rats; ALA administered at 31.5 mg/kg (low dose) or 63 mg/kg (high dose), 3 weeks | ↓ Dyskinesia; ↓ MDA, ↑ glutathione/GSH activity; ↓ Iba-1-positive cells; ↓ apoptotic markers, including cleaved caspase-3 and PARP overexpression in the substantia nigra | [80] |
| ALA—Parkinson’s disease (L-DOPA-induced dyskinesia in 6-OHDA-lesioned rats) | Male rats; 6-OHDA lesion; ALA (31.5 or 63 mg/kg, i.p.) co-administered with L-DOPA over the treatment period (chronic administration model) | ↓ Abnormal involuntary movements (dyskinesia); ↓ oxidative stress (↓ MDA, ↑ GSH); ↓ neuroinflammation and apoptosis in substantia nigra | [81] |
| Nanomicellar complex of carnosine and ALA (CLA)—Rotenone-induced Parkinson’s disease model | Male rats; rotenone-induced PD model; nanomicellar CLA administered intraperitoneally (25 or 50 mg/kg) daily for 18 days | ↓ Muscle rigidity; ↑ locomotor activity; ↑ total brain antioxidant activity; ↑ neuronal density in substantia nigra; ↑ striatal dopamine levels (neuroprotective and antioxidant effects) | [82] |
| Long-term relapsing-remitting EAE (Experimental Autoimmune Encephalomyelitis) | ALA (100 mg/kg/day) intraperitoneally, evaluated at 7 days and 180 days post-onset | ↓ MBP; ↓ β-APP expression; down-regulated TNF-α and up-regulated TGF-β 7 days after onset; ↓ MDA; ↑ SOD; ↑ Treg levels at 7 days after onset | [85] |
| Focal-cortical EAE model | C57BL/6 mice with focal cortical EAE induced by intracortical TNF-α/IFN-γ injection; ALA administered subcutaneously (100 mg/kg/day), with lesions analyzed 3 days after induction | ↓ Cortical CD4+ inflammatory infiltrates; ↓ galectin-3+ activated microglia/macrophages; ↓ CD45+ infiltrating cells | [86] |
| Transgenic ALS mice carrying the G93A Cu/Zn superoxide dismutase (SOD1) mutation | ALA administration: Dietary supplementation. Dose: NR (not specified in abstract). Duration: Chronic, until end-stage/survival analysis (exact duration not specified) | ↑ Survival (statistically significant improvement vs. untreated G93A mice); ↓ Disease progression (inferred from survival extension); ↓ Oxidative stress; ↑ Mitochondrial/redox support; Neuroprotection consistent with antioxidant and metabolic modulation rather than GAPDH-dependent anti-apoptotic signaling | [89] |
| Drosophila models harboring mutant hSOD1 (G85R, G93A) + NSC34 Motor neuron-like cell line expressing mutant hSOD1 | Transgenic Drosophila fed ALA-supplemented food at 2 mM concentration for 30 days post-eclosion; NSC-34 cells expressing hSOD1 mutants treated with 100 µM ALA for 24 h | ↑ Climbing ability and lifespan in hSOD1-mutant flies; ↓ ROS accumulation; restored antioxidant enzyme activity; ↓ H2O2-induced cytotoxicity in NSC-34 cells; ↑ cell viability; activated ERK/Akt signaling pathway | [90] |
| Transgenic mouse models of Huntington’s disease (R6/2 and N171-82Q) | Diet 0.005% (100 mg/kg/day), began at four weeks and continued throughout life | ↑ Survival in both HD mouse lines; delayed weight loss and disease progression | [94] |
| Rat model of Huntington’s disease induced by 3-nitropropionic acid (3-NP) | Rats received ALA (100 mg/kg i.p.) and ALCAR (100 mg/kg i.p.) daily for 28 days, starting with 3-NP exposure | Restored mitochondrial complex II and IV activity; ↑ ATP levels; ↓ ROS; ↓ MDA, protein carbonyls; ↑ SOD; ↑ catalase; ↑ learning and memory; inhibition of apoptotic pathways | [95] |
| Mechanistic/in vitro or non-ND models | |||
| In vitro HDAC assays and mammalian cell models | In vitro; lipoic acid/lipoamide evaluated for HDAC inhibition and associated acetylation changes | Supports an epigenetically relevant regulatory layer beyond redox and metabolic actions | [17] |
| H. pylori-infected gastric epithelial AGS cells | ALA pretreatment (5 µM), co-treatment with H. pylori (strain NCTC11637) for 8 h in AGS cells | ↑ Nuclear Nrf2/↑ HO-1 expression → ↓ ROS levels → ↓ IL-8 expression and secretion in H. pylori-infected cells | [16] |
| BV-2 microglia (LPS model) | Pretreatment with ALA (50–400 µM), LPS (1 µg/mL) for 30 min and ALA treatment for 24 h | ↓ pro-inflammatory cytokines (TNF-α, IL-6); ↓ ROS and NO production; ↓ ERK/p38 phosphorylation; ↓ NF-κB and NLRP3 inflammasome activation; ↑ M2 phenotype-associated genes (MRC1, ARG1); ↓ M1 phenotype-associated genes (IL-1β, ICAM-1) | [18] |
| Subarachnoid hemorrhage (SAH) in SD rats | Sprague-Dawley rats; SAH induced by endovascular perforation; ALA 100 mg/kg i.p. administered 30 min before SAH and then daily for 3 days | ↓ STING-NLRP3 inflammasome activation, ↓ IL-1β and IL-18, ↓ neuronal apoptosis, ↓ brain edema, ↑ neurological scores → neuroprotection via suppression of STING-mediated innate immune signaling | [19] |
| Human PBMCs, Jurkat T cells | In vitro study; ALA 10–100 µM (1–24 h) in activated human PBMCs and Jurkat T cells | ↑ cAMP levels → activation of PKA → phosphorylation of CREB (Ser133) → ↓ TNF-α, IFN-γ, IL-2 production; anti-inflammatory effect independent of the EP2 receptor | [20] |
| Male CD-1 mice subjected to permanent middle cerebral artery occlusion | ALA (50 mg/kg i.p.), 30 min prior to ischemia; evaluation at 24 h post-ischemia | ↓ Neurological deficit score; ↓ infarct volume; ↓ brain edema → ↑ SIRT1 expression; ↑ PGC-1α expression; ↑ SOD activity → neuroprotection via SIRT1-dependent PGC-1α up-regulation | [22] |
| Primary rat astrocyte cultures exposed to induced oxidative stress and iron overload | Pretreatment with ALA (50 µM) and Vitamin D3 (10 nM) → cells challenged with H2O2 (200 µM) + Fe2+ (100 µM) for 24 h | ↓ ROS production; ↓ intracellular iron accumulation; ↑ glutathione (GSH); ↑ mitochondrial membrane potential; ↓ astrocyte senescence markers → protective effect via combined antioxidant and iron-homeostasis modulation | [25] |
| Rat Schwann cells (high-glucose exposure) | ALA (1, 10, 50, 100 µM) → 24–48 h exposure under CHG (30 mM) or IHG cycling (30 ↔ 5.5 mM glucose) | ↓ ROS and oxidative stress → attenuation of mitochondrial apoptotic pathway activation (↓ Bax, ↓ cytochrome c release/AIF translocation) → ↓ caspase-3/9 activation and PARP cleavage; ↑ Bcl-2 expression → improved Schwann cell survival | [26] |
| NG108-15 neuronal hybrid cells | Pretreatment with R-LA (50 μM, 2 h) followed by H2O2 (400 μM, 24 h) | ↑ neuroprotection via PI3K–Akt/GSK-3β activation and NF-κB/cytokine suppression (↓ ROS, ↓ apoptosis) | [27] |
| Diabetic peripheral neuropathy in Sprague–Dawley rats | In vivo; STZ-induced diabetes + HFD; ALA 60 mg/kg/day for 12 weeks | ↑ Motor and sensory NCV; ↑ GSH; ↓ MDA; ↑ p-AMPK → ↑ Nrf2/HO-1/NQO1; ↓ FoxO3a/Bim → ↓ oxidative stress and apoptosis in DRG | [30] |
| Pilocarpine-induced seizures in Wistar rats | In vivo; pilocarpine 400 mg/kg i.p.; ALA 20 mg/kg i.p. 30 min before seizure; evaluated 24 h post-seizure | ↓ Hippocampal cell death; inhibition of caspase-dependent (cyt c → casp-3) and independent (AIF translocation) pathways → neuroprotection against epileptic damage | [63] |
| Traumatic brain injury (TBI) in Sprague—Dawley rats | In vivo; cortical impact TBI model; ALA 100 mg/kg i.p., once daily for 3 days post-injury | ↓ Neuronal apoptosis; ↓ cytochrome c release; ↓ caspase-9/-3 activation; ↑ Bcl-2/Bax ratio; preservation of mitochondrial membrane potential → neuroprotection via inhibition of mitochondrial apoptotic pathway | [64] |
| SH-SY5Y-APP695 cells and SH-SY5Y-MOCK (cellular Alzheimer’s model vs. control) | In vitro; ALA at 100 µM and 1 mM for 24 h (and 1 h pre-treatment for rotenone challenge) in APP695 and MOCK cells | ↑ ATP; ↑ MMP in control cells; ↓ ROS in both cell types; → enhanced mitochondrial function, modest protection against rotenone-induced stress | [69] |
| BV-2 mouse microglial cells (Aβ1–42 oligomers exposure) | In vitro; BV-2 cells pretreated with ALA (10–100 µM, 2 h) then exposed to Aβ1–42 oligomers (2 µM, 24 h) | ↑ Phagocytosis of Aβ1–42; ↓ NF-κB, NLRP3, p-p38 MAPK, and pro-inflammatory cytokines (IL-1β, TNF-α, IL-6); activation of PPAR-γ and PI3K/Akt → shift toward a neuroprotective microglial phenotype | [73] |
| Biotin (Vitamin B7) | |||
| Preclinical in vivo studies (Neurodegenerative models) | |||
| Drosophila melanogaster expressing human mutant tau (R406W) | In vivo; dietary biotin 10 µM from L1 larval stage to 10-day-old adults (~15–18 days total) | Restored mitochondrial membrane potential and morphology; ↑ ATP, ↓ ROS, ↓ neuronal degeneration; improved locomotor performance → neuroprotection via enhanced mitochondrial metabolism and biotin-dependent carboxylase activity | [49] |
| Rat model of Alzheimer’s disease (Aβ injection into lateral ventricle) | In vivo; oral biotin 10 mg/kg/day × 28 days (+/− swimming training) before and after Aβ injection | ↑ Learning and memory; ↓ anxiety and depression-like behavior; ↓ MDA; ↑ total thiols; ↓ Aβ plaques and neuronal loss (CA1) → antioxidant and neuroprotective effects | [75] |
| Drosophila melanogaster model of manganese (Mn) toxicity-human derived midbrain dopaminergic neurons | Biotin supplementation in diet of flies; human derived dopaminergic neurons cultured with biotin and Mn exposure | ↑ Locomotor/climbing behavior in flies; preserved dopaminergic neurons (TH+); mitigated mitochondrial dysfunction and neuronal loss | [83] |
| Dysmyelinating shiverer mice (murine oligodendrocytes) + human oligodendrocyte progenitor cell grafts | Shiverer (Shi/Shi:Rag2-/-) mice treated via maternal MD1003 in diet (5 mg/kg/day) starting P0; pups exposed in utero + via milk; grafted with human NPC-derived OPCs at P1; analyses at 12 and 20 weeks post treatment/graft | ↑ Number and differentiation potential of endogenous murine oligodendroglia (↑ OLIG2+/CC1+) and of grafted human oligodendroglia; without significantly increasing axonal myelination | [88] |
| Biotin (Mg-biotin complex)/LPC-induced hippocampal demyelination in rats | In vivo; rats given biotin at 0.9 mg/rat/day (B1) or 9 mg/rat/day (B2) and MgB at 0.9 mg/rat/day (MgB1) or 9 mg/rat/day (MgB2); duration post-LPC demyelination (remyelination phase) | ↑ Remyelination; ↑ spatial memory; ↓ NF-κB p65; ↑ BDNF, GAP43, ICAM -1; ↓ GFAP gliosis → dose-dependent beneficial effect of biotin/MgB on remyelination and neuronal transmission | [89] |
| Huntington’s disease mouse model (R6/1 HD mice) with OL and OPC maturation deficits | Daily combined thiamine 50 mg/kg + biotin 20 mg/kg, i.p. started at 8 weeks of age, continued for 7 weeks, euthanized/analyzed at 15 weeks | Rescue of OL/OPC maturation state; normalization of dysregulated maturation signatures and OL-lineage DEGs; ↓ neuronal pathology | [97] |
| Mechanistic/in vitro, ex vivo or non-ND models | |||
| Biotin (maternal supplementation)/Female rat offspring (post-weaning fructose) | Maternal global caloric restriction during gestation + biotin 2 mg/kg for dams; offspring exposed to 20% fructose in drinking water for 16 weeks after weaning | ↓ cardiometabolic risk markers, including hypertriglyceridemia, hypercholesterolemia, hepatic steatosis, glucose intolerance, insulin resistance, hypertension, and vascular hyperresponsiveness | [37] |
| Pancreatic rat islets | In vitro/ex vivo; isolated rat islets treated with biotin (50 µM) for 2–24 h; measurement of mRNA, ATP content | ↑ Glucokinase mRNA; ↑ ATP content; effect blocked by sGC or PKG inhibitors → Mechanism: sGC/PKG signaling → ↑ ATP → autocrine insulin release → PI3K/Akt activation | [38] |
| Wistar rat aortic rings | Ex vivo; isolated rat aortic rings; biotin 10 nM incubation with or without antihypertensive drugs (BMY 7378 100 nM; captopril 1 µM; nitrendipine 100 nM) during vasoconstriction assays | ↑ Relaxation/enhanced vasodilatory response with antihypertensive drugs; ↓ phenylephrine- and Ca2+-induced contraction → vascular protective/vasorelaxant effect of biotin | [39] |
| Murine oligodendrocytes (158N) | Pre- and co-treatment with biotin (10–100 μM) for 24–48 h before/with 7β-OHC challenge | ↑ Cell survival via attenuation of oxidative stress, preservation of mitochondrial function (ΔΨm ↑, ATP ↑), and normalization of lipid metabolism under 7β-OHC-induced toxicity | [44] |
| Male BALB/cAnN Hsd mice | Dietary supplementation: control diet 1.76 mg free biotin/kg diet vs. biotin-supplemented diet 97.7 mg free biotin/kg diet; 8 weeks post-weaning | ↓ Serum triglycerides; ↓ hepatic triglycerides; ↑ Hepatic cGMP; ↑ p-AMPK and ↑ p-ACC-1; ↓ Mature SREBP-1c and ↓ FAS → ↓ lipogenesis | [45] |
| Murine adipocyte cell line (3T3-L1) | Control media vs. biotin-supplemented media (1 μM biotin); 8 days (main experimental condition; dose–response evaluated but downstream assays performed at 1 μM) | ↑ p-AMPK (T172); ↑ p-ACC-1 (S79) and ↑ p-ACC-2 (S212); ↓ Fatty acid synthesis; ↑ Fatty acid uptake; FA transport/activation genes: ↑ Fatp1 mRNA, ↑ Acsl1 mRNA; ↑ GPAT-3 protein; Triglyceride content ↔; ↑ Lipid droplet number with droplet area/size | [46] |
| Abcd1− mouse model of X-ALD | In vivo; high-dose pharmaceutical-grade biotin (MD1003) in Abcd1 knockout mice; treated for months (starting at ~13 months old mice, followed until ~18 months) | ↑ NRF2-driven antioxidant response; ↑ mitochondrial biogenesis (PGC-1α) and ATP; ↓ axonal degeneration and locomotor impairment; normalized SREBP-1c/mTORC1 lipid-metabolism program → restored redox, energy and lipid homeostasis | [47] |
| Post-natal rat-derived oligodendrocyte lineage cells | OPCs (A2B5+) cultured; biotin (2.5 to 250 µg/mL), ensheathment assessed on nanofiber assay (3 days) | ↓ Cell death under glucose-free conditions; ↑myelin-like ensheathment; ↑ % ensheathing cells, number and length of segments; ↑ ATP production | [49] |
| Human PBMCs obtained from healthy adults | In vivo supplementation, pre–post design: biotin 3.1 μmol/day (≈0.75 mg/day) for 14 days; blood/urine collected pre- vs. post-supplementation; PBMC stimulated ex vivo with concanavalin A (up to 3 days) to mimic antigenic activation | ↓ PBMC proliferation; IL-1β secretion ↓; IL-2 secretion ↓; PBMC subset proportions (CD markers) ↔ no change after 14 days | [51] |
| Human PBMC (stimulated ex vivo with concanavalin A (ConA) to mimic antigenic activation | In vivo supplementation (pre–post): biotin 8.8 μmol/day for 21 days; PBMC isolated before vs. after supplementation; ex vivo ConA stimulation 21 h | After biotin vs. before biotin; ↑ fold-changes: IFN-γ mRNA (~4.3×); ↑ IL-1β mRNA (~5.6×); ↑ 3-Methylcrotonyl-CoA carboxylase mRNA (~8.9×); ↓ IL-4 mRNA (reported as ~6.8× higher before supplementation) → reduced after | [52] |
| Male Wistar rats with propionic acid (PPA)-induced autism-like features | PPA: 500 mg/kg/day, s.c., 5 days (autism-like induction); MgB (oral gavage): 160.7 µg/day (MgBI), 1606.9 µg/day (MgBII), 8034.4 µg/day (MgBIII), 2 weeks (doses derived from human-equivalent 10/100/500 mg biotin) | ↑ Social behavior and anxiety-like metrics; ↑ learning/memory; ↓ MDA (serum/brain); ↑ CAT; ↑ SOD; ↑ GPx; ↑ GSH; ↓ TNF-α; ↓ IL-6; ↓ IL-17; ↓ CCL-3, ↓ CCL-5; ↓ CXCL-16; ↑ counter-regulators OPG, ↑ MMP-9; Neurotransmission/trophic markers improved: ↑ serotonin, ↑ dopamine; ↑ BDNF, ↑ GAP-43, ↑ ICAM-1, ↑ PSD-93, ↑ PSD-95; ↓ GFAP; ↑ Purkinje cell number/size/density; ↓ hippocampal disorganization; ↑ Carboxylase-related “neurodevelopment markers”: ↑ ACC-1, ↑ ACC-2, ↑ PC, ↑ PCC, ↑ MCC | [53] |
| Murine models of intestinal inflammation | Oral biotin supplementation (≈1–10 mg/kg/day; duration 7–21 days, depending on model) | ↓ NF-κB activation and downstream pro-inflammatory cytokines (TNF-α, IL-1β, IL-6); ↑ intestinal barrier integrity and epithelial tight-junction preservation → anti-inflammatory | [54] |
| Dextran sulfate sodium (DSS)-induced colitis in mice | In vivo; DSS-induced colitis model in mice with oral biotin supplementation (≈1–5 mg/kg/day) administered throughout DSS exposure (5–7 days) and recovery phase (up to 14 days) | ↓ NF-κB activation; ↓ pro-inflammatory cytokine expression (TNF-α, IL-1β, IL-6); ↑ intestinal barrier integrity; ↓ disease activity index → anti-inflammatory and barrier-protective | [56] |
| High-fructose diet-induced metabolic syndrome in Wistar rats | In vivo; male Wistar rats fed 30% fructose in drinking water for 12 weeks, then biotin i.p. 2 mg/kg/day for 4 weeks | ↓ Plasma TG, cholesterol, LDL-c, transaminases, blood pressure; ↑ HDL-c; improved glucose and insulin tolerance; ↓ hepatic steatosis and oxidative stress | [61] |
| Type/Population/Model | Design/Dose/Duration | Primary Outcome | Comment (Quality) | Ref. |
|---|---|---|---|---|
| Alpha-Lipoic Acid (ALA)—Clinical studies | ||||
| Alzheimer’s disease (probable AD), age ≥ 55; MMSE 15–26 | Randomized, double-blind, placebo-controlled, 3-arm pilot (n = 39; 13/group); ω-3 fish oil 3 g/day (DHA 675 mg/day + EPA 975 mg/day) ± alpha-lipoic acid 600 mg/day; 12 months | Primary: urinary F2-isoprostanes ↔ (no between-group difference at 12 months). Secondary: ↓ MMSE decline in ω-3 + ALA vs. placebo; ↓ IADL decline in ω-3 and ω-3 + ALA vs. placebo; ↔ ADAS-Cog; ADL | Moderate internal validity (randomized, double-blind, placebo-controlled; 12-month follow-up; prespecified primary endpoint). Limitations: small pilot sample (underpowered for clinical endpoints); baseline imbalance in F2-isoprostanes; no ALA-only arm, so synergy vs. ALA effect alone cannot be isolated. | [99] |
| Prospective observational study/Alzheimer’s disease (mild–moderate; MMSE 12–26), stratified by type 2 diabetes presence vs. absence | ALA 600 mg/day (oral) given adjunctively (with standard antidementia care), comparing outcomes in AD + DM (Group A) vs. AD without DM (Group B); follow-up up to 16 months | ↑ Cognition/function within groups, with greater improvement in AD + DM: ↑ proportion with MMSE improvement (43% in AD + DM vs. 23% without DM); ↓ ADAS-Cog (improved), ↑ CIBIC(+)(improved), ↑ ADFACS (improved) effects reported as greater in AD + DM. ↓ Insulin resistance over follow-up, proposed as a contributor | Low–moderate evidence strength: not randomized, no placebo arm, group comparison is by DM status (not treatment assignment), substantial confounding risk. The authors explicitly note that the study is observational and cannot establish definitive efficacy | [100] |
| Randomized, double-blind, placebo-controlled/Mild–moderate Alzheimer’s disease | 3-arm randomization for 16 weeks: Vitamin E 800 IU/day + Vitamin C 500 mg/day + α-lipoic acid 900 mg/day (E/C/ALA) vs. CoQ10 400 mg TID vs. placebo | CSF F2-isoprostanes ↓ (~19%) in the E/C/ALA arm; CSF Aβ42 ↔, tau ↔, p-tau181 ↔. Cognition: MMSE decline ↑ (faster decline) in the E/C/ALA arm | Strong internal validity (randomized, double-blind, placebo-controlled; biomarker-focused). Limitations: short duration (16 weeks); biomarker endpoints may not translate to clinical benefit; signal of faster cognitive decline in E/C/ALA warrants caution | [102] |
| Clinical trial, Phase 1/2, open-label/Probable progressive supranuclear palsy (PSP; atypical parkinsonism), single-site small cohort | Open-label daily supplementation with ALA 600 mg/day combined with L-acetyl-carnitine 1.5 g/day for approximately 6 months in a small exploratory cohort (n ≈ 11) | Primary: safety/tolerability. Common adverse events included restlessness, dizziness, insomnia, and seizures. Exploratory efficacy and biomarker outcomes were listed but were not adequately reported or remain unpublished | Very low evidence strength due to exploratory design, very small sample (~11 participants), lack of randomization and placebo/control group, and absence of peer-reviewed outcome data. Informative mainly as a feasibility and safety signal rather than evidence of clinical efficacy | [107] |
| Clinical trial, randomized placebo-controlled pilot/MS subjects (n = 37) | Randomized to placebo or oral ALA: 600 mg BID, 1200 mg QD, or 1200 mg BID for 14 days | ↓ Biomarkers of immune cell migration: ↓ serum MMP-9 (higher peak ALA exposure associated with greater MMP-9 reduction) and ↓ sICAM-1 with a dose–response relationship; tolerability generally acceptable | Strengths: randomized placebo arm; clear dosing arms; objective PK + biomarker endpoints. Limitations: very short duration (14 days); not powered for clinical disability outcomes; biomarker changes are surrogate (clinical benefit not established). | [110] |
| Clinical trial, randomized, double-blind, placebo-controlled pilot/SPMS participants | Oral alpha-lipoic acid 1200 mg/day vs. placebo; 2 years | ↑ Walking performance (modest): ↓ Timed Up and Go (TUG) time (medium effect at 2 years); in lower-disability subgroup (EDSS < 6, no assistive device), ↓ turning time (significant). ↔ no between-group differences in balance metrics | Solid design (double-blind RCT) with objective sensor-based outcomes, but pilot sample size and small cohort limit precision; subgroup finding is hypothesis-generating; clinical disability endpoints not primary | [111] |
| Mechanistic pharmacokinetic study/Healthy controls, RRMS, and SPMS | Single-dose, within-subject (pre–post) design: after standardized breakfast + baseline draw, participants ingested racemic ALA 1200 mg PO (4 × 300 mg capsules); blood collected at 0, 1, 2, 3, 4 h (acute exposure) | ↑ PBMC cAMP at 2 and 4 h in healthy controls and SPMS; ↓ PBMC cAMP in RRMS (divergent response). PK: ↔ plasma ALA (no significant between-group PK differences). PGE2: ↓ in female RRMS, plasma PGE2 at 4 h vs. female HC/SPMS | Strong for mechanism-of-action inference (objective PK + cellular second messenger readout), but limited clinical inference: no placebo/control, acute single-dose window, and endpoints are surrogate biomarkers rather than disability/relapse outcomes. | [111] |
| Clinical trial, randomized parallel-group/ALS patients (planned n = 150; China; age 20–75 years; revised El Escorial criteria; disease onset < 2 years; baseline FVC ≥ 70%) | Randomized to lipoic acid vs. control; 6 treatment courses (~5 months), with assessments at baseline, course 3, and course 6. Dose: NR in accessible registry summaries | Planned outcomes included motor function and disease progression (ALSFRS-R, ROADS, UMN scale, muscle strength scale, EMG) and respiratory function (including FVC); planned safety outcomes included routine blood and urine testing, liver and kidney function, and coagulation indices | Protocol-level evidence only. It is appropriate for describing registered clinical testing, but no conclusions regarding efficacy or safety can be drawn until results are posted or published. Randomization supports internal validity in principle, although risk of reporting bias remains in the absence of available outcome data | [119] |
| Pilot RCT/SPMS adults (single-site; total randomized n = 51) | Double-blind, randomized, placebo-controlled; alpha-lipoic acid (ALA) 1200 mg/day orally vs. placebo for 2 years | Brain atrophy ↓: annualized percent change brain volume (PCBV) significantly less negative with ALA (reported as ~68% reduction vs. placebo); exploratory clinical outcomes suggested possible benefit, but primary signal is MRI atrophy. Safety: serious renal events ↑ (reported in ALA arm) | Good internal validity for a pilot study (randomized, double-blind; Class I evidence for primary MRI endpoint). Limits: small sample/single center; clinical endpoints underpowered; safety signal (renal events) warrants caution. | [124] |
| Biotin—Clinical studies | ||||
| Clinical trial, phase 3 (pivotal) randomized, double-blind, placebo-controlled trial/SPMS and PPMS | MD1003 (biotin 100 mg) orally TID (300 mg/day) vs. placebo for 12 months, then extension phase with MD1003 for all patients (trial included 154 randomized) | ↑ Disability reversal at month 9 confirmed at month 12: 12.6% MD1003 vs. 0% placebo (p = 0.005). Disability reversal defined as EDSS ↓ ≥1.0 (or ≥0.5 if EDSS 6–7) or T25FW ↓ ≥20% vs. best baseline. Secondary: ↓ EDSS progression and ↑ clinician global impression improved vs. placebo | Strong design (double-blind RCT; prespecified composite primary endpoint). Limitations: modest responder proportion; endpoint combines EDSS/T25FW (functional heterogeneity). Safety reported as similar to placebo over the blinded phase. Potential COI reported | [35] |
| Prospective cohort/UK Biobank adults (40–69 years at baseline); outcome: incident all-cause dementia and Alzheimer’s disease | Exposure: dietary biotin intake estimated via 24 h recall and categorized into quartiles (Q1–Q4). Follow-up: median 11.25 years; 1256 incident dementia cases. (No intervention dose; this is intake, not supplementation | ↓ Dementia risk with higher dietary biotin: vs. Q1, ↓ all-cause dementia HR (Q2 0.75, Q3 0.68, Q4 0.67). ↓ AD risk ↓ (Q2 0.74; Q3 0.65; nonlinear association reported). Inflammation mediation: SII partially mediates the association | Large sample, prospective design, extensive covariate adjustment + sensitivity analyses. Key limitations: observational residual confounding, dietary recall measurement error, and dementia defined via algorithmic linkage (not uniform clinical adjudication). Cannot infer causality or supplementation efficacy | [103] |
| Clinical, pilot proof-of-concept; uncontrolled, non-blinded/Progressive MS (PPMS + SPMS); n = 23 | High-dose biotin 100–300 mg/day (oral); treatment 2–36 months (mean ~9 months) | Visual function improved in a subset of patients with optic-nerve involvement (↑ visual acuity; VEP P100 responses improved or reappeared in some cases). Exploratory metabolic imaging findings were also reported | Low evidence strength; uncontrolled observational series. Findings derive from uncontrolled clinical experience rather than from a randomized trial and should be interpreted cautiously | [113] |
| Retrospective real-world case series/Progressive MS (n ≈ 43) | Pharmaceutical-grade biotin 300 mg/day (oral); follow-up up to ~1 year | No sustained disability improvement; EDSS unchanged overall and clinical worsening observed in ~38–43% of patients, with some individuals improving after treatment discontinuation. Safety laboratory parameters remained largely stable | Retrospective real-world clinical experience without randomization or placebo control; results reflect observational outcomes rather than controlled efficacy evidence | [113] |
| Randomized, double-blind, placebo-controlled (RCT)/MS patients with chronic visual loss after optic neuritis: either AON (acute optic neuritis) or PON (progressive optic neuropathy) | Randomized 2:1 to MD1003 (biotin 300 mg/day, oral; 100 mg TID) vs. placebo for 6 months, then 6-month open-label extension | Primary (month 6): visual acuity at 100% contrast (logMAR) ↔ overall. Subgroup: PON showed a trend toward benefit (↑ 100% contrast visual acuity), with ↑ RNFL thickness ↔/trend and ↑ health outcomes ↔/trend; ↔ AON | Strong internal validity (double-blind RCT; prespecified primary endpoint). Limitations: primary endpoint negative; subgroup findings are exploratory; sensitivity of visual outcomes and heterogeneity (AON vs. PON) may dilute effects. Funding by MedDay Pharmaceuticals (potential COI). | [114] |
| Phase 3 RCT/Progressive MS (SPMS and PPMS); multicenter (90 sites, 13 countries); randomized MD1003 n = 326 vs. placebo n = 316 | Randomized, double-blind, parallel-group, placebo-controlled. MD1003 (biotin) 100 mg TID = 300 mg/day (oral) vs. placebo. Primary endpoint assessed at month 12 with confirmation at month 15. Mean time in placebo-controlled phase 20.1 months (range 15–27) | Disability improvement ↔ (not significant): improved at month 12 (confirmed at month 15) in 12% (39/326) MD1003 vs. 9% (29/316) placebo; OR 1.35 (95% CI 0.81–2.26). No significant benefit on disability or walking outcomes overall | High-quality design (large Phase 3 double-blind RCT). However, primary endpoint negative; conclusion is that MD1003 cannot be recommended for progressive MS. Also highlights risk of harmful consequences from laboratory-test interference with high-dose biotin. Funded by MedDay Pharmaceuticals (potential conflict of interest) | [115] |
| Prospective cohort (observational)/UK Biobank participants (n = 122,959) | Dietary biotin intake estimated from 24 h dietary recall, analyzed by intake categories (quartiles). Follow-up: long-term (median ~11 years). No supplementation dose (this is intake, not an intervention) | Higher dietary biotin intake associated with ↓ dementia risk and ↓ AD risk (dose–response/nonlinear patterns reported) | Strong for association (large sample, prospective design), but cannot infer causality (residual confounding, dietary measurement error). Not evidence for therapeutic high-dose biotin efficacy | [116] |
| Clinical trial, Phase 2b open-label, uncontrolled pilot/15 patients with chronic demyelinating peripheral neuropathy: CIDP (n = 5), anti-MAG neuropathy (n = 5), CMT1A/1B (n = 5) | High-dose pharmaceutical-grade biotin 100 mg PO TID = 300 mg/day, up to 52 weeks | Primary endpoint: predefined electrophysiology improvement ↔ (primary endpoint not met): required ≥10% relative improvement in 2/4 variables (MNCV ↑, DML ↓, F-wave latency ↓, CMAP duration ↓). Secondary/exploratory: several sensory/motor parameters ↑, gait measures ↑, nerve excitability measures ↑; tolerability acceptable with AE present but serious AE not judged related. | Low–moderate evidence for efficacy: open-label, no control group, very small sample and heterogeneous etiologies → high risk of bias/confounding; best viewed as proof-of-concept/signal-generating | [117] |
| Clinical trial, pilot RCT/Adults with probable or definite ALS, age 25–80 (n = 30; MD1003 n = 20, placebo n = 10) | Randomized (2:1), double-blind, placebo-controlled; MD1003 (pharmaceutical-grade biotin) 300 mg/day PO vs. placebo for 24 weeks (6 months) | Primary (safety/tolerability): AE ↔ vs. placebo (both 60%); deaths: MD1003 2 vs. placebo 1. Efficacy (exploratory): ALSFRS-R change at month 6 ↔ (no significant difference; p = 0.49) | Small pilot with baseline imbalance (MD1003 group had faster pre-screening ALSFRS-R decline and more upper-limb onset), limiting efficacy inference; supports safety signal, efficacy not established → larger trials warranted. | [119] |
| Clinical trial, Phase 2/ALS patients | Randomized, double-blind, 2:1 allocation; biotin 300 mg/day PO vs. placebo; 6 months, visits at baseline, 3, and 6 months | Primary: adverse effects (safety/tolerability) NR. Secondary: motor disability by ALS-FRS, pulmonary function (FEV1, FVC), body weight NR | Protocol-level evidence only (no results → no conclusions on benefit). Trial design is stronger than observational studies (randomized, blinded), but risk of non-publication/reporting bias remains until results are posted. | [120] |
| Clinical trial, interventional/Huntington’s disease (mild–moderate stages) | Combined oral thiamine + biotin; dose escalation with lab monitoring during escalation; follow-up ~12 months. Exact mg doses: NR in the accessible registry summaries. | Primary: safety/tolerability, NR (no posted results). Key biomarker endpoint: CSF thiamine monophosphate (TMP) ↑ (planned) as the main CNS biological marker. Additional planned exploratory outcomes include progression-related biomarkers/neuroimaging (e.g., caudate/WM/cortical measures) and CSF neurofilaments (planned) | Protocol-level evidence only (no outcomes yet → no conclusions). Strength: prospective interventional design with CNS biomarker focus. Limitation: until results are available, efficacy/safety remain undetermined and susceptible to reporting bias | [122] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Aguilera-Méndez, A.; Aguilera-Manuel, K.; Saavedra-Molina, A.; Ríos-Chávez, P.; Villafaña, S.; Nieto-Aguilar, R.; Godínez-Hernández, D.; Ortega-Cuellar, D.; Palomera-Sanchez, Z.; Gauthereau-Torres, M. Alpha-Lipoic Acid and Biotin in Neurodegenerative Diseases: Convergent Mechanistic Insights from Preclinical Models to Clinical Perspectives. Neurol. Int. 2026, 18, 64. https://doi.org/10.3390/neurolint18040064
Aguilera-Méndez A, Aguilera-Manuel K, Saavedra-Molina A, Ríos-Chávez P, Villafaña S, Nieto-Aguilar R, Godínez-Hernández D, Ortega-Cuellar D, Palomera-Sanchez Z, Gauthereau-Torres M. Alpha-Lipoic Acid and Biotin in Neurodegenerative Diseases: Convergent Mechanistic Insights from Preclinical Models to Clinical Perspectives. Neurology International. 2026; 18(4):64. https://doi.org/10.3390/neurolint18040064
Chicago/Turabian StyleAguilera-Méndez, Asdrubal, Karel Aguilera-Manuel, Alfredo Saavedra-Molina, Patricia Ríos-Chávez, Santiago Villafaña, Renato Nieto-Aguilar, Daniel Godínez-Hernández, Daniel Ortega-Cuellar, Zoraya Palomera-Sanchez, and Marcia Gauthereau-Torres. 2026. "Alpha-Lipoic Acid and Biotin in Neurodegenerative Diseases: Convergent Mechanistic Insights from Preclinical Models to Clinical Perspectives" Neurology International 18, no. 4: 64. https://doi.org/10.3390/neurolint18040064
APA StyleAguilera-Méndez, A., Aguilera-Manuel, K., Saavedra-Molina, A., Ríos-Chávez, P., Villafaña, S., Nieto-Aguilar, R., Godínez-Hernández, D., Ortega-Cuellar, D., Palomera-Sanchez, Z., & Gauthereau-Torres, M. (2026). Alpha-Lipoic Acid and Biotin in Neurodegenerative Diseases: Convergent Mechanistic Insights from Preclinical Models to Clinical Perspectives. Neurology International, 18(4), 64. https://doi.org/10.3390/neurolint18040064