
Late-Life Blood Pressure and Cerebral Amyloid Angiopathy: Findings from the U.S. National Alzheimer’s Coordinating Center Uniform Dataset

 ,
,  and
and
Abstract
1. Introduction
2. Methods
2.1. Data and Participants
2.2. Identification of Cases and Non-Cases
2.3. Study Exposure
2.4. Covariates
2.5. Statistical Analyses
3. Results
3.1. Participant Characteristics
3.2. Blood Pressure and CAA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chalmers, K.; Wilcock, G.K.; Love, S. APOE epsilon 4 influences the pathological phenotype of alzheimer’s disease by favouring cerebrovascular over parenchymal accumulation of A beta protein. Neuropathol. Appl. Neurobiol. 2003, 29, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Love, S.; Miners, S.; Palmer, J.; Chalmers, K.; Kehoe, P. Insights into the pathogenesis and pathogenicity of cerebral amyloid angiopathy. Front. Biosci. 2009, 14, 4778–4792. [Google Scholar] [CrossRef] [PubMed]
- Esiri, M.M.; Wilcock, G.K. Cerebral amyloid angiopathy in dementia and old age. J. Neurol. Neurosurg. Psychiatry 1986, 49, 1221–1226. [Google Scholar] [CrossRef] [PubMed]
- Love, S.; Nicoll, J.A.R.; Hughes, A.; Wilcock, G.K. APOE and cerebral amyloid angiopathy in the elderly. Neuroreport 2003, 14, 1535–1536. [Google Scholar] [CrossRef] [PubMed]
- Charidimou, A.; Martinez-Ramirez, S.; Shoamanesh, A.; Oliveira-Filho, J.; Frosch, M.; Vashkevich, A.; Ayres, A.; Rosand, J.; Gurol, M.E.; Greenberg, S.M.; et al. Cerebral amyloid angiopathy with and without hemorrhage: Evidence for different disease phenotypes. Neurology 2015, 84, 1206–1212. [Google Scholar] [CrossRef]
- Banerjee, G.; Carare, R.; Cordonnier, C.; Greenberg, S.M.; Schneider, J.A.; Smith, E.E.; Van Buchem, M.; Van Der Grond, J.; Verbeek, M.M.; Werring, D.J.; et al. The increasing impact of cerebral amyloid angiopathy: Essential new insights for clinical practice. J. Neurol. Neurosurg. Psychiatry 2017, 88, 982–994. [Google Scholar] [CrossRef] [PubMed]
- Biffi, A.; Bailey, D.; Anderson, C.D.; Ayres, A.M.; Gurol, E.M.; Greenberg, S.M.; Rosand, J.; Viswanathan, A. Risk factors associated with early vs. delayed dementia after intracerebral hemorrhage. JAMA Neurol. 2016, 73, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Planton, M.; Saint-Aubert, L.; Raposo, N.; Branchu, L.; Lyoubi, A.; Bonneville, F.; Albucher, J.F.; Olivot, J.M.; Péran, P.; Pariente, J. High prevalence of cognitive impairment after intracerebral hemorrhage. PLoS ONE 2017, 12, e0178886. [Google Scholar] [CrossRef]
- Keep, R.F.; Hua, Y.; Xi, G. Intracerebral hemorrhage: Mechanisms of injurty and therapeutic targets. Lancet Neurol. 2021, 11, 720–731. [Google Scholar] [CrossRef]
- Zhang, Z.; Lim, M.J.R. Incident dementia after spontaneous intracerebral hemorrhage. J. Alzheimers Dis. 2024, 99, 41–51. [Google Scholar] [CrossRef]
- Arima, H.; Anderson, C.; Omae, T.; Woodward, M.; MacMahon, S.; Mancia, G.; Bousser, M.G.; Tzourio, C.; Rodgers, A.; Neal, B.; et al. Effects of blood pressure lowering on intracranial and extracranial bleeding in patients on antithrombotic therapy: The PROGRESS trial. Stroke 2012, 43, 1675–1677. [Google Scholar] [CrossRef] [PubMed]
- Jandke, S.; Garz, C.; Schwanke, D.; Sendtner, M.; Heinze, H.J.; Carare, R.O.; Schreiber, S. The association between hypertensive arteriopathy and cerebral amyloid angiopathy in spontaneously hypertensive stroke-prone rats. Brain Pathol. 2018, 28, 844–859. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.L.; Gao, Q.I.; Nyunt, M.S.Z.; Gong, L.; Lunaria, J.B.; Lim, M.L.; Ling, A.; Lam, C.S.P.; Richards, A.M.; Ling, L.H.; et al. Vascular health indices and cognitive domain function: Singapore longitudinal ageing studies. J. Alzheimers Dis. 2016, 50, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Langbaum, J.B.; Chen, K.; Launer, L.J.; Fleisher, A.S.; Lee, W.; Liu, X.; Protas, H.D.; Reeder, S.A.; Bandy, D.; Yu, M.; et al. Blood pressure is associated with higher brain amyloid burden and lower glucose metabolism in healthy late middle-age persons. Neurobiol. Aging 2012, 33, 827.e11–827.e19. [Google Scholar] [CrossRef] [PubMed]
- University of Washington. About NACC. Available online: https://naccdata.org/nacc-collaborations/about-nacc (accessed on 22 July 2024).
- Morris, J.C.; Weintraub, S.; Chui, H.C.; Cummings, J.; DeCarli, C.; Ferris, S.; Foster, N.L.; Galasko, D.; Graff-Radford, N.; Peskind, E.R.; et al. The uniform data set (UDS): Clinical and cognitive variables and descriptive data from alzheimer disease centers. Alzheimer Dis. Assoc. Disord. 2006, 20, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Besser, L.M.; Kukull, W.A.; Teylan, M.A.; Bigio, E.H.; Cairns, N.J.; Kofler, J.K.; Montine, T.J.; Schneider, J.A.; Nelson, P.T. The revised national alzheimer’s coordinating center’s neuropathology form-available data and new analyses. J. Neuropathol. Exp. Neurol. 2018, 77, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Beekly, D.L.; Ramos, E.M.; van Belle, G.; Deitrich, W.; Clark, A.D.; Jacka, M.E.; Kukull, W.A. The national alzheimer’s coordinating center (NACC) database: An alzheimer disease database. Alzheimer Dis. Assoc. Disord. 2004, 18, 270–277. [Google Scholar] [PubMed]
- Yakupova, E.I.; Bobyleva, L.G.; Vikhlyantseve, I.M.; Bobyleve, A.G. Congo red amyloids: History and relationship. Biosci. Rep. 2019, 39, BSR20181415. [Google Scholar] [CrossRef] [PubMed]
- Chobanian, A.V.; Bakris, G.L.; Black, H.R.; Cushman, W.C.; Green, L.A.; Izzo, J.L., Jr.; Jones, D.W.; Materson, B.J.; Oparil, S.; Wright, J.T., Jr.; et al. The seventh report of the joint national committee on prevention, detection, evaluation, and treatment of high blood pressure: The JNC 7 report. JAMA 2003, 289, 2560–2572. [Google Scholar] [CrossRef]
- Mirra, S.S.; Heyman, A.; McKeel, D.; Sumi, S.M.; Crain, B.J.; Brownlee, L.M.; Vogel, F.S.; Hughes, J.P.; Belle, G.V.; Berg, L.; et al. The consortium to establish a registry for alzheimer’s disease (CERAD). part II. standardization of the neuropathologic assessment of alzheimer’s disease. Neurology 1991, 41, 479–486. [Google Scholar] [CrossRef]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef] [PubMed]
- Sonnen, J.A.; Larson, E.B.; Crane, P.K.; Haneuse, S.; Li, G.; Schellenberg, G.D.; Craft, S.; Leverenz, J.B.; Montine, T.J. Pathological correlates of dementia in a longitudinal, population-based sample of aging. Ann Neurol. 2007, 62, 406–413. [Google Scholar] [CrossRef] [PubMed]
- McCullagh, P. Regression models for ordinal data. J. R. Stat. Soc. Ser. B 1980, 42, 109–142. Available online: https://www.stat.uchicago.edu/~pmcc/pubs/paper2.pdf (accessed on 22 July 2024). [CrossRef]
- Hosmer, D.W.; Lameshow, S. Applies Logistic Regression; John Wiley &Sons: Hoboken, NJ, USA, 2000. [Google Scholar]
- Brant, R. Assessing proportionality in the proportional odds model for ordinal logistic regression. Biometrics 1990, 46, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Lipsitz, S.R.; Fitzmaurice, G.M.; Molenberghs, G. Goodness-of fit tests for ordinal response regression models. J. R. Stat. Soc. 1996, 45, 175–190. [Google Scholar] [CrossRef]
- The R Foundation. The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 10 June 2024).
- Venables, W.N.; Ripley, B.D. Modern Applied Statistics with S. Fourth Edition. Available online: https://www.stats.ox.ac.uk/pub/MASS4/ (accessed on 10 June 2024).
- Jonckheere, A.R. A distribution-free k-sample test against ordered alternatives. Biometrika 1954, 41, 133–145. [Google Scholar] [CrossRef]
- Lunneborg, C.E. Jonckheere–Terpstra Test. Available online: https://onlinelibrary.wiley.com/doi/10.1002/0470013192.bsa324 (accessed on 22 July 2024).
- Sin, M.; Cheng, Y.; Roseman, J.M.; Zamrini, E.; Ahmed, A. Relationships between cerebral vasculopathies and microinfarcts in a community-based cohort of older adults. J. Clin. Med. 2023, 12, 3807. [Google Scholar] [CrossRef] [PubMed]
- Tanskanen, M.; Mäkelä, M.; Myllykangas, L.; Notkola, I.L.; Polvikoski, T.; Sulkava, R.; Kalimo, H.; Paetau, A. Prevalence and severity of cerebral amyloid angiopathy: A population-based study on very elderly finns (vantaa 85+). Neuropathol. Appl. Neurobiol. 2012, 38, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Vidoni, E.D.; Yeh, H.W.; Morris, J.K.; Newell, K.L.; Alqahtani, A.; Burns, N.C.; Burns, J.M.; Billinger, S.A. Cerebral β-amyloid angiopathy is associated with earlier dementia onset in alzheimer’s disease. Neurodegener. Dis. 2016, 16, 218–224. [Google Scholar] [CrossRef]
- Hughes, T.M.; Sink, K.M. Hypertension and its role in cognitive function: Current evidence and challenges for the future. Am. J. Hypertens. 2016, 29, 149–157. [Google Scholar] [CrossRef]
- Xu, T.Y.; Staessen, J.A.; Wei, F.F.; Xu, J.; Li, F.H.; Fan, W.X.; Gao, P.J.; Wang, J.G.; Li, Y. Blood flow pattern in the middle cerebral artery in relation to indices of arterial stiffness in the systemic circulation. Am. J. Hypertens. 2012, 25, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Nation, D.A.; Edland, S.D.; Bondi, M.W.; Salmon, D.P.; Delano-Wood, L.; Peskind, E.R.; Quinn, J.F.; Galasko, D.R. Pulse pressure is associated with alzheimer biomarkers in cognitively normal older adults. Neurology 2013, 81, 2024–2027. [Google Scholar] [CrossRef] [PubMed]
- Nation, D.A.; Edmonds, E.C.; Bangen, K.J.; Delano-Wood, L.; Scanlon, B.K.; Han, S.D.; Edland, S.D.; Salmon, D.P.; Galasko, D.R.; Bondi, M.W. Pulse pressure in relation to tau-mediated neurodegeneration, cerebral amyloidosis, and progression to dementia in very old adults. JAMA Neurol. 2015, 72, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Rivier, C.A.; Kamel, H.; Sheth, K.N.; Iadecola, C.; Gupta, A.; de Leon, M.J.; Ross, E.; Falcone, G.J.; Murthy, S.B. Cerebral amyloid angiopathy and risk of isolated nontraumatic subdural hemorrhage. JAMA Neurol. 2023, 81, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, Z.; Zheng, A.; Yuan, R.; Shu, Y.; Zhang, S.; Lei, P.; Wu, B.; Liu, M. Blood pressure and outcomes in patients with different etiologies of intracerebral hemorrhage: A multicenter cohort study. J. Am. Heart Assoc. 2020, 9, e016766. [Google Scholar] [CrossRef] [PubMed]
- Olichney, J.M.; Hansen, L.A.; Galasko, D.; Saitoh, T.; Hofstetter, C.R.; Katzman, R.; Thal, L.J. The apolipoprotein E epsilon 4 allele is associated with increased neuritic plaques and cerebral amyloid angiopathy in alzheimer’s disease and lewy body variant. Neurology 1996, 47, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Premkumar, D.R.; Cohen, D.L.; Hedera, P.; Friedland, R.P.; Kalaria, R.N. Apolipoprotein E-epsilon4 alleles in cerebral amyloid angiopathy and cerebrovascular pathology associated with alzheimer’s disease. Am. J. Pathol. 1996, 148, 2083–2095. [Google Scholar] [PubMed]
- Ba, M.; Kong, M.; Li, X.; Ng, K.P.; Rosa-Neto, P.; Gauthier, S. Is ApoE ɛ 4 a good biomarker for amyloid pathology in late onset alzheimer’s disease? Transl. Neurodegener. 2016, 5, 20. [Google Scholar] [CrossRef]
- Rabin, J.S.; Nichols, E.; La Joie, R.; Casaletto, K.B.; Palta, P.; Dams-O’Connor, K.; Kumar, R.G.; George, K.M.; Satizabal, C.L.; Schneider, J.A.; et al. Cerebral amyloid angiopathy interacts with neuritic amyloid plaques to promote tau and cognitive decline. Brain 2022, 145, 2823–2833. [Google Scholar] [CrossRef] [PubMed]
- McCorkindale, A.N.; Mundell, H.D.; Guennewig, B.; Sutherland, G.T. Vascular dysfunction is central to alzheimer’s disease pathogenesis in APOE e4 carriers. Int. J. Mol. Sci. 2022, 23, 7106. [Google Scholar] [CrossRef]
- Malek-Ahmadi, M.; Perez, S.E.; Chen, K.; Mufson, E.J. Braak stage, cerebral amyloid angiopathy, and cognitive decline in early alzheimer’s disease. J. Alzheimers Dis. 2020, 74, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Koemans, E.A.; Castello, J.P.; Rasing, I.; Abramson, J.R.; Voigt, S.; Perosa, V.; Van Harten, T.W.; Van Zwet, E.W.; Terwindt, G.M.; Gurol, M.E.; et al. Sex differences in onset and progression of cerebral amyloid angiopathy. Stroke 2023, 54, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.; Bryant, M.; Clark, L.; Garces, A.; Rhodin, J. Estrogen and raloxifene activities on amyloid-beta-induced inflammatory reaction. Microvasc. Res. 2001, 61, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, R.; Teixeira, D.; Calhau, C. Estrogen signaling in metabolic inflammation. Mediat. Inflamm. 2014, 2014, 615917. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Murray, M.E.; Frank, R.D.; Shinohara, M.; DeTure, M.; Yamazaki, Y.; Tachibana, M.; Atagi, Y.; Davis, M.D.; Liu, C.C.; et al. Impact of sex and APOE4 on cerebral amyloid angiopathy in alzheimer’s disease. Acta Neuropathol. 2016, 132, 225–234. [Google Scholar] [CrossRef]
- Pinho, J.; Almeida, F.C.; Araújo, J.M.; Machado, Á.; Costa, A.S.; Silva, F.; Francisco, A.; Quintas-Neves, M.; Ferreira, C.; Soares-Fernandes, J.P.; et al. Sex-specific patterns of cerebral atrophy and enlarged perivascular spaces in patients with cerebral amyloid angiopathy and dementia. AJNR Am. J. Neuroradiol. 2023, 44, 792–798. [Google Scholar] [CrossRef]

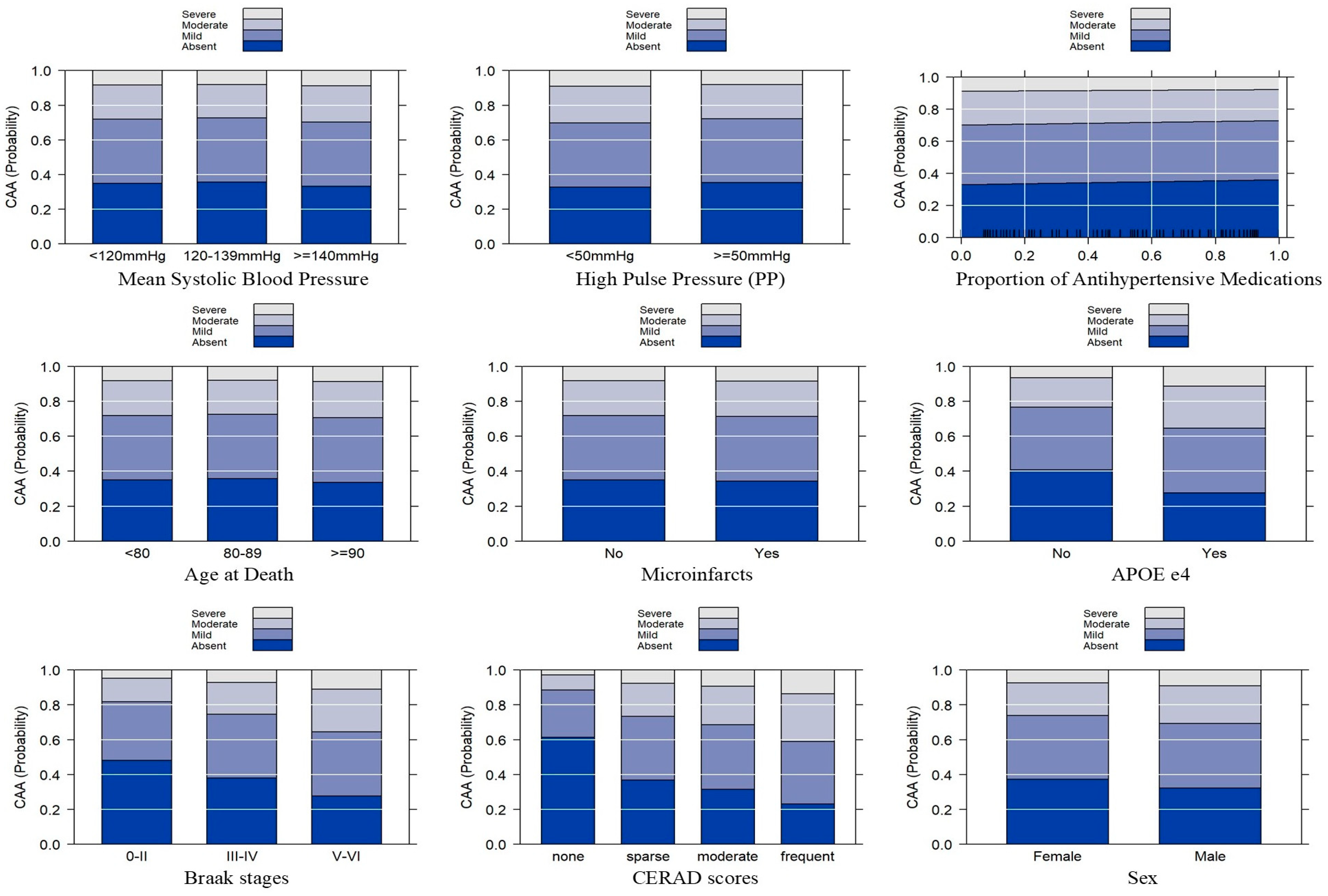

{kind=link}
{kind=link}
| Variables | Total Sample (n = 2510) | CAA None (n = 930) | Mild CAA (n = 759) | Moderate CAA (n = 529) | Severe CAA (n = 292) | p-Value a | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| N | % | N | % | N | % | N | % | N | % | ||
| Age at death | |||||||||||
| <80 | 621 | 24.7% | 196 | 21.1% | 184 | 24.2% | 155 | 29.3% | 86 | 29.5% | <0.001 |
| 80–89 | 895 | 35.7% | 383 | 41.2% | 266 | 35.0% | 158 | 29.9% | 88 | 30.1% | |
| ≥90 | 994 | 39.6% | 351 | 37.7% | 309 | 40.7% | 216 | 40.8% | 118 | 40.4% | |
| Apoe e4 carrier | |||||||||||
| No e4 allele | 1422 | 56.7% | 691 | 74.3% | 409 | 53.9% | 213 | 40.3% | 109 | 37.3% | <0.001 |
| 1 or 2 copies of e4 allele | 1088 | 43.3% | 239 | 25.7% | 350 | 46.1% | 316 | 59.7% | 183 | 62.7% | |
| Braak Stage | |||||||||||
| 0–II | 549 | 21.9% | 372 | 40.0% | 118 | 15.5% | 40 | 7.6% | 19 | 6.5% | <0.001 |
| III–IV | 724 | 28.8% | 326 | 35.1% | 226 | 29.8% | 126 | 23.8% | 46 | 15.8% | |
| V–VI | 1237 | 49.3% | 232 | 24.9% | 415 | 54.7% | 363 | 68.6% | 227 | 77.7% | |
| CERAD | |||||||||||
| Absent neuritic plaques | 571 | 22.7% | 417 | 44.8% | 111 | 14.6% | 31 | 5.9% | 12 | 4.1% | <0.001 |
| Sparse neuritic plaques | 394 | 15.7% | 166 | 17.8% | 132 | 17.4% | 74 | 14.0% | 22 | 7.5% | |
| Moderate | 525 | 20.9% | 165 | 17.7% | 180 | 23.7% | 129 | 24.4% | 51 | 17.5% | |
| Frequent | 1020 | 40.6% | 182 | 19.6% | 336 | 44.3% | 295 | 55.8% | 207 | 70.9% | |
| Race | |||||||||||
| White | 2363 | 94.1% | 877 | 94.3% | 713 | 93.9% | 496 | 93.8% | 277 | 94.9% | 0.453 |
| Black or African American | 120 | 4.8% | 44 | 4.7% | 37 | 4.9% | 27 | 5.1% | 12 | 4.1% | |
| American Indian or Alaska Native | 2 | 0.1% | 0 | 0.0% | 1 | 0.1% | 0 | 0.0% | 1 | 0.3% | |
| Asian | 20 | 0.8% | 9 | 1.0% | 7 | 0.9% | 3 | 0.6% | 1 | 0.3% | |
| Other | 5 | 0.2% | 0 | 0.0% | 1 | 0.1% | 3 | 0.6% | 1 | 0.3% | |
| Sex | |||||||||||
| Female | 1219 | 48.6% | 468 | 50.3% | 385 | 50.7% | 248 | 46.9% | 118 | 40.4% | 0.012 |
| Male | 1291 | 51.4% | 462 | 49.7% | 374 | 49.3% | 281 | 53.1% | 174 | 59.6% | |
| Micro infarcts | |||||||||||
| No | 1874 | 74.7% | 682 | 73.3% | 567 | 74.7% | 407 | 76.9% | 218 | 74.7% | 0.509 |
| Yes | 636 | 25.3% | 248 | 26.7% | 192 | 25.3% | 122 | 23.1% | 74 | 25.3% | |
| Mean Late-Life Systolic Blood Pressure | |||||||||||
| Low: <120 mmHg | 435 | 17.3% | 153 | 16.5% | 135 | 17.8% | 94 | 17.8% | 53 | 18.2% | 0.482 |
| Normal: 120–139 mmHg | 1335 | 53.2% | 516 | 55.5% | 405 | 53.4% | 272 | 51.4% | 142 | 48.6% | |
| High: ≥140 mmHg | 740 | 29.5% | 261 | 28.1% | 219 | 28.9% | 163 | 30.8% | 97 | 33.2% | |
| Mean Late-Life Pulse Pressure | |||||||||||
| Normal: <50 mmHg | 487 | 19.4% | 175 | 18.8% | 138 | 18.2% | 103 | 19.5% | 71 | 24.3% | 0.143 |
| High: ≥50 mmHg | 2023 | 80.6% | 755 | 81.2% | 621 | 81.8% | 426 | 80.5% | 221 | 75.7% | |
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | ||
| Time gap (last BP to death in years) | 3.029 | 2.318 | 2.825 | 2.296 | 3.032 | 2.337 | 3.217 | 2.255 | 3.336 | 2.395 | 0.001 |
| Proportion of Antihypertensive Medication Use Across Time | 0.636 | 0.404 | 0.679 | 0.383 | 0.635 | 0.404 | 0.599 | 0.421 | 0.567 | 0.423 | <0.001 |
| Predictor | Model 1 (Excludes Late-Life Mean PP) | Model 2 (Excludes Late-Life Mean SBP) | Model 3 (Includes Both Late-Life Mean SBP and PP) | |||
|---|---|---|---|---|---|---|
| aOR (95% CI) | p-Value | aOR (95% CI) | p-Value | aOR (95% CI) | p-Value | |
| Late-life mean SBP | ||||||
| 120–139 mmHg vs. <120 | 0.906 (0.737, 1.115) | 0.3510 | - | - | 0.966 (0.754, 1.238) | 0.783 |
| ≥140 mmHg vs. <120 | 1.002 (0.797, 1.260) | 0.9896 | - | - | 1.082 (0.816, 1.435) | 0.586 |
| Late-life mean PP | ||||||
| ≥50 mmHg vs. <50 mmHg | - | - | 0.910 (0.750, 1.104) | 0.3379 | 0.893 (0.701, 1.138) | 0.359 |
| Proportion of Antihypertensive Medications | 0.869 (0.718, 1.051) | 0.056 | 0.877 (0.725, 1.061) | 0.1754 | 0.872 (0.721, 1.056) | 0.160 |
| Age at Death | ||||||
| 80–89 vs. <80 | 0.951 (0.783, 1.155) | 0.6123 | 0.966 (0.795, 1.175) | 0.7310 | 0.962 (0.791, 1.170) | 0.696 |
| ≥90 vs. <80 | 1.055 (0.850, 1.311) | 0.6256 | 1.069 (0.860, 1.329) | 0.5480 | 1.068 (0.859, 1.328) | 0.555 |
| Microinfarcts | ||||||
| Yes vs. No | 1.028 (0.862, 1.224) | 0.7612 | 1.034 (0.868, 1.232) | 0.7058 | 1.028 (0.862, 1.224) | 0.762 |
| APOE e4 carrier | ||||||
| Yes vs. No | 1.795 (1.526, 2.111) | <0.001 | 1.797 (1.528, 2.114) | <0.001 | 1.796 (1.527, 2.113) | <0.001 |
| Braak stage | ||||||
| III–IV vs. 0–II | 1.509 (1.172, 1.943) | 0.0014 | 1.513 (1.176, 1.949) | 0.0013 | 1.510 (1.173, 1.945) | 0.001 |
| V–VI vs. 0–II | 2.427 (1.824, 3.233) | <0.001 | 2.424 (1.821, 3.228) | <0.001 | 2.425 (1.822, 3.230) | <0.001 |
| CERAD stage | ||||||
| Sparse vs. Absent | 2.720 (2.068, 3.583) | <0.001 | 2.744 (2.087, 3.613) | <0.001 | 2.734 (2.079, 3.603) | <0.001 |
| Moderate vs. Absent | 3.456 (2.607, 4.592) | <0.001 | 3.471 (2.619, 4.612) | <0.001 | 3.460 (2.610, 4.598) | <0.001 |
| Frequent vs. Absent | 5.233 (3.885, 7.070) | <0.001 | 5.289 (3.927, 7.145) | <0.001 | 5.251 (3.897, 7.094) | <0.001 |
| Sex | ||||||
| Male vs. Female | 1.255 (1.079, 1.461) | 0.0033 | 1.246 (1.071, 1.450) | 0.0044 | 1.253 (1.076, 1.458) | 0.004 |
| Deviance | 5881.29 | 5882.08 | 5880.45 | |||
| AAIC | 5913 | 5912 | 5914 | |||
| Lipsitz goodness- of- fit test (LR statistic, p-value) | 10.92, p = 0.281 | 8.99, p = 0.438 | 14.487, p = 0.110 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sin, M.-K.; Dowling, N.M.; Roseman, J.M.; Ahmed, A.; Zamrini, E. Late-Life Blood Pressure and Cerebral Amyloid Angiopathy: Findings from the U.S. National Alzheimer’s Coordinating Center Uniform Dataset. Neurol. Int. 2024, 16, 821-832. https://doi.org/10.3390/neurolint16040061
Sin M-K, Dowling NM, Roseman JM, Ahmed A, Zamrini E. Late-Life Blood Pressure and Cerebral Amyloid Angiopathy: Findings from the U.S. National Alzheimer’s Coordinating Center Uniform Dataset. Neurology International. 2024; 16(4):821-832. https://doi.org/10.3390/neurolint16040061
Chicago/Turabian StyleSin, Mo-Kyung, N. Maritza Dowling, Jeffrey M. Roseman, Ali Ahmed, and Edward Zamrini. 2024. "Late-Life Blood Pressure and Cerebral Amyloid Angiopathy: Findings from the U.S. National Alzheimer’s Coordinating Center Uniform Dataset" Neurology International 16, no. 4: 821-832. https://doi.org/10.3390/neurolint16040061
APA StyleSin, M.-K., Dowling, N. M., Roseman, J. M., Ahmed, A., & Zamrini, E. (2024). Late-Life Blood Pressure and Cerebral Amyloid Angiopathy: Findings from the U.S. National Alzheimer’s Coordinating Center Uniform Dataset. Neurology International, 16(4), 821-832. https://doi.org/10.3390/neurolint16040061