
Monomethyl Fumarate (MMF, Bafiertam) for the Treatment of Relapsing Forms of Multiple Sclerosis (MS)

 , ,
, , 
 ,
,  ,
,
Abstract
1. Introduction
2. General Information about MS
2.1. Multiple Sclerosis Classifications
2.2. Disease Progression
2.3. Pathophysiology
2.4. Risk Factors
2.5. Diagnostic Criteria
3. Current Treatment Options
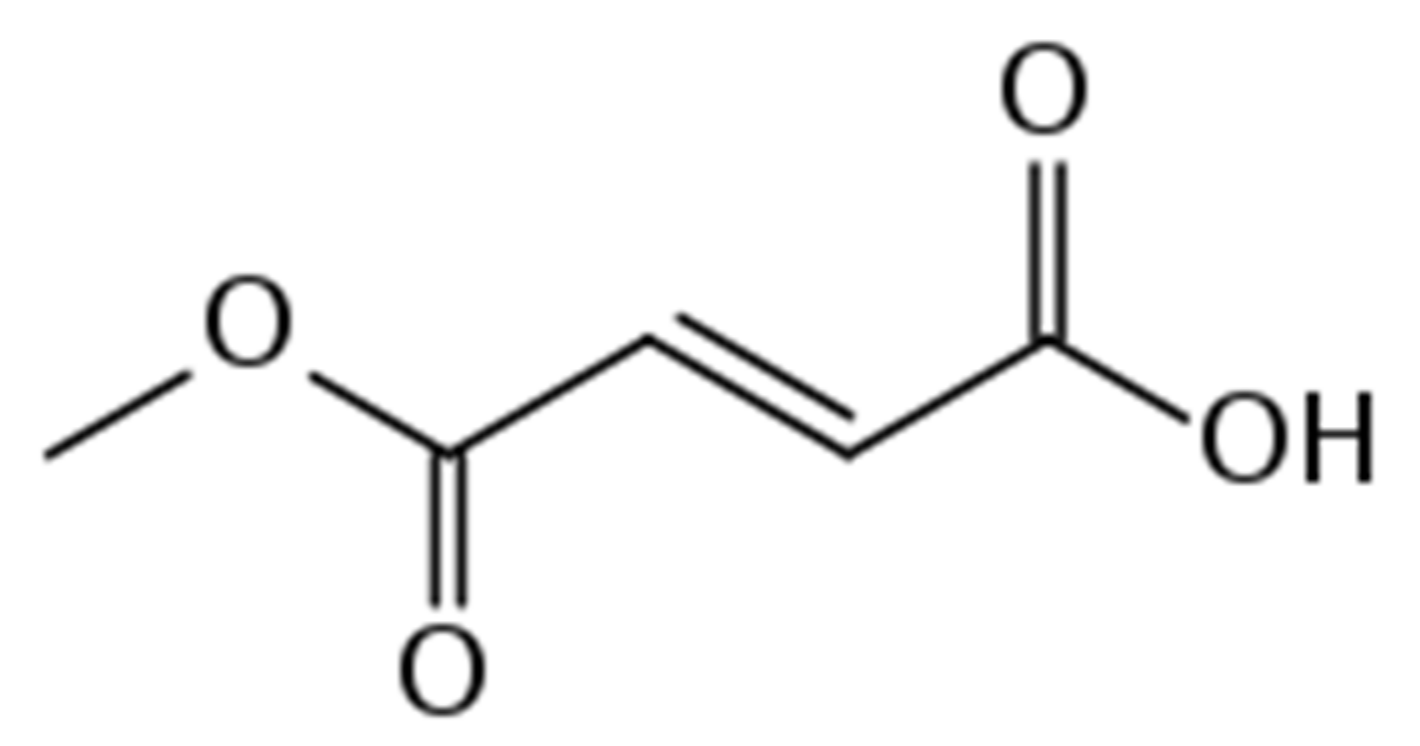
4. Monomethyl Fumarate (MMF, Bafiertam)
4.1. Pharmacology of MMF
4.2. Side Effects/Adverse Events of MMF
5. Important Clinical Studies Involving MMF and MS
Safety, Adverse Events
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Thompson, A.J.; Banwell, B.L.; Barkhof, F.; Carroll, W.M.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Freedman, M.S.; et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018, 15, 162–173. [Google Scholar] [CrossRef]
- Milo, R.; Kahana, E. Multiple sclerosis: Geoepidemiology, genetics and the environment. Autoimmun. Rev. 2010, 9, A387–A394. [Google Scholar] [CrossRef] [PubMed]
- Harbo, H.F.; Gold, R.; Tintoré, M. Sex and gender issues in multiple sclerosis. Ther. Adv. Neurol. Disord. 2013, 6, 237–248. [Google Scholar] [CrossRef]
- Goodin, D.S. The epidemiology of multiple sclerosis. In Neuroparasitology and Tropical Neurology; Elsevier BV: Amsterdam, The Netherlands, 2014; Volume 122, pp. 231–266. [Google Scholar]
- The U.S. Food and Drug Administration. Highlights of Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/210296s000lbl.pdf (accessed on 1 March 2021).
- Albrecht, P.; Bouchachia, I.; Goebels, N.; Henke, N.; Hofstetter, H.H.; Issberner, A.; Kovacs, Z.; Lewerenz, J.; Lisak, D.; Maher, P.; et al. Effects of dimethyl fumarate on neuroprotection and immunomodulation. J. Neuroinflamm. 2012, 8, 163. [Google Scholar] [CrossRef]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sørensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 80, 278–286. [Google Scholar] [CrossRef]
- Dobson, R.; Giovannoni, G. Multiple sclerosis – A review. Eur. J. Neurol. 2019, 26, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, W.J.; Hardy, A.T.; Fazekas, F.; Miller, D.H. Diagnosis of multiple sclerosis: Progress and challenges. Lancet 2017, 389, 1336–1346. [Google Scholar] [CrossRef]
- Weinshenker, B.G. Natural history of multiple sclerosis. Ann. Neurol. 1994, 36, S6–S11. [Google Scholar] [CrossRef]
- Rae-Grant, A.; Day, G.S.; Marrie, R.A.; Rabinstein, A.; Cree, B.A.; Gronseth, G.S.; Haboubi, M.; Halper, J.; Hosey, J.P.; Jones, D.E.; et al. Practice guideline recommendations summary: Disease-modifying therapies for adults with multiple sclerosis. Neurolology 2018, 90, 777–788. [Google Scholar] [CrossRef]
- Eriksson, M.; Andersen, O.; Runmarker, B. Long-term follow up of patients with clinically isolated syndromes, relapsing-remitting and secondary progressive multiple sclerosis. Mult. Scler. J. 2003, 9, 260–274. [Google Scholar] [CrossRef]
- Loma, I. Multiple Sclerosis: Pathogenesis and Treatment. Curr. Neuropharmacol. 2011, 9, 409–416. [Google Scholar] [CrossRef]
- Rice, C.M.; Cottrell, D.; Wilkins, A.; Scolding, N.J. Primary progressive multiple sclerosis: Progress and challenges. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1100–1106. [Google Scholar] [CrossRef]
- Goldenberg, M.M. Multiple Sclerosis Review. P T 2012, 37, 175–184. [Google Scholar]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Weiner, H.L. Multiple Sclerosis Is an Inflammatory T-Cell–Mediated Autoimmune Disease. Arch. Neurol. 2004, 61, 1613–1615. [Google Scholar] [CrossRef] [PubMed]
- Frohman, E.M.; Racke, M.K.; Raine, C.S. Multiple Sclerosis — The Plaque and Its Pathogenesis. N. Engl. J. Med. 2006, 354, 942–955. [Google Scholar] [CrossRef]
- Popescu, B.F.G.; Pirko, I.; Lucchinetti, C.F. Pathology of Multiple Sclerosis. Contin. Lifelong Learn. Neurol. 2013, 19, 901–921. [Google Scholar] [CrossRef]
- Popescu, B.F.; Lucchinetti, C.F. Pathology of Demyelinating Diseases. Annu. Rev. Pathol. Mech. Dis. 2012, 7, 185–217. [Google Scholar] [CrossRef] [PubMed]
- De Barcelos, I.P.; Troxell, R.M.; Graves, J.S. Mitochondrial Dysfunction and Multiple Sclerosis. Biology 2019, 8, 37. [Google Scholar] [CrossRef] [PubMed]
- Kutzelnigg, A.; Lassmann, H. Pathology of multiple sclerosis and related inflammatory demyelinating diseases. In Neurocutaneous Syndromes; Elsevier BV: Amsterdam, The Netherlands, 2014; Volume 122, pp. 15–58. [Google Scholar]
- Nourbakhsh, B.; Mowry, E.M. Multiple Sclerosis Risk Factors and Pathogenesis. Contin. Lifelong Learn. Neurol. 2019, 25, 596–610. [Google Scholar] [CrossRef]
- Sundqvist, E.; Sundstrom, P.; Van Der Linden, M.; Hedström, A.K.; Aloisi, F.; Hillert, J.; Kockum, I.; Alfredsson, L.; Olsson, T. Epstein-Barr virus and multiple sclerosis: Interaction with HLA. Genes Immun. 2011, 13, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Alotaibi, S. Epstein-Barr Virus in Pediatric Multiple Sclerosis. JAMA 2004, 291, 1875–1879. [Google Scholar] [CrossRef] [PubMed]
- Levin, L.I.; ScD, K.L.M.; O’Reilly, E.J.; Falk, K.I.; Ascherio, A. Primary infection with the epstein-barr virus and risk of multiple sclerosis. Ann. Neurol. 2010, 67, 824–830. [Google Scholar] [CrossRef] [PubMed]
- Handel, A.E.; Williamson, A.J.; Disanto, G.; Handunnetthi, L.; Giovannoni, G.; Ramagopalan, S.V. An Updated Meta-Analysis of Risk of Multiple Sclerosis following Infectious Mononucleosis. PLoS ONE 2010, 5, e12496. [Google Scholar] [CrossRef]
- Mokry, L.E.; Ross, S.; Ahmad, O.S.; Forgetta, V.; Smith, G.D.; Leong, A.; Greenwood, C.M.T.; Thanassoulis, G.; Richards, J.B. Vitamin D and Risk of Multiple Sclerosis: A Mendelian Randomization Study. PLoS Med. 2015, 12, e1001866. [Google Scholar] [CrossRef]
- Langer-Gould, A.; Lucas, R.; Xiang, A.H.; Chen, L.H.; Wu, J.; Gonzalez, E.; Haraszti, S.; Smith, J.B.; Quach, H.; Barcellos, L.F. MS Sunshine Study: Sun Exposure but Not Vitamin D Is Associated with Multiple Sclerosis Risk in Blacks and Hispanics. Nutrients 2018, 10, 268. [Google Scholar] [CrossRef]
- Hedström, A.K.; Bomfim, I.L.; Barcellos, L.; Gianfrancesco, M.; Schaefer, C.; Kockum, I.; Olsson, T.; Alfredsson, L. Interaction between adolescent obesity and HLA risk genes in the etiology of multiple sclerosis. Neurology 2014, 82, 865–872. [Google Scholar] [CrossRef]
- Handel, A.E.; Williamson, A.J.; Disanto, G.; Dobson, R.; Giovannoni, G.; Ramagopalan, S.V. Smoking and Multiple Sclerosis: An Updated Meta-Analysis. PLoS ONE 2011, 6, e16149. [Google Scholar] [CrossRef] [PubMed]
- Hedström, A.K.; Hillert, J.; Olsson, T.; Alfredsson, L. Nicotine might have a protective effect in the etiology of multiple sclerosis. Mult. Scler. J. 2013, 19, 1009–1013. [Google Scholar] [CrossRef] [PubMed]
- Ebers, G.C.; Sadovnick, A.D.; Risch, N.J. A genetic basis for familial aggregation in multiple sclerosis. Nat. Cell Biol. 1995, 377, 150–151. [Google Scholar] [CrossRef]
- Briggs, F.B.; Yu, J.C.; Davis, M.F.; Jiangyang, J.; Fu, S.; Parrotta, E.; Gunzler, D.D.; Ontaneda, D. Multiple sclerosis risk factors contribute to onset heterogeneity. Mult. Scler. Relat. Disord. 2019, 28, 11–16. [Google Scholar] [CrossRef]
- The International Multiple Sclerosis Genetics Consortium. Class II HLA interactions modulate genetic risk for multiple sclerosis. Nat. Genet. 2015, 47, 1107–1113. [Google Scholar] [CrossRef] [PubMed]
- International Multiple Sclerosis Genetics Consortium. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019, 365, eaav7188. [Google Scholar] [CrossRef] [PubMed]
- Baranzini, S.E.; Oksenberg, J.R. The Genetics of Multiple Sclerosis: From 0 to 200 in 50 Years. Trends Genet. 2017, 33, 960–970. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Vidal-Jordana, A.; Montalban, X. Multiple sclerosis: Clinical aspects. Curr. Opin. Neurol. 2018, 31, 752–759. [Google Scholar] [CrossRef]
- Omerhoca, S.; Akkas, S.Y.; Icen, N.K. Multiple sclerosis: Diagnosis and Differrential Diagnosis. Arch. Neuropsychiatry 2018, 55, S1–S9. [Google Scholar] [CrossRef] [PubMed]
- Brand, J.; Köpke, S.; Kasper, J.; Rahn, A.; Backhus, I.; Poettgen, J.; Stellmann, J.-P.; Siemonsen, S.; Heesen, C. Magnetic Resonance Imaging in Multiple Sclerosis – Patients’ Experiences, Information Interests and Responses to an Education Programme. PLoS ONE 2014, 9, e113252. [Google Scholar] [CrossRef]
- Ntranos, A.; Lublin, F. Diagnostic Criteria, Classification and Treatment Goals in Multiple Sclerosis: The Chronicles of Time and Space. Curr. Neurol. Neurosci. Rep. 2016, 16, 90. [Google Scholar] [CrossRef]
- Frohman, E.M.; Shah, A.; Eggenberger, E.; Metz, L.; Zivadinov, R.; Stüve, O. Corticosteroids for multiple sclerosis: I. Application for treating exacerbations. Neurother. 2007, 4, 618–626. [Google Scholar] [CrossRef]
- Tumani, H. Corticosteroids and plasma exchange in multiple sclerosis. J. Neurol. 2008, 255, 36–42. [Google Scholar] [CrossRef]
- Goodin, D.S. Glucocorticoid Treatment of Multiple Sclerosis; Elsevier BV: Amsterdam, The Netherlands, 2014; Volume 122, pp. 455–464. [Google Scholar]
- Filippini, G.; Brusaferri, F.; Sibley, W.A.; Citterio, A.; Ciucci, G.; Midgard, R.; Candelise, L. Corticosteroids or ACTH for acute exacerbations in multiple sclerosis. Cochrane Database Syst. Rev. 2000, 4, CD001331. [Google Scholar] [CrossRef]
- Wingerchuk, D.M.; Carter, J.L. Multiple Sclerosis: Current and Emerging Disease-Modifying Therapies and Treatment Strategies. Mayo Clin. Proc. 2014, 89, 225–240. [Google Scholar] [CrossRef] [PubMed]
- Beck, R.W.; Cleary, P.A.; Anderson, M.M.; Keltner, J.L.; Shults, W.T.; Kaufman, D.I.; Buckley, E.G.; Corbett, J.J.; Kupersmith, M.J.; Miller, N.R.; et al. A Randomized, Controlled Trial of Corticosteroids in the Treatment of Acute Optic Neuritis. N. Engl. J. Med. 1992, 326, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Linker, R.A.; Chan, A.; Sommer, M.; Koziolek, M.; Müller, G.-A.; Paulus, W.; Gold, R. Plasma exchange therapy for steroid-refractory superimposed relapses in secondary progressive multiple sclerosis. J. Neurol. 2007, 254, 1288–1289. [Google Scholar] [CrossRef] [PubMed]
- Weinshenker, B.G.; O’Brien, P.C.; Petterson, T.M.; Noseworthy, J.H.; Lucchinetti, C.F.; Dodick, D.W.; Pineda, A.A.; Stevens, L.N.; Rodriguez, M. A randomized trial of plasma exchange in acute central nervous system inflammatory demyelinating disease. Ann. Neurol. 1999, 46, 878–886. [Google Scholar] [CrossRef]
- Multiple Sklerose Therapie Konsensus Gruppe (MSTKG); Rieckmann, P. Immunmodulatorische Stufentherapie der Multiplen Sklerose. Der Nervenarzt 2006, 77, 1506–1518. [Google Scholar] [CrossRef]
- Dhib-Jalbut, S. Mechanisms of action of interferons and glatiramer acetate in multiple sclerosis. Neurology 2002, 58, S3–S9. [Google Scholar] [CrossRef]
- Stockman, J. Effect of early versus delayed interferon beta-1b treatment on disability after a first clinical event suggestive of multiple sclerosis: A 3-year follow-up analysis of the BENEFIT study. Yearb. Pediatr. 2009, 2009, 394–396. [Google Scholar] [CrossRef]
- Kappos, L.; Polman, C.H.; Freedman, M.S.; Edan, G.; Hartung, H.P.; Miller, D.H.; Montalban, X.; Barkhof, F.; Bauer, L.; Jakobs, P.; et al. Treatment with interferon beta-1b delays conversion to clinically definite and McDonald MS in patients with clinically isolated syndromes. Neurology 2006, 67, 1242–1249. [Google Scholar] [CrossRef]
- Jacobs, L.D.; Beck, R.W.; Simon, J.H.; Kinkel, R.P.; Brownscheidle, C.M.; Murray, T.J.; Simonian, N.A.; Slasor, P.J.; Sandrock, A.W. Intramuscular Interferon Beta-1A Therapy Initiated during a First Demyelinating Event in Multiple Sclerosis. N. Engl. J. Med. 2000, 343, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Comi, G.; Martinelli, V.; Rodegher, M.; Moiola, L.; Bajenaru, O.; Carra, A.; Elovaara, I.; Fazekas, F.; Hartung, H.; Hillert, J.; et al. Effect of glatiramer acetate on conversion to clinically definite multiple sclerosis in patients with clinically isolated syndrome (PreCISe study): A randomised, double-blind, placebo-controlled trial. Lancet 2009, 374, 1503–1511. [Google Scholar] [CrossRef]
- Kappos, L.; Weinshenker, B.; Pozzilli, C.; Thompson, A.J.; Dahlke, F.; Beckmann, K.; Polman, C.; McFarland, H. Interferon beta-1b in secondary progressive MS: A combined analysis of the two trials. Neurology 2004, 63, 1788–1795. [Google Scholar] [CrossRef] [PubMed]
- The North American Study Group on Interferon beta-1b in Secondary Progressive MS. Interferon beta-1b in secondary progressive MS: Results from a 3-year controlled study. Neurology 2004, 63, 1788–1795. [Google Scholar] [CrossRef]
- Secondary Progressive Efficacy Clinical Trial of Recombinant Interferon-Beta-1a in MS (SPECTRIMS) Study Group. Randomized controlled trial of interferon- beta-1a in secondary progressive MS: Clinical results. Neurology 2001, 56, 1496–1504. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. Natalizumab for Multiple Sclerosis. N. Engl. J. Med. 2007, 356, 2622–2629. [Google Scholar] [CrossRef] [PubMed]
- Polman, C.H.; O’Connor, P.W.; Havrdova, E.; Hutchinson, M.; Kappos, L.; Miller, D.H.; Phillips, J.T.; Lublin, F.D.; Giovannoni, G.; Wajgt, A.; et al. A Randomized, Placebo-Controlled Trial of Natalizumab for Relapsing Multiple Sclerosis. N. Engl. J. Med. 2006, 354, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Rudick, R.A.; Stuart, W.H.; Calabresi, P.A.; Confavreux, C.; Galetta, S.L.; Radue, M.S.E.R.S.E.; Lublin, F.D.; Weinstock-Guttman, B.; Wynn, D.R.; Lynn, F.; et al. Natalizumab plus Interferon Beta-1a for Relapsing Multiple Sclerosis. N. Engl. J. Med. 2006, 354, 911–923. [Google Scholar] [CrossRef] [PubMed]
- Freedman, M.S. Present and Emerging Therapies for Multiple Sclerosis. Contin. Lifelong Learn. Neurol. 2013, 19, 968–991. [Google Scholar] [CrossRef]
- Graff-Radford, J.; Robinson, M.T.; Warsame, R.M.; Matteson, E.L.; Eggers, S.D.Z.; Keegan, B.M. Progressive Multifocal Leukoencephalopathy in a Patient Treated With Etanercept. Neurology 2012, 18, 85–87. [Google Scholar] [CrossRef]
- Kleinschmidt-DeMasters, B.; Tyler, K.L. Progressive Multifocal Leukoencephalopathy Complicating Treatment with Natalizumab and Interferon Beta-1a for Multiple Sclerosis. N. Engl. J. Med. 2005, 353, 369–374. [Google Scholar] [CrossRef]
- Major, E.O. Progressive Multifocal Leukoencephalopathy in Patients on Immunomodulatory Therapies. Annu. Rev. Med. 2010, 61, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Claussen, M.C.; Korn, T. Immune mechanisms of new therapeutic strategies in MS — Teriflunomide. Clin. Immunol. 2012, 142, 49–56. [Google Scholar] [CrossRef]
- Tanasescu, R.; Constantinescu, C.S. Pharmacokinetic evaluation of fingolimod for the treatment of multiple sclerosis. Expert Opin. Drug Metab. Toxicol. 2014, 10, 621–630. [Google Scholar] [CrossRef]
- Oh, J.; O’Connor, P.W. Teriflunomide. Neurol. Clin. Pract. 2013, 3, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Radue, E.-W.; O’Connor, P.; Polman, C.; Hohlfeld, R.; Calabresi, P.; Selmaj, K.; Agoropoulou, C.; Leyk, M.; Zhang-Auberson, L.; et al. A Placebo-Controlled Trial of Oral Fingolimod in Relapsing Multiple Sclerosis. N. Engl. J. Med. 2010, 362, 387–401. [Google Scholar] [CrossRef]
- Cohen, J.A.; Barkhof, F.; Comi, G.; Hartung, H.-P.; Khatri, B.O.; Montalban, X.; Pelletier, J.; Capra, R.; Gallo, P.; Izquierdo, G.; et al. Oral Fingolimod or Intramuscular Interferon for Relapsing Multiple Sclerosis. N. Engl. J. Med. 2010, 362, 402–415. [Google Scholar] [CrossRef]
- Cohen, J.A.; Chun, J. Mechanisms of fingolimod’s efficacy and adverse effects in multiple sclerosis. Ann. Neurol. 2011, 69, 759–777. [Google Scholar] [CrossRef] [PubMed]
- Noda, H.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Fingolimod phosphate promotes the neuroprotective effects of microglia. J. Neuroimmunol. 2013, 256, 13–18. [Google Scholar] [CrossRef]
- Linker, R.A.; Lee, D.-H.; Ryan, S.; Van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011, 134, 678–692. [Google Scholar] [CrossRef]
- Fox, R.J.; Miller, D.H.; Phillips, J.T.; Hutchinson, M.; Havrdova, E.; Kita, M.; Yang, M.; Raghupathi, K.; Novas, M.; Sweetser, M.T.; et al. Placebo-Controlled Phase 3 Study of Oral BG-12 or Glatiramer in Multiple Sclerosis. N. Engl. J. Med. 2012, 367, 1087–1097. [Google Scholar] [CrossRef]
- Gold, R.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Tornatore, C.; Sweetser, M.T.; Yang, M.S.M.; Sheikh, S.I.; et al. Placebo-Controlled Phase 3 Study of Oral BG-12 for Relapsing Multiple Sclerosis. N. Engl. J. Med. 2012, 367, 1098–1107. [Google Scholar] [CrossRef]
- Hartung, H.-P.; Gonsette, R.; Konig, N.; Kwiecinski, H.; Guseo, A.; Morrissey, S.P.; Krapf, H.; Zwingers, T. Mitoxantrone in progressive multiple sclerosis: A placebo-controlled, double-blind, randomised, multicentre trial. Lancet 2002, 360, 2018–2025. [Google Scholar] [CrossRef]
- Marriott, J.J.; Miyasaki, J.M.; Gronseth, G.; O’Connor, P.W. Evidence Report: The efficacy and safety of mitoxantrone (Novantrone) in the treatment of multiple sclerosis: Report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2010, 74, 1463–1470. [Google Scholar] [CrossRef]
- Gerardi, C.; Bertele’, V.; Rossi, S.; Garattini, S.; Banzi, R. Preapproval and postapproval evidence on drugs for multiple sclerosis. Neurology 2018, 90, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Brennan, M.S.; Matos, M.F.; Richter, K.E.; Li, B.; Scannevin, R.H. The NRF2 transcriptional target, OSGIN1, contributes to monomethyl fumarate-mediated cytoprotection in human astrocytes. Sci. Rep. 2017, 7, srep42054. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.L.; Van Der Pol, S.M.; Di Dio, F.; Hof, B.V.H.; Kooij, G.; De Vries, H.E.; Van Horssen, J. Protective effects of monomethyl fumarate at the inflamed blood–brain barrier. Microvasc. Res. 2016, 105, 61–69. [Google Scholar] [CrossRef]
- Mazzola, M.A.; Raheja, R.; Regev, K.; Beynon, V.; Von Glehn, F.; Paul, A.; Pierre, I.; Kivisakk, P.; Weiner, H.L.; Gandhi, R. Monomethyl fumarate treatment impairs maturation of human myeloid dendritic cells and their ability to activate T cells. Mult. Scler. J. 2019, 25, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Reeta, K.; Sharma, U.; Jagannathan, N.; Dinda, A.; Gupta, Y. Neuro-protective effect of monomethyl fumarate on ischemia reperfusion injury in rats: Role of Nrf2/HO1 pathway in peri-infarct region. Neurochem. Int. 2019, 126, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Hartsock, M.J.; Xu, Z.; He, M.; Duh, E.J. Monomethyl fumarate promotes Nrf2-dependent neuroprotection in retinal ischemia-reperfusion. J. Neuroinflamm. 2015, 12, 1–12. [Google Scholar] [CrossRef]
- Horton, M.; Rudick, R.A.; Hara-Cleaver, C.; Marrie, R.A. Validation of a Self-Report Comorbidity Questionnaire for Multiple Sclerosis. Neuroepidemiology 2010, 35, 83–90. [Google Scholar] [CrossRef]
- Kappos, L.; Gold, R.; Miller, D.H.; MacManus, D.G.; Havrdova, E.; Limmroth, V.; Polman, C.H.; Schmierer, K.; Yousry, A.T.; Yang, M.; et al. Efficacy and safety of oral fumarate in patients with relapsing-remitting multiple sclerosis: A multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet 2008, 372, 1463–1472. [Google Scholar] [CrossRef]
- Gold, R.; Arnold, D.L.; Bar-Or, A.; Hutchinson, M.; Kappos, L.; Havrdova, E.; MacManus, D.G.; Yousry, A.T.; Pozzilli, C.; Selmaj, K.; et al. Long-term effects of delayed-release dimethyl fumarate in multiple sclerosis: Interim analysis of ENDORSE, a randomized extension study. Mult. Scler. J. 2016, 21, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Gold, R.; Miller, D.H.; MacManus, D.G.; Havrdova, E.; Limmroth, V.; Polman, C.H.; Schmierer, K.; Yousry, A.T.; Eraksoy, M.; et al. Effect of BG-12 on contrast-enhanced lesions in patients with relapsing– remitting multiple sclerosis: Subgroup analyses from the phase 2b study. Mult. Scler. J. 2011, 18, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Gold, R.; Giovannoni, G.; Phillips, J.T.; Fox, R.J.; Zhang, A.; Meltzer, L.; Kurukulasuriya, N.C. Efficacy and safety of delayed-release dimethyl fumarate in patients newly diagnosed with relapsing–remitting multiple sclerosis (RRMS). Mult. Scler. J. 2014, 21, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Kita, M.; Fox, R.J.; Phillips, J.T.; Hutchinson, M.; Havrdova, E.K.; Sarda, S.P.; Agarwal, S.; Kong, J.; Zhang, A.; Viglietta, V.; et al. Effects of BG-12 (dimethyl fumarate) on health-related quality of life in patients with relapsing–remitting multiple sclerosis: Findings from the CONFIRM study. Mult. Scler. J. 2013, 20, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Gold, R.; Arnold, D.L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Sarda, S.P.; Agarwal, S.; Zhang, A.; Sheikh, S.I.; et al. Quality of life outcomes with BG-12 (dimethyl fumarate) in patients with relapsing–remitting multiple sclerosis: The DEFINE study. Mult. Scler. J. 2014, 20, 243–252. [Google Scholar] [CrossRef]
- Bar-Or, A.; Gold, R.; Kappos, L.; Arnold, D.L.; Giovannoni, G.; Selmaj, K.; O’Gorman, J.; Stephan, M.; Dawson, K.T. Clinical efficacy of BG-12 (dimethyl fumarate) in patients with relapsing–remitting multiple sclerosis: Subgroup analyses of the DEFINE study. J. Neurol. 2013, 260, 2297–2305. [Google Scholar] [CrossRef]
- Gold, R.; Arnold, D.L.; Bar-Or, A.; Fox, R.J.; Kappos, L.; Chen, C.; Parks, B.; Miller, C. Safety and efficacy of delayed-release dimethyl fumarate in patients with relapsing-remitting multiple sclerosis: 9 years’ follow-up of DEFINE, CONFIRM, and ENDORSE. Ther. Adv. Neurol. Disord. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Prosperini, L.; Pontecorvo, S. Dimethyl fumarate in the management of multiple sclerosis: Appropriate patient selection and special considerations. Ther. Clin. Risk Manag. 2016, 12, 339–350. [Google Scholar] [CrossRef]
- Wynn, D.; Lategan, T.W.; Sprague, T.N.; Rousseau, F.S.; Fox, E.J. Monomethyl fumarate has better gastrointestinal tolerability profile compared with dimethyl fumarate. Mult. Scler. Relat. Disord. 2020, 45, 102335. [Google Scholar] [CrossRef] [PubMed]
- Wehr, A.; Hard, M.; Yu, M.; Leigh-Pemberton, R.; von Moltke, L. Relative Bioavailability of Monomethyl Fumarate after Administration of ALKS 8700 and Dimethyl Fumarate in Healthy Subjects. Neurology 2018, 90, P1.403. [Google Scholar]
- Schimrigk, S.; Brune, N.; Hellwig, K.; Lukas, C.; Bellenberg, B.; Rieks, M.; Hoffmann, V.; Pohlau, D.; Przuntek, H. Oral fumaric acid esters for the treatment of active multiple sclerosis: An open-label, baseline-controlled pilot study. Eur. J. Neurol. 2006, 11, 604–610. [Google Scholar] [CrossRef] [PubMed]
- MacManus, D.G.; Miller, D.H.; Kappos, L.; Gold, R.; Havrdova, E.; Limmroth, V.; Polman, C.H.; Schmierer, K.; Yousry, T.A.; Eraksoy, M.; et al. BG-12 reduces evolution of new enhancing lesions to T1-hypointense lesions in patients with multiple sclerosis. J. Neurol. 2011, 258, 449–456. [Google Scholar] [CrossRef]
- Ryerson, L.Z.; Green, R.; Confident, G.; Pandey, K.; Richter, B.; Bacon, T.; Sammarco, C.; Laing, L.; Kalina, J.; Kister, I. Efficacy and tolerability of dimethyl fumarate in White-, African- and Hispanic- Americans with multiple sclerosis. Ther. Adv. Neurol. Disord. 2016, 9, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Hersh, C.M.; Love, T.E.; Cohn, S.; Hara-Cleaver, C.; Bermel, R.A.; Fox, R.J.; Cohen, J.A.; Ontaneda, D. Comparative efficacy and discontinuation of dimethyl fumarate and fingolimod in clinical practice at 12-month follow-up. Mult. Scler. Relat. Disord. 2016, 10, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Alroughani, R.; Ahmed, S.F.; Behbehani, R.; Al-Hashel, J. Effectiveness and Safety of Dimethyl Fumarate Treatment in Relapsing Multiple Sclerosis Patients: Real-World Evidence. Neurol. Ther. 2017, 6, 189–196. [Google Scholar] [CrossRef][Green Version]
- Vollmer, B.; Ontaneda, D.; Bandyopadhyay, A.; Cohn, S.; Nair, K.; Sillau, S.; Bermel, R.A.; Corboy, J.R.; Fox, R.J.; Vollmer, T.; et al. Discontinuation and comparative effectiveness of dimethyl fumarate and fingolimod in 2 centers. Neurol. Clin. Pract. 2018, 8, 292–301. [Google Scholar] [CrossRef]
- Laplaud, D.-A.; Casey, R.; Barbin, L.; Debouverie, M.; De Sèze, J.; Brassat, D.; Wiertlewski, S.; Brochet, B.; Pelletier, J.; Vermersch, P.; et al. Comparative effectiveness of teriflunomide vs dimethyl fumarate in multiple sclerosis. Neurology 2019, 93, e635–e646. [Google Scholar] [CrossRef]
- Lanzillo, R.; Moccia, M.; Palladino, R.; Signoriello, E.; Carotenuto, A.; Maniscalco, G.; Saccà, F.; Bonavita, S.; Russo, C.; Iodice, R.; et al. Clinical predictors of Dimethyl Fumarate response in multiple sclerosis: A real life multicentre study. Mult. Scler. Relat. Disord. 2020, 38, 101871. [Google Scholar] [CrossRef]
- Fox, E.J.; Vasquez, A.; Grainger, W.; Ma, T.S.; Von Hehn, C.; Walsh, J.; Li, J.; Zambrano, J. Gastrointestinal Tolerability of Delayed-Release Dimethyl Fumarate in a Multicenter, Open-Label Study of Patients with Relapsing Forms of Multiple Sclerosis (MANAGE). Int. J. MS Care 2016, 18, 9–18. [Google Scholar] [CrossRef] [PubMed]
- E Longbrake, E.; Naismith, R.T.; Parks, B.J.; Wu, G.F.; Cross, A.H. Dimethyl fumarate-associated lymphopenia: Risk factors and clinical significance. Mult. Scler. J. Exp. Transl. Clin. 2015, 1, 205521731559699. [Google Scholar] [CrossRef]
- Berkovich, R.; Weiner, L.P. Effects of dimethyl fumarate on lymphocyte subsets. Mult. Scler. Relat. Disord. 2015, 4, 339–341. [Google Scholar] [CrossRef]
- Van Oosten, B.W.; Killestein, J.; Barkhof, F.; Polman, C.H.; Wattjes, M.P. PML in a Patient Treated with Dimethyl Fumarate from a Compounding Pharmacy. N. Engl. J. Med. 2013, 368, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Nieuwkamp, D.J.; Cremers, C.H.; Frijlink, D.W.; Van Oosten, B.W.; Murk, J.-L.; Killestein, J.; Viveen, M.C.; Van Hecke, W.; Wattjes, M.P. PML in a Patient without Severe Lymphocytopenia Receiving Dimethyl Fumarate. N. Engl. J. Med. 2015, 372, 1474–1476. [Google Scholar] [CrossRef]
- Sabin, J.; DMF Study Group; Urtiaga, S.; Pilo, B.; Thuissard, I.; Galan, V.; De La Maza, S.S.; Costa-Frossard, L.; Gómez-Moreno, M.; Díaz-Díaz, J.; et al. Tolerability and safety of dimethyl fumarate in relapsing multiple sclerosis: A prospective observational multicenter study in a real-life Spanish population. J. Neurol. 2020, 267, 2362–2371. [Google Scholar] [CrossRef]
- A Muñoz, M.; Kulick, C.G.; Kortepeter, C.M.; Levin, R.L.; I Avigan, M. Liver injury associated with dimethyl fumarate in multiple sclerosis patients. Mult. Scler. J. 2017, 23, 1947–1949. [Google Scholar] [CrossRef]
- Gold, R.; Phillips, J.T.; Havrdova, E.; Bar-Or, A.; Kappos, L.; Kim, N.; Thullen, T.; Valencia, P.; Oliva, L.; Novas, M.; et al. Delayed-Release Dimethyl Fumarate and Pregnancy: Preclinical Studies and Pregnancy Outcomes from Clinical Trials and Postmarketing Experience. Neurol. Ther. 2015, 4, 93–104. [Google Scholar] [CrossRef]
- Everage, N.J.; Jones, C.C.; Hellwig, K.; Rog, D.; Liu, S.; Mou, J.; Prada, C.; Hanna, J. Pregnancy Outcomes from an International Registry of Patients Treated with Delayed-release Dimethyl Fumarate. Neurology 2019, 92, P4.2-095. [Google Scholar] [CrossRef]
- Mallucci, G.; Annovazzi, P.; Miante, S.; Torri-Clerici, V.; Matta, M.; La Gioia, S.; Cavarretta, R.; Mantero, V.; Costantini, G.; D’Ambrosio, V.; et al. Two-year real-life efficacy, tolerability and safety of dimethyl fumarate in an Italian multicentre study. J. Neurol. 2018, 265, 1850–1859. [Google Scholar] [CrossRef] [PubMed]
- Naismith, R.T.; The EVOLVE-MS-2 Study Group; Wundes, A.; Ziemssen, T.; Jasinska, E.; Freedman, M.S.; Lembo, A.J.; Selmaj, K.; Bidollari, I.; Chen, H.; et al. Diroximel Fumarate Demonstrates an Improved Gastrointestinal Tolerability Profile Compared with Dimethyl Fumarate in Patients with Relapsing-Remitting Multiple Sclerosis: Results from the Randomized, Double-Blind, Phase III EVOLVE-MS-2 Study. CNS Drugs 2020, 34, 185–196. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Author (Year) | Groups Studied and Intervention | Results and Findings | Conclusions |
|---|---|---|---|
| Schimrigk et al. 2006 [97] Phase I | 10 patients with definite RRMS, relapse in the year prior to enrollment, active lesion on MRI, active EDSS score 2.0–6.0; oral FAE * (Fumaderm initial®, Fumaderm forte®) 720 mg/day for 18 weeks, followed by 360 mg/day for 48 weeks | Reductions in mean number and total volume of gadolinium enhancing lesions (GdE) on T1 MRI after 18 weeks of treatment | Fumaric acid esters reduced radiologic progression of MS lesions in a small group of patients. Some FAE preparations contain more than 55% DMF and may be useful for RRMS patients. |
| Kappos et al. 2008 [86] Phase IIb | 257 patients with RRMS; 120 mg of DMF QD, 120 mg of DMF TID, 240 mg of DMF TID, or placebo. | DMF 240 TID reduced mean number of GdE lesions (69%) over a 12 week period, number of new or enlarging T2-hyperintense lesions, new T1-hypointense lesions, and annual relapse rate (32%). | DMF can reduce radiologic progression of disease in RRMS patients. Consider DMF 240 mg TID for prevention of radiological progression of disease in RRMS patients. |
| Gold et al. 2012 [76] DEFINE Phase III | 1234 RRMS patients; 240 mg DMF twice daily, 240 mg DMF thrice daily, or placebo. | Significantly lower estimated proportion of relapse: 27% in patients taking BID DMF, 26% taking TID DMF, and 46% with placebo. Annualized relapse rate at 2 years: 0.17 with BID, 0.19 with TID, and 0.36 with placebo. Rate of disability progression: 16% with BID, 18% with TID, and 27% with placebo. Significant reduction in number of Gd+ T2 MRI hyperintensities with each DMF regimen compared to placebo. | DMF regimens reduced MS relapses and imaging findings compared to placebo. Consider 240 mg DMF twice or thrice daily for MS patients unable to tolerate other MS treatments. |
| Malllucci et al. 2018 [114] Phase IV | Records of 720 MS patients (478 female) treated with DMF: 25.8% treatment-naïve; 19.5% discontinued another DMF treatment >12 months prior; 54.6% switched from another disease-modifying treatment (DMT): (IFN (45.8%), GA (27.2%), TFU (5.8%), FTY (7.3%), NTZ (6.6%). Median DMF exposure 17 months. | Reduction in ARR by 63.2% (mean ARR before DMF vs. mean ARR at least follow-up). 85% of patients relapse-free at 12 months, 76% of patients relapse-free at 24 months. 89% continued DMF at 12 months, and 70% continued DMF at 24 months. | DMF may be considered in patients who must switch from another disease-modifying therapy due to tolerance issues, lack of efficacy, or safety concerns. |
| Sabin et al. 2020 [110] Phase IV | 886 MS patients (629 female) treated with DMF: 25.3% treatment-naïve; 74.7% switched from another DMT. Median exposure 39.5 months. 56.2% completed at least 3 years DMF treatment. | Tolerability and safety study. 71.2% experienced adverse events (flushing 44.1%, grade III lymphopenia 5.4%). 11.7% discontinued in the first year. No safety problems reported. | DMF may be considered a generally safe alternative to existing DMT for RRMS patients. Acknowledge that adverse effects are relatively common and there may occasionally be the need for discontinuation. |
| Gold et al. 2020 [93] ENDORSE Phase IV | 1736 patients taking 240 mg of DMF who completed CONFIRM/DEFINE. Patients having taken GA or TID DMF excluded. Median follow-up 8.5 years (range 2.0–11.3). | ARR remained low (<0.20) over ~9 year treatment period. Approximately 70% patients had no new or enlarging MRI lesions compared to baseline after 7 years of DMF treatment. Of 2470 patients had ≥ lymphocyte assessment, 53 developed severe prolonged lymphopenia. | There is support for long-term safety and efficacy of DMF in RRMS patients. |
| Author (Year) | Groups Studied and Intervention | Results and Findings | Conclusions |
|---|---|---|---|
| Fox et al. 2012 [75] CONFIRM Phase III | 1417 RRMS patients; DMF 240 mg BID, DMF 240 mg TID, glatiramer acetate subcutaneous 20 mg daily, or placebo. | Annualized relapse rate at 2 years: 0.22 with BID DMF, 0.20 with TID DMF, 0.29 with glatiramer acetate, and 0.40 with placebo. Significant reduction in number of new or enlarging T2 hyperintensities, and new T1 hypointensities. No significant reductions in disability progression comparing DMF regimens with glatiramer acetate. | Both DMF and glatiramer acetate reduced relapse rates and neuroradiologic progression of disease compared to placebo. No significant difference |
| Gold et al. 2017 [87] ENDORSE Phase IV | 1736 patients who completed CONFIRM/DEFINE: All dose combinations represented (DMF BID and TID, placebo, and GA). | Cumulative ARR during 0–5 years for patients taking BID/BID was 0.163, versus patients taking placebo/BID 0.240. For the GA/BID patients, cumulative ARR was 0.199. | DMF treatment is associated with sustained low clinical disease activity and MRI progression. Treatment benefit may be sustained and safety profile may be favorable long-term. |
| Wehr et al. 2018 [96] Phase I | Direct pharmacokinetic comparison of monomethyl fumarate (MMF) and DMF. 35 healthy fasting volunteers, a single dose of 462 mg of MMF versus a single dose of 240 mg of DMF. | MMF was well-tolerated. Comparable mean concentrations of MMF and DMF over time. Adverse events 45.7% with MMF, and 54.3% with DMF. | The pharmacokinetic profiles of MMF and DMF are similar; the substances may be considered bioequivalent. |
| Naismith et al. 2020 [115] Phase III | 504 patients with RRMS, randomized to diroximel fumarate (DRF) or DMF. BID 231 mg of DRF or BID 120 mg of DMF for 1 week; then BID 462 mg of DRF and BID 240 mg of DMF for 4 weeks. Tolerability and symptoms assessed by patient self-report. | Significantly reduction (46%) in Individual Gastrointestinal Symptom and Impact Scale (IGISIS) scores with DRF compared to DMF. Fewer gastrointestinal adverse events with DRF (34.8%) than with DMF (49.0%), and fewer discontinued DRF (1.6%) compared to DMF (5.6%). | DRF may have better short-term gastrointestinal tolerability than DMF. |
| Laplaud et al. 2019 [103] phase IV | 1770 RRMS patients: 713 patients taking teriflunomide (TRF), 1057 taking DMF, evaluated at 2 years of treatment. | Adjusted proportion of patients with at least one new T2 lesion after 2 years of treatment, 60.8% for DMF, 72.2% for TRF. More patients were withdrawn from TRF (14.5%) than from DMF (8.5%) due to lack of effectiveness. | Class III evidence that TRF and DMF have similar clinical effectiveness for RRMS patients at 2 years. The larger patient population of this study may better reflect real-life MRI progression with DMF treatment. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berger, A.A.; Sottosanti, E.R.; Winnick, A.; Izygon, J.; Berardino, K.; Cornett, E.M.; Kaye, A.D.; Varrassi, G.; Viswanath, O.; Urits, I. Monomethyl Fumarate (MMF, Bafiertam) for the Treatment of Relapsing Forms of Multiple Sclerosis (MS). Neurol. Int. 2021, 13, 207-223. https://doi.org/10.3390/neurolint13020022
Berger AA, Sottosanti ER, Winnick A, Izygon J, Berardino K, Cornett EM, Kaye AD, Varrassi G, Viswanath O, Urits I. Monomethyl Fumarate (MMF, Bafiertam) for the Treatment of Relapsing Forms of Multiple Sclerosis (MS). Neurology International. 2021; 13(2):207-223. https://doi.org/10.3390/neurolint13020022
Chicago/Turabian StyleBerger, Amnon A., Emily R. Sottosanti, Ariel Winnick, Jonathan Izygon, Kevin Berardino, Elyse M. Cornett, Alan D. Kaye, Giustino Varrassi, Omar Viswanath, and Ivan Urits. 2021. "Monomethyl Fumarate (MMF, Bafiertam) for the Treatment of Relapsing Forms of Multiple Sclerosis (MS)" Neurology International 13, no. 2: 207-223. https://doi.org/10.3390/neurolint13020022
APA StyleBerger, A. A., Sottosanti, E. R., Winnick, A., Izygon, J., Berardino, K., Cornett, E. M., Kaye, A. D., Varrassi, G., Viswanath, O., & Urits, I. (2021). Monomethyl Fumarate (MMF, Bafiertam) for the Treatment of Relapsing Forms of Multiple Sclerosis (MS). Neurology International, 13(2), 207-223. https://doi.org/10.3390/neurolint13020022