
Desmosomal Versus Non-Desmosomal Arrhythmogenic Cardiomyopathies: A State-of-the-Art Review

 , , , ,
, , , ,  , ,
, ,  ,
, 
Abstract
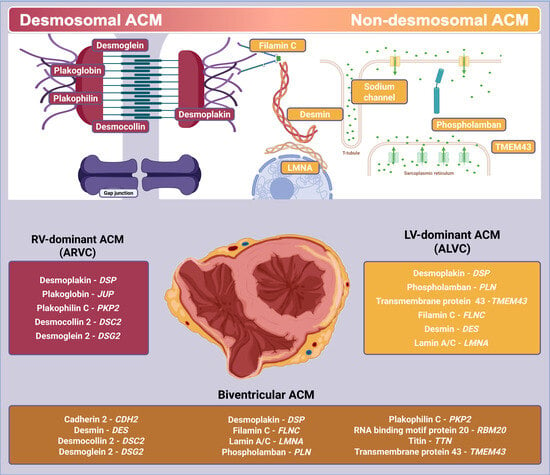
1. Introduction
2. Definitions
3. Genetic History
4. Pathophysiology
4.1. From Histopathology to Disease Manifestation
4.2. Translational Science Insights
5. Genotype–Phenotype Correlation
5.1. Desmosomal Variants
5.2. Non-Desmosomal Variants
5.3. Genotype-Negative–Phenotype-Positive Individuals
5.4. Genetically Undefined Forms and Polygenic Risk Scores
6. Therapeutic Management
6.1. Exercise Prescription
6.2. Current Management: From Drugs to Heart Transplant
6.3. Disease-Specific Therapies and Ongoing Trials
7. Prognosis
7.1. Desmosomal Genes
- DSG2: Zhang et al. reported that DSG2 pathogenic variants lead to cardiomyocyte loss and fibrosis, with early LV involvement, extensive necrosis, and persistent immune cell infiltration [54].
- PKP2: A study on 56 Polish patients found that PKP2 rare variant carriers had an earlier diagnosis (mean age 32 ± 11 years) compared to non-carriers (mean age 42 ± 12 years) [104].
- DSP: Smith et al. described a DSP cardiomyopathy cohort characterized by a higher prevalence in females, an average diagnostic age of 36 ± 16 years, and a predominantly LV phenotype, with increased clinical penetrance compared to PKP2-related ACM [105]. Recent data from the DSP-ERADOS Network, involving twenty-six academic institutions across nine countries, highlighted the distinct phenotype of DSP cardiomyopathy. Patients with P/LP-DSP genetic variants exhibit higher rates of sustained VAs and heart failure hospitalizations. Key adverse outcome predictors include prior sustained or NSVTs, TWI in ≥3 leads, LVEF ≤ 50%, and myocardial injury events [106].
7.2. Non-Desmosomal Genes
- LMNA: LMNA-associated cardiomyopathy, often linked to conduction disturbances and malignant VAs, is the second most common genetic cause of DCM after TTN [107]. A study by Wahbi et al. on 444 LMNA variant carriers identified male sex, missense mutations, first-degree or higher AV block, NSVTs, and ventricular dysfunction as predictors of life-threatening arrhythmic events [108].
- TMEM43: The TMEM43 1073C→T variant has been linked to a severe ACM phenotype with complete penetrance by the age of 63 years in males and 76 years in females, with males experiencing twice the disease risk [26]. Hodgkinson et al. reported that ARVC patients carrying the p.S358L TMEM43 variant benefited significantly from ICD implantation, particularly males. Over a median 6.3-year follow-up, sixty-five of eighty control males experienced ventricular tachycardia, fibrillation, or SCD versus thirteen of sixty-eight females, while among ICD-implanted males, the median time to first appropriate discharge was 11.1 years, whereas it was not reached for females [109].
- FLNC truncating variants: FLNC truncating variants are primarily associated with LV involvement, regardless of the expressed cardiomyopathy phenotype. Ortiz-Genga et al. described twenty-eight unrelated patients with FLNC truncating variants presenting with DCM, ACM, or restrictive cardiomyopathy, with predominant myocardial fibrosis in the LV wall and VAs in 82% of cases, including >500 premature ventricular contractions per day and NSVTs [110]. Gigli et al. reported eighty-five FLNC truncating variant carriers, 49% with DCM and 28% with ACM (25% ALVC, 3% ARVC), characterized by LGE distribution predominantly in the LV [111].
- PLN: The PLN gene is associated with both ACM and DCM. van der Zwaag et al. analyzed ninety-seven PLN R14del variant carriers with ARVC-like expression, noting a higher prevalence of arrhythmic events (appropriate ICD interventions or family history of SCD) in mutation carriers compared to non-carriers, although statistical significance was not reached [112].
7.3. Arrhythmic Risk Stratification
8. Gaps in Knowledge and Future Directions
9. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AAD | antiarrhythmic drug | HF | heart failure |
| AAV | adeno-associated viral | HRS | Heart Rhythm Society |
| ACM | arrhythmogenic cardiomyopathy | ICD | implantable cardioverter defibrillator |
| AHA | American Heart Association | JUP | plakoglobin |
| AI | artificial intelligence | LBBB | left bundle branch block |
| AJs | adherens junctions | LGE | late gadolinium enhancement |
| ALVC | arrhythmogenic left ventricular cardiomyopathy | LMNA | lamin A/C |
| ARVC | arrhythmogenic right ventricular cardiomyopathy | LV | left ventricular |
| ARVD | arrhythmogenic right ventricular dysplasia | MET-Hr/year | metabolic equivalent hours per year |
| BivACM | biventricular arrhythmogenic cardiomyopathy | miRNA | microRNA |
| βBs | beta-blockers | ncRNA | noncoding RNA |
| CA | catheter ablation | NDLVC | non-dilated left ventricular cardiomyopathy |
| CMR | cardiovascular magnetic resonance | NSVT | non-sustained ventricular tachycardia |
| Cx43 | connexin-43 | P | phenotype |
| DCM | dilated cardiomyopathy | P/LP | pathogenic/likely pathogenic |
| DES | desmin | PKP2 | plakophilin 2 |
| DSC2 | desmocollin 2 | PLN | phospholamban |
| DSG2 | desmoglein 2 | PPARγ | peroxisome proliferator-activated receptor-γ |
| DSP | desmoplakin | PRS | polygenic risk score |
| EMB | endomyocardial biopsy | RV | right ventricular |
| ESC | European Association of Cardiology | SCD | sudden cardiac death |
| FLNC | filamin C | TMEM43 | transmembrane protein 43 |
| G | genotype | TWI | T-wave inversion |
| GJs | gap junctions | VAs | ventricular arrhythmias |
| hiPSC-CMs | human induced pluripotent stem cell-derived cardiomyocytes | YAP | Yes-associated protein |
References
- Corrado, D.; Basso, C.; Judge, D.P. Arrhythmogenic Cardiomyopathy. Circ. Res. 2017, 121, 784–802. [Google Scholar] [CrossRef]
- Corrado, D.; Link, M.S.; Calkins, H. Arrhythmogenic Right Ventricular Cardiomyopathy. N. Engl. J. Med. 2017, 376, 61–72. [Google Scholar] [CrossRef]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.; Daubert, J.; et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed modification of the task force criteria. Circulation 2010, 121, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Sen-Chowdhry, S.; Syrris, P.; Prasad, S.K.; Hughes, S.E.; Merrifield, R.; Ward, D.; Pennell, D.J.; McKenna, W.J. Left-dominant arrhythmogenic cardiomyopathy: An under-recognized clinical entity. J. Am. Coll. Cardiol. 2008, 52, 2175–2187. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Basso, C.; Thiene, G.; McKenna, W.J.; Davies, M.J.; Fontaliran, F.; Nava, A.; Silvestri, F.; Blomstrom-Lundqvist, C.; Fontaine, G.; et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: A multicenter study. J. Am. Coll. Cardiol. 1997, 30, 1512–1520. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Graziano, F.; Bauce, B.; Bueno Marinas, M.; Calore, C.; Celeghin, R.; Cipriani, A.; De Gaspari, M.; De Lazzari, M.; Migliore, F.; et al. The ‘Padua classification’ of cardiomyopathies into three groups: Hypertrophic/restrictive, dilated/hypokinetic, and scarring/arrhythmogenic. Eur. Heart J. Suppl. 2025, 27 (Suppl. S1), i73–i82. [Google Scholar] [CrossRef]
- Basso, C.; Thiene, G.; Corrado, D.; Angelini, A.; Nava, A.; Valente, M. Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation 1996, 94, 983–991. [Google Scholar] [CrossRef]
- Thiene, G.; Nava, A.; Corrado, D.; Rossi, L.; Pennelli, N. Right Ventricular Cardiomyopathy and Sudden Death in Young People. N. Engl. J. Med. 1988, 318, 129–133. [Google Scholar] [CrossRef]
- Basso, C.; Bauce, B.; Corrado, D.; Thiene, G. Pathophysiology of arrhythmogenic cardiomyopathy. Nat. Rev. Cardiol. 2012, 9, 223–233. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C.; Pilichou, K.; Thiene, G. Molecular biology and clinical management of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart 2011, 97, 530–539. [Google Scholar] [CrossRef]
- Peters, S.; Trümmel, M.; Meyners, W. Prevalence of right ventricular dysplasia-cardiomyopathy in a non-referral hospital. Int. J. Cardiol. 2004, 97, 499–501. [Google Scholar] [CrossRef]
- Bomma, C.; Rutberg, J.; Tandri, H.; Nasir, K.; Roguin, A.; Tichnell, C.; Rodriguez, R.; James, C.; Kasper, E.; Spevak, P.; et al. Misdiagnosis of Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. J. Cardiovasc. Electrophysiol. 2004, 15, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Li, G.L.; Saguner, A.M.; Fontaine, G.H. Naxos disease: From the origin to today. Orphanet J. Rare Dis. 2018, 13, 74. [Google Scholar] [CrossRef]
- Norgett, E.E.; Hatsell, S.J.; Carvajal-Huerta, L.; Cabezas, J.C.; Common, J.; Purkis, P.E.; Whittock, N.; Leigh, I.M.; Stevens, H.P.; Kelsell, D.P. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum. Mol. Genet. 2000, 9, 2761–2766. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, M.; Song, H.; Wang, B.; Chen, H.; Wang, J.; Wang, W.; Feng, S.; Zhang, F.; Ju, W.; et al. Comprehensive analysis of desmosomal gene mutations in Han Chinese patients with arrhythmogenic right ventricular cardiomyopathy. Eur. J. Med. Genet. 2015, 58, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Hendricks, N.; Watkins, D.A.; Mayosi, B.M. Lessons from the first report of the Arrhythmogenic Right Ventricular Cardiomyopathy Registry of South Africa. Cardiovasc. J. Afr. 2010, 21, 129–130. [Google Scholar] [CrossRef]
- Marcus, F.I.; Fontaine, G.H.; Guiraudon, G.; Frank, R.; Laurenceau, J.L.; Malergue, C.; Grosgogeat, Y. Right ventricular dysplasia: A report of 24 adult cases. Circulation 1982, 65, 384–398. [Google Scholar] [CrossRef]
- Fontaine, G.; Frank, R.; Tonet, J.L.; Guiraudon, G.; Cabrol, C.; Chomette, G.; Grosgogeat, Y. Arrhythmogenic right ventricular dysplasia a clinical model for the study of chronic ventricular tachycardia. Jpn. Circ. J. 1984, 48, 515–538. [Google Scholar] [CrossRef]
- McKenna, W.J.; Thiene, G.; Nava, A.; Fontaliran, F.; Blomstrom-Lundqvist, C.; Fontaine, G.; Camerini, F. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Heart 1994, 71, 215–218. [Google Scholar] [CrossRef]
- Corrado, D.; Perazzolo Marra, M.; Zorzi, A.; Beffagna, G.; Cipriani, A.; De Lazzari, M.; Migliore, F.; Pilichou, K.; Rampazzo, A.; Rigato, I.; et al. Diagnosis of arrhythmogenic cardiomyopathy: The Padua criteria. Int. J. Cardiol. 2020, 319, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Anastasakis, A.; Basso, C.; Bauce, B.; Blomström-Lundqvist, C.; Bucciarelli-Ducci, C.; Cipriani, A.; De Asmundis, C.; Gandjbakhch, E.; Jimenez-Jaimez, J.; et al. Proposed diagnostic criteria for arrhythmogenic cardiomyopathy: European Task Force consensus report. Int. J. Cardiol. 2024, 395, 131447. [Google Scholar] [CrossRef] [PubMed]
- Nava, A.; Thiene, G.; Canciani, B.; Scognamiglio, R.; Daliento, L.; Buja, G.; Martini, B.; Stritoni, P.; Fasoli, G. Familial occurrence of right ventricular dysplasia: A study involving nine families. J. Am. Coll. Cardiol. 1988, 12, 1222–1228. [Google Scholar] [CrossRef]
- McKoy, G.; Protonotarios, N.; Crosby, A.; Tsatsopoulou, A.; Anastasakis, A.; Coonar, A.; Norman, M.; Baboonian, C.; Jeffrey, S.; McKenna, W.J. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 2000, 355, 2119–2124. [Google Scholar] [CrossRef]
- Paul, M.; Wichter, T.; Fabritz, L.; Waltenberger, J.; Schulze-Bahr, E.; Kirchhof, P. Arrhythmogenic right ventricular cardiomyopathy: An update on pathophysiology, genetics, diagnosis, and risk stratification. Herzschrittmacherther. Elektrophysiol. 2012, 23, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Beffagna, G.; Occhi, G.; Nava, A.; Vitiello, L.; Ditadi, A.; Basso, C.; Bauce, B.; Carraro, G.; Thiene, G.; Towbin, J.A.; et al. Regulatory mutations in transforming growth factor-?3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc. Res. 2005, 65, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Merner, N.D.; Hodgkinson, K.A.; Haywood, A.F.M.; Connors, S.; French, V.M.; Drenckhahn, J.D.; Kupprion, C.; Ramadanova, K.; Thierfelder, L.; McKenna, W.J.; et al. Arrhythmogenic Right Ventricular Cardiomyopathy Type 5 Is a Fully Penetrant, Lethal Arrhythmic Disorder Caused by a Missense Mutation in the TMEM43 Gene. Am. J. Human Genet. 2008, 82, 809–821. [Google Scholar] [CrossRef]
- Delmar, M.; McKenna, W.J. The cardiac desmosome and arrhythmogenic cardiomyopathies: From gene to disease. Circ. Res. 2010, 107, 700–714. [Google Scholar] [CrossRef]
- Zhao, G.; Qiu, Y.; Zhang, H.M.; Yang, D. Intercalated discs: Cellular adhesion and signaling in heart health and diseases. Heart Fail. Rev. 2019, 24, 115–132. [Google Scholar] [CrossRef]
- Li, K.; Jiang, Y.; Zeng, Y.; Zhou, Y. Advances in Ion Channel, Non-Desmosomal Variants and Autophagic Mechanisms Implicated in Arrhythmogenic Cardiomyopathy. Curr. Issues Mol. Biol. 2023, 45, 2186–2200. [Google Scholar] [CrossRef]
- Vimalanathan, A.K.; Ehler, E.; Gehmlich, K. Genetics of and pathogenic mechanisms in arrhythmogenic right ventricular cardiomyopathy. Biophys. Rev. 2018, 10, 973–982. [Google Scholar] [CrossRef]
- Shaikh, T.; Nguyen, D.; Dugal, J.K.; DiCaro, M.V.; Yee, B.; Houshmand, N.; Lei, K.; Namazi, A. Arrhythmogenic Right Ventricular Cardiomyopathy: A Comprehensive Review. J. Cardiovasc. Dev. Dis. 2025, 12, 71. [Google Scholar] [CrossRef]
- Austin, K.M.; Trembley, M.A.; Chandler, S.F.; Sanders, S.P.; Saffitz, J.E.; Abrams, D.J.; Pu, W.T. Molecular mechanisms of arrhythmogenic cardiomyopathy. Nat. Rev. Cardiol. 2019, 16, 519–537. [Google Scholar] [CrossRef]
- Gerull, B.; Brodehl, A. Insights Into Genetics and Pathophysiology of Arrhythmogenic Cardiomyopathy. Curr. Heart Fail. Rep. 2021, 18, 378–390. [Google Scholar] [CrossRef]
- Hariharan, V.; Asimaki, A.; Michaelson, J.E.; Plovie, E.; MacRae, C.A.; Saffitz, J.E.; Huang, H. Arrhythmogenic right ventricular cardiomyopathy mutations alter shear response without changes in cell–cell adhesion. Cardiovasc. Res. 2014, 104, 280–289. [Google Scholar] [CrossRef]
- Cerrone, M.; Marrón-Liñares, G.M.; van Opbergen, C.J.M.; Costa, S.; Bourfiss, M.; Pérez-Hernández, M.; Schlamp, F.; Sanchis-Gomar, F.; Mailkani, K.; Drenkova, K.; et al. Role of plakophilin-2 expression on exercise-related progression of arrhythmogenic right ventricular cardiomyopathy: A translational study. Eur. Heart J. 2022, 43, 1251–1264. [Google Scholar] [CrossRef] [PubMed]
- Asatryan, B.; Asimaki, A.; Landstrom, A.P.; Khanji, M.Y.; Odening, K.E.; Cooper, L.T.; Marchlinkski, F.E.; Gelzer, A.R.; Semsarian, C.; Reichlin, T.; et al. Inflammation and Immune Response in Arrhythmogenic Cardiomyopathy: State-of-the-Art Review. Circulation 2021, 144, 1646–1655. [Google Scholar] [CrossRef]
- Chatterjee, D.; Fatah, M.; Akdis, D.; Spears, D.A.; Koopmann, T.T.; Mittal, K.; Rafiq, M.A.; Cattanach, B.M.; Zhao, Q.; Healey, J.S.; et al. An autoantibody identifies arrhythmogenic right ventricular cardiomyopathy and participates in its pathogenesis. Eur. Heart J. 2018, 39, 3932–3944. [Google Scholar] [CrossRef] [PubMed]
- Lota, A.S.; Hazebroek, M.R.; Theotokis, P.; Wassall, R.; Salmi, S.; Halliday, B.P.; Tayal, U.; Verdonschot, J.; Meena, D.; Owen, R.; et al. Genetic Architecture of Acute Myocarditis and the Overlap with Inherited Cardiomyopathy. Circulation 2022, 146, 1123–1134. [Google Scholar] [CrossRef]
- Lutokhina, Y.; Zaklyazminskaya, E.; Kogan, E.; Nartov, A.; Nartova, V.; Blagova, O. Incidence and Impact of Myocarditis in Genetic Cardiomyopathies: Inflammation as a Potential Therapeutic Target. Genes 2025, 16, 51. [Google Scholar] [CrossRef]
- Liu, X.; Liu, L.; Zhao, J.; Wang, H.; Li, Y. Mechanotransduction regulates inflammation responses of epicardial adipocytes in cardiovascular diseases. Front. Endocrinol. 2022, 13, 1080383. [Google Scholar] [CrossRef] [PubMed]
- Mestroni, L.; Sbaizero, O. Arrhythmogenic Cardiomyopathy. Circulation 2018, 137, 1611–1613. [Google Scholar] [CrossRef]
- Rouhi, L.; Fan, S.; Cheedipudi, S.M.; Braza-Boïls, A.; Molina, M.S.; Yao, Y.; Robertson, M.J.; Coarfa, C.; Gimeno, J.R.; Molina, P.; et al. The EP300/TP53 pathway, a suppressor of the Hippo and canonical WNT pathways, is activated in human hearts with arrhythmogenic cardiomyopathy in the absence of overt heart failure. Cardiovasc. Res. 2022, 118, 1466–1478. [Google Scholar] [CrossRef] [PubMed]
- Vencato, S.; Romanato, C.; Rampazzo, A.; Calore, M. Animal Models and Molecular Pathogenesis of Arrhythmogenic Cardiomyopathy Associated with Pathogenic Variants in Intercalated Disc Genes. Int. J. Mol. Sci. 2024, 25, 6208. [Google Scholar] [CrossRef] [PubMed]
- Fabritz, L.; Hoogendijk, M.G.; Scicluna, B.P.; van Amersfoorth, S.C.M.; Fortmueller, L.; Wolf, S.; Laakman, S.; Kreienkamp, N.; Piccini, I.; Breithardt, G.; et al. Load-Reducing Therapy Prevents Development of Arrhythmogenic Right Ventricular Cardiomyopathy in Plakoglobin-Deficient Mice. J. Am. Coll. Cardiol. 2011, 57, 740–750. [Google Scholar] [CrossRef] [PubMed]
- Fabritz, L.; Fortmueller, L.; Gehmlich, K.; Kant, S.; Kemper, M.; Kucerova, D.; Syeda, F.; Faber, C.; Leube, R.E.; Kirchhof, P.; et al. Endurance Training Provokes Arrhythmogenic Right Ventricular Cardiomyopathy Phenotype in Heterozygous Desmoglein-2 Mutants: Alleviation by Preload Reduction. Biomedicines 2024, 12, 985. [Google Scholar] [CrossRef]
- Schinner, C.; Xu, L.; Franz, H.; Zimmermann, A.; Wanuske, M.T.; Rathod, M.; Hanns, P.; Geier, F.; Pelczar, P.; Liang, Y.; et al. Defective Desmosomal Adhesion Causes Arrhythmogenic Cardiomyopathy by Involving an Integrin-αVβ6/TGF-β Signaling Cascade. Circulation 2022, 146, 1610–1626. [Google Scholar] [CrossRef]
- Pitsch, M.; Kant, S.; Mytzka, C.; Leube, R.E.; Krusche, C.A. Autophagy and Endoplasmic Reticulum Stress during Onset and Progression of Arrhythmogenic Cardiomyopathy. Cells 2021, 11, 96. [Google Scholar] [CrossRef]
- Caspi, O.; Huber, I.; Gepstein, A.; Arbel, G.; Maizels, L.; Boulos, M.; Gepstein, L. Modeling of Arrhythmogenic Right Ventricular Cardiomyopathy with Human Induced Pluripotent Stem Cells. Circ. Cardiovasc. Genet. 2013, 6, 557–568. [Google Scholar] [CrossRef]
- Bauce, B.; Basso, C.; Rampazzo, A.; Beffagna, G.; Daliento, L.; Frigo, G.; Malacrida, S.; Settimo, L.; Danieli, G.; Thiene, G.; et al. Clinical profile of four families with arrhythmogenic right ventricular cardiomyopathy caused by dominant desmoplakin mutations. Eur. Heart J. 2005, 26, 1666–1675. [Google Scholar] [CrossRef]
- Kapplinger, J.D.; Landstrom, A.P.; Salisbury, B.A.; Callis, T.E.; Pollevick, G.D.; Tester, D.J.; Cox, M.G.P.J.; Bhuiyan, Z.; Bikker, H.; Wiesfeld, A.C.P.; et al. Distinguishing Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia–Associated Mutations from Background Genetic Noise. J. Am. Coll. Cardiol. 2011, 57, 2317–2327. [Google Scholar] [CrossRef]
- James, C.A.; Jongbloed, J.D.H.; Hershberger, R.E.; Morales, A.; Judge, D.P.; Syrris, P.; Pilichou, K.; Domingo, A.M.; Murray, B.; Cadrin-Tourigny, J.; et al. International Evidence Based Reappraisal of Genes Associated with Arrhythmogenic Right Ventricular Cardiomyopathy Using the Clinical Genome Resource Framework. Circ. Genom. Precis. Med. 2021, 14, 273–284. [Google Scholar] [CrossRef]
- Lombardi, R.; da Graca Cabreira-Hansen, M.; Bell, A.; Fromm, R.R.; Willerson, J.T.; Marian, A.J. Nuclear Plakoglobin Is Essential for Differentiation of Cardiac Progenitor Cells to Adipocytes in Arrhythmogenic Right Ventricular Cardiomyopathy. Circ. Res. 2011, 109, 1342–1353. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Hernández, M.; van Opbergen, C.J.M.; Bagwan, N.; Vissing, C.R.; Marrón-Liñares, G.M.; Zhang, M.; Torres Vega, E.; Sorrentino, A.; Drici, L.; Sulek, K.; et al. Loss of Nuclear Envelope Integrity and Increased Oxidant Production Cause DNA Damage in Adult Hearts Deficient in PKP2: A Molecular Substrate of ARVC. Circulation 2022, 146, 851–867. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wu, Y.; Yang, X.; Xiang, Y.; Yang, B. Molecular insight into arrhythmogenic cardiomyopathy caused by DSG2 mutations. Biomed. Pharmacother. 2023, 167, 115448. [Google Scholar] [CrossRef]
- Vite, A.; Gandjbakhch, E.; Hery, T.; Fressart, V.; Gary, F.; Simon, F.; Varnous, S.; Hidden Lucet, F.; Charron, P.; Villard, E. Desmoglein-2 mutations in propeptide cleavage-site causes arrhythmogenic right ventricular cardiomyopathy/dysplasia by impairing extracellular 1-dependent desmosomal interactions upon cellular stress. EP Eur. 2020, 22, 320–329. [Google Scholar] [CrossRef]
- Micolonghi, C.; Perrone, F.; Fabiani, M.; Caroselli, S.; Savio, C.; Pizzuti, A.; Germani, A.; Visco, V.; Petrucci, S.; Rabattu, S.; et al. Unveiling the Spectrum of Minor Genes in Cardiomyopathies: A Narrative Review. Int. J. Mol. Sci. 2024, 25, 9787. [Google Scholar] [CrossRef]
- Salavati, A.; van der Wilt, C.N.; Calore, M.; van Es, R.; Rampazzo, A.; van der Harst, P.; van Steenbeek, F.G.; van Tintelen, J.P.; Harakalova, M.; Te Riele, A.S.J.M. Artificial Intelligence Advancements in Cardiomyopathies: Implications for Diagnosis and Management of Arrhythmogenic Cardiomyopathy. Curr. Heart Fail. Rep. 2025, 22, 5. [Google Scholar] [CrossRef]
- Carabetta, N.; Siracusa, C.; Leo, I.; Panuccio, G.; Strangio, A.; Sabatino, J.; Torella, D.; De Rosa, S. Cardiomyopathies: The Role of Non-Coding RNAs. Noncoding RNA 2024, 10, 53. [Google Scholar] [CrossRef]
- Chen, L.; Hu, Y.; Saguner, A.M.; Bauce, B.; Liu, Y.; Shi, A.; Guan, F.; Chen, Z.; Bueno Marines, M.; Wu, L.; et al. Natural History and Clinical Outcomes of Patients with DSG2/DSC2 Variant-Related Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation 2025, 151, 1213–1230. [Google Scholar] [CrossRef]
- Xu, Z.; Zhu, W.; Wang, C.; Huang, L.; Zhou, Q.; Hu, J.; Cheng, X.; Hong, K. Genotype-phenotype relationship in patients with arrhythmogenic right ventricular cardiomyopathy caused by desmosomal gene mutations: A systematic review and meta-analysis. Sci. Rep. 2017, 7, 41387. [Google Scholar] [CrossRef] [PubMed]
- van Tintelen, J.P.; Entius, M.M.; Bhuiyan, Z.A.; Jongbloed, R.; Wiesfeld, A.C.P.; Wilde, A.A.M.; van der Smagt, J.; Boven, L.G.; Mannens, M.M.A.M.; van Langen, I.M.; et al. Plakophilin-2 Mutations Are the Major Determinant of Familial Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Circulation 2006, 113, 1650–1658. [Google Scholar] [CrossRef]
- van Lint, F.H.M.; Murray, B.; Tichnell, C.; Zwart, R.; Amat, N.; Lekanne Deprez, R.H.; Dittmann, S.; Stallmeyer, B.; Calkins, H.; van der Smagt, J.J.; et al. Arrhythmogenic Right Ventricular Cardiomyopathy-Associated Desmosomal Variants Are Rarely De Novo. Circ. Genom. Precis. Med. 2019, 12, e002467. [Google Scholar] [CrossRef]
- Pergola, V.; Trancuccio, A.; Kukavica, D.; Mazzanti, A.; Napolitano, C.; Scilabra, G.G.; Steele, K.; Memmi, M.; Gambelli, P.; Sugamiele, A.; et al. Genotype-Specific Outcomes of Desmosomal Cardiomyopathies. Circulation. Circ. 2025, 152, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Borrego, E.; Bermúdez-Jiménez, F.J.; Gasperetti, A.; Tandri, H.; Sánchez-Millán, P.J.; Molina-Lerma, M.; Roca-Luque, I.; Vasquez-Calvo, S.; Compagnucci, P.; Casella, M.; et al. Electrophysiological Phenotype-Genotype Study of Sustained Monomorphic Ventricular Tachycardia in Inherited, High Arrhythmic Risk, Left Ventricular Cardiomyopathy. Circ. Arrhythm. Electrophysiol. 2024, 17, e013145. [Google Scholar] [CrossRef]
- Nielsen, J.C.; Lin, Y.J.; de Oliveira Figueiredo, M.J.; Sepehri Shamloo, A.; Alfie, A.; Boveda, S.; Dagres, N.; Di Toro, D.; Eckhardt, L.L.; Ellenbogen, K.; et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) expert consensus on risk assessment in cardiac arrhythmias: Use the right tool for the right outcome, in the right population. Heart Rhythm 2020, 17, e269–e316. [Google Scholar]
- Schmitt, J.P.; Kamisago, M.; Asahi, M.; Li, G.H.; Ahmad, F.; Mende, U.; Kranias, E.G.; MacLennan, D.H.; Seidman, J.G.; Seidman, C.E. Dilated Cardiomyopathy and Heart Failure Caused by a Mutation in Phospholamban. Science 2003, 299, 1410–1413. [Google Scholar] [CrossRef]
- Young, S.G.; Jung, H.J.; Lee, J.M.; Fong, L.G. Nuclear Lamins and Neurobiology. Mol. Cell. Biol. 2014, 34, 2776–2785. [Google Scholar] [CrossRef]
- Quarta, G.; Syrris, P.; Ashworth, M.; Jenkins, S.; Zuborne Alapi, K.; Morgan, J.; Muir, A.; Pantazis, A.; McKenna, W.J.; Elliott, P.M. Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2012, 33, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Josephs, K.S.; Roberts, A.M.; Theotokis, P.; Walsh, R.; Ostrowski, P.J.; Edwards, M.; Fleming, A.; Thaxton, C.; Roberts, J.D.; Care, M.; et al. Beyond gene-disease validity: Capturing structured data on inheritance, allelic requirement, disease-relevant variant classes, and disease mechanism for inherited cardiac conditions. Genome Med. 2023, 15, 86. [Google Scholar] [CrossRef] [PubMed]
- Zorzi, A.; Rigato, I.; Pilichou, K.; Perazzolo Marra, M.; Migliore, F.; Mazzotti, E.; Gregori, D.; Thiene, G.; Daliento, L.; Iliceto, S.; et al. Phenotypic expression is a prerequisite for malignant arrhythmic events and sudden cardiac death in arrhythmogenic right ventricular cardiomyopathy. Europace 2016, 18, 1086–1094. [Google Scholar] [CrossRef]
- Christensen, A.H.; Platonov, P.G.; Jensen, H.K.; Chivulescu, M.; Svensson, A.; Dahlberg, P.; Madsen, T.; Frederiksen, T.C.; Helio, T.; Haugen Lie, O.; et al. Genotype–phenotype correlation in arrhythmogenic right ventricular cardiomyopathy—Risk of arrhythmias and heart failure. J. Med. Genet. 2022, 59, 858–864. [Google Scholar] [CrossRef]
- Lippi, M.; Chiesa, M.; Ascione, C.; Pedrazzini, M.; Mushtaq, S.; Rovina, D.; Rigio, D.; Di Blasio, A.M.; Biondi, M.L.; Pompilio, G.; et al. Spectrum of Rare and Common Genetic Variants in Arrhythmogenic Cardiomyopathy Patients. Biomolecules 2022, 12, 1043. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hedge, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Wilde, A.A.M.; Semsarian, C.; Márquez, M.F.; Sepehri Shamloo, A.; Ackerman, M.J.; Ashley, E.A.; Back Sternick, E.; Barajas-Martinez, H.; Behr, E.R.; Bezzina, C.R.; et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the State of Genetic Testing for Cardiac Diseases. Heart Rhythm 2022, 19, e1–e60. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Yang, Z.; Vatta, M.; Rampazzo, A.; Beffagna, G.; Pillichou, K.; Scherer, S.E.; Saffitz, J.; Kravitz, J.; Zareba, W.; et al. Compound and Digenic Heterozygosity Contributes to Arrhythmogenic Right Ventricular Cardiomyopathy. J. Am. Coll. Cardiol. 2010, 55, 587–597. [Google Scholar] [CrossRef]
- James, C.A.; Syrris, P.; van Tintelen, J.P.; Calkins, H. The role of genetics in cardiovascular disease: Arrhythmogenic cardiomyopathy. Eur. Heart J. 2020, 41, 1393–1400. [Google Scholar] [CrossRef] [PubMed]
- Protonotarios, A.; Bariani, R.; Cappelletto, C.; Pavlou, M.; García-García, A.; Cipriani, A.; Protonotarius, I.; Rivas, A.; Wittenberg, R.; Graziosi, M.; et al. Corrigendum to: Importance of genotype for risk stratification in arrhythmogenic right ventricular cardiomyopathy using the 2019 ARVC risk calculator. Eur. Heart J. 2022, 43, 4373. [Google Scholar] [CrossRef]
- Gasperetti, A.; Carrick, R.; Protonotarios, A.; Laredo, M.; van der Schaaf, I.; Syrris, P.; Murray, B.; Tichnell, C.; Cappelletto, C.; Gigli, M.; et al. Long-Term Arrhythmic Follow-Up and Risk Stratification of Patients with Desmoplakin-Associated Arrhythmogenic Right Ventricular Cardiomyopathy. JACC Adv. 2024, 3, 100832. [Google Scholar] [CrossRef]
- Schunkert, H.; Di Angelantonio, E.; Inouye, M.; Patel, R.S.; Ripatti, S.; Widen, E.; Sanderson, S.C.; Kaski, J.P.; McEvoy, J.W.; Vardas, P.; et al. Clinical utility and implementation of polygenic risk scores for predicting cardiovascular disease. Eur. Heart J. 2025, 46, 1372–1383. [Google Scholar] [CrossRef]
- Corrado, D.; Wichter, T.; Link, M.S.; Hauer, R.; Marchlinski, F.; Anastasakis, A.; Bauce, B.; Basso, C.; Brunckhorst, C.; Tsatsopoulou, A.; et al. Treatment of arrhythmogenic right ventricular cardiomyopathy/dysplasia: An international task force consensus statement. Eur. Heart J. 2015, 36, 3227–3237. [Google Scholar] [CrossRef]
- Al-Khatib, S.M.; Stevenson, W.G.; Ackerman, M.J.; Bryant, W.J.; Callans, D.J.; Curtis, A.B.; Deal, B.J.; Dickfeld, T.; Filed, M.E.; Fonarow, G.C.; et al. 2017 AHA/ACC/HRS Guideline for Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death. Circulation 2018, 138, e91–e220. [Google Scholar]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.C.; Daubert, J.P.; de Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm 2019, 16, e301–e372. [Google Scholar] [CrossRef] [PubMed]
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; de Boer, R.A.; et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur. Heart J. 2023, 44, 3503–3626. [Google Scholar] [CrossRef] [PubMed]
- Cruz, F.M.; Sanz-Rosa, D.; Roche-Molina, M.; García-Prieto, J.; García-Ruiz, J.M.; Pizarro, G.; Jimenez-Borreguero, L.J.; Torres, M.; Bernad, A.; Ruiz-Cabello, J.; et al. Exercise Triggers ARVC Phenotype in Mice Expressing a Disease-Causing Mutated Version of Human Plakophilin-2. J. Am. Coll. Cardiol. 2015, 65, 1438–1450. [Google Scholar] [CrossRef]
- Corrado, D.; Thiene, G.; Nava, A.; Rossi, L.; Pennelli, N. Sudden death in young competitive athletes: Clinicopathologic correlations in 22 cases. Am. J. Med. 1990, 89, 588–596. [Google Scholar] [CrossRef]
- Pelliccia, A.; Sharma, S.; Gati, S.; Bäck, M.; Börjesson, M.; Caselli, S.; Collet, J.P.; Corrado, D.; Drezner, J.A.; Halle, M.; et al. 2020 ESC Guidelines on sports cardiology and exercise in patients with cardiovascular disease. Eur. Heart J. 2021, 42, 17–96. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Tichnell, C.; Murray, B.A.; Agafonova, J.; Cadrin-Tourigny, J.; Chelko, S.; Tandri, H.; Calkins, H.; James, C.A. Exercise restriction is protective for genotype-positive family members of arrhythmogenic right ventricular cardiomyopathy patients. EP Eur. 2020, 22, 1270–1278. [Google Scholar] [CrossRef]
- van Lint, F.H.M.; Hassanzada, F.; Verstraelen, T.E.; Wang, W.; Bosman, L.P.; van der Zwaag, P.A.; Oomen, T.; Calkins, H.; Murray, B.; Tichnell, C.; et al. Exercise does not influence development of phenotype in PLN p.(Arg14del) cardiomyopathy. Neth. Heart J. 2023, 31, 291–299. [Google Scholar] [CrossRef]
- Corrado, D.; Leoni, L.; Link, M.S.; Bella, P.; Della Gaita, F.; Curnis, A.; Salerno, J.U.; Igidbashian, D.; Raviele, A.; Disertori, M.; et al. Implantable Cardioverter-Defibrillator Therapy for Prevention of Sudden Death in Patients with Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia. Circulation 2003, 108, 3084–3091. [Google Scholar] [CrossRef]
- Marcus, G.M.; Glidden, D.V.; Polonsky, B.; Zareba, W.; Smith, L.M.; Cannom, D.S.; Mark Estes, N.A., 3rd; Marcus, F.; Scheinman, M.M. Efficacy of Antiarrhythmic Drugs in Arrhythmogenic Right Ventricular Cardiomyopathy. J. Am. Coll. Cardiol. 2009, 54, 609–615. [Google Scholar] [CrossRef]
- Al-Aidarous, S.; Protonotarios, A.; Elliott, P.M.; Lambiase, P.D. Management of arrhythmogenic right ventricular cardiomyopathy. Heart 2024, 110, 156–162. [Google Scholar] [CrossRef]
- Verma, A.; Kilicaslan, F.; Schweikert, R.A.; Tomassoni, G.; Rossillo, A.; Marrouche, N.F.; Ozduran, V.; Wazni, O.M.; Elayi, S.C.; Saenz, L.C.; et al. Short- and Long-Term Success of Substrate-Based Mapping and Ablation of Ventricular Tachycardia in Arrhythmogenic Right Ventricular Dysplasia. Circulation 2005, 111, 3209–3216. [Google Scholar] [CrossRef] [PubMed]
- Dalal, D.; Jain, R.; Tandri, H.; Dong, J.; Eid, S.M.; Prakasa, K.; Tichnell, C.; James, C.; Abraham, T.; Russell, S.D.; et al. Long-Term Efficacy of Catheter Ablation of Ventricular Tachycardia in Patients with Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. J. Am. Coll. Cardiol. 2007, 50, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.F.L. Implantable Cardioverter Defibrillators in Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Circ. Arrhythm. Electrophysiol. 2013, 6, 562–568. [Google Scholar] [CrossRef]
- Tedford, R.J.; James, C.; Judge, D.P.; Tichnell, C.; Murray, B.; Bhonsale, A.; Philips, B.; Abraham, T.; Dalal, D.; Halushka, M.K.; et al. Cardiac Transplantation in Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. J. Am. Coll. Cardiol. 2012, 59, 289–290. [Google Scholar] [CrossRef]
- DePasquale, E.C.; Cheng, R.K.; Deng, M.C.; Nsair, A.; McKenna, W.J.; Fonarow, G.C.; Jacoby, D.L. Survival After Heart Transplantation in Patients with Arrhythmogenic Right Ventricular Cardiomyopathy. J. Card. Fail. 2017, 23, 107–112. [Google Scholar] [CrossRef]
- Venturiello, D.; Tiberi, P.G.; Perulli, F.; Nardoianni, G.; Guida, L.; Barsali, C.; Terrone, C.; Cianca, A.; Lustri, C.; Sclafani, M.; et al. Unveiling the Future of Cardiac Care: A Review of Gene Therapy in Cardiomyopathies. Int. J. Mol. Sci. 2024, 25, 13147. [Google Scholar] [CrossRef]
- Sheikh, F.; Zhang, J.; Wang, J.; Bradford, W.H.; Nair, A.; Fargnoli, A.; Selvan, N.; Gutierrez, S.; Law, K.; Fenn, T.; et al. Abstract 13599: LX2020, an Adeno Associated Viral-Based Plakophilin 2 Gene Therapy Stabilizes Cardiac Disease Phenotype in a Severe Mouse Model of Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation 2022, 146 (Suppl. S1), A13599. [Google Scholar] [CrossRef]
- Bradford, W.H.; Zhang, J.; Gutierrez-Lara, E.J.; Liang, Y.; Do, A.; Wang, T.M.; Nguyen, L.; Mataraarachchi, N.; Wang, J.; Gu, Y.; et al. Plakophilin 2 gene therapy prevents and rescues arrhythmogenic right ventricular cardiomyopathy in a mouse model harboring patient genetics. Nat. Cardiovasc. Res. 2023, 2, 1246–1261. [Google Scholar] [CrossRef]
- Argiro, A.; Bui, Q.; Hong, K.N.; Ammirati, E.; Olivotto, I.; Adler, E. Applications of Gene Therapy in Cardiomyopathies. JACC Heart Fail. 2024, 12, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Dalal, D.; Nasir, K.; Bomma, C.; Prakasa, K.; Tandri, H.; Piccini, J.; Roguin, A.; Tichnell, C.; James, C.; Russell, S.D.; et al. Arrhythmogenic Right Ventricular Dysplasia. Circulation 2005, 112, 3823–3832. [Google Scholar] [CrossRef]
- Bueno Marinas, M.; Cason, M.; Bariani, R.; Celeghin, R.; De Gaspari, M.; Pinci, S.; Cipriani, A.; Rigato, I.; Zorzi, A.; Rizzo, S.; et al. A Comprehensive Analysis of Non-Desmosomal Rare Genetic Variants in Arrhythmogenic Cardiomyopathy: Integrating in Padua Cohort Literature-Derived Data. Int. J. Mol. Sci. 2024, 25, 6267. [Google Scholar] [CrossRef]
- Ammirati, E.; Raimondi, F.; Piriou, N.; Sardo Infirri, L.; Mohiddin, S.A.; Mazzanti, A.; Shenoy, C.; Cavallari, U.A.; Imazio, M.; Aquaro, G.D.; et al. Acute Myocarditis Associated with Desmosomal Gene Variants. JACC Heart Fail. 2022, 10, 714–727. [Google Scholar] [CrossRef]
- Biernacka, E.K.; Borowiec, K.; Franaszczyk, M.; Szperl, M.; Rampazzo, A.; Woźniak, O.; Roszczynko, M.; Smigielski, W.; Lutynska, A.; Hoffman, P. Pathogenic variants in plakophilin-2 gene (PKP2) are associated with better survival in arrhythmogenic right ventricular cardiomyopathy. J. Appl. Genet. 2021, 62, 613–620. [Google Scholar] [CrossRef]
- Smith, E.D.; Lakdawala, N.K.; Papoutsidakis, N.; Aubert, G.; Mazzanti, A.; McCanta, A.C.; Agarwal, P.P.; Arscott, P.; Dellefave-Castillo, L.M.; Vorovich, E.E.; et al. Desmoplakin Cardiomyopathy, a Fibrotic and Inflammatory Form of Cardiomyopathy Distinct from Typical Dilated or Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation 2020, 141, 1872–1884. [Google Scholar] [CrossRef]
- Gasperetti, A.; Carrick, R.T.; Protonotarios, A.; Murray, B.; Laredo, M.; van der Schaaf, I.; Lekanna, R.H.; Syrris, P.; Cannie, D.; Tichnell, C.; et al. Clinical features and outcomes in carriers of pathogenic desmoplakin variants. Eur. Heart J. 2025, 46, 362–376. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, J.; Lv, Z.; Liang, P.; Li, Q.; Li, Y.; Guo, Y. LMNA-related cardiomyopathy: From molecular pathology to cardiac gene therapy. J. Adv. Res. 2025, in press. [Google Scholar] [CrossRef] [PubMed]
- Wahbi, K.; Ben Yaou, R.; Gandjbakhch, E.; Anselme, F.; Gossios, T.; Lakdawala, N.K.; Stalens, C.; Sacher, F.; Babuty, D.; Trochu, J.N.; et al. Development and Validation of a New Risk Prediction Score for Life-Threatening Ventricular Tachyarrhythmias in Laminopathies. Circulation 2019, 140, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Hodgkinson, K.A.; Howes, A.J.; Boland, P.; Shen, X.S.; Stuckless, S.; Young, T.L.; Curtis, F.; Collier, A.; Parfrey, P.S.; Connors, S.P.; et al. Long-Term Clinical Outcome of Arrhythmogenic Right Ventricular Cardiomyopathy in Individuals with a p.S358L Mutation in TMEM43 Following Implantable Cardioverter Defibrillator Therapy. Circ. Arrhythm. Electrophysiol. Electrophysiol. 2016, 9, e003589. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Genga, M.F.; Cuenca, S.; Dal Ferro, M.; Zorio, E.; Salgado-Aranda, R.; Climent, V.; Padron-Barthe, L.; Duro-Aguado, I.; Jimenez-Jaimez, J.; Hidalgo-Olivares, V.M.; et al. Truncating FLNC Mutations Are Associated with High-Risk Dilated and Arrhythmogenic Cardiomyopathies. J. Am. Coll. Cardiol. 2016, 68, 2440–2451. [Google Scholar] [CrossRef]
- Gigli, M.; Stolfo, D.; Graw, S.L.; Merlo, M.; Gregorio, C.; Nee Chen, S.; Dal Ferro, M.; Paldino, A.; De Angelis, G.; Brun, F.; et al. Phenotypic Expression, Natural History, and Risk Stratification of Cardiomyopathy Caused by Filamin C Truncating Variants. Circulation 2021, 144, 1600–1611. [Google Scholar] [CrossRef]
- van der Zwaag, P.A.; van Rijsingen, I.A.W.; Asimaki, A.; Jongbloed, J.D.H.; van Veldhuisen, D.J.; Wiesfeld, A.C.P.; Cox, M.G.P.J.; van Lochem, L.T.; de Boer, R.A.; Hofstra, R.M.W.; et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Heart Fail. 2012, 14, 1199–1207. [Google Scholar] [CrossRef]
- Iezzi, L.; Sorella, A.; Galanti, K.; Gallina, S.; Chahal, A.A.; Bauce, B.; Cipriani, A.; Providencia, R.; Lopes, L.R.; Ricci, F.; et al. Arrhythmogenic cardiomyopathy diagnosis and management: A systematic review of clinical practice guidelines and recommendations with insights for future research. Eur. Heart J. Qual. Care Clin. Outcomes 2025. ahead of the print. [Google Scholar] [CrossRef]
- Asatryan, B.; Murray, B.; Tadros, R.; Rieder, M.; Shah, R.A.; Sharaf Dabbagh, G.; Landstrom, A.P.; Dobner, S.; Munroe, P.B.; Haggerty, C.M.; et al. Promise and Peril of a Genotype-First Approach to Mendelian Cardiovascular Disease. J. Am. Heart Assoc. 2024, 13, e033557. [Google Scholar] [CrossRef]
- Asatryan, B.; Shah, R.A.; Sharaf Dabbagh, G.; Landstrom, A.P.; Darbar, D.; Khanji, M.Y.; Lopes, L.R.; van Duijvenboden, S.; Muser, D.; Lee, A.M.; et al. Predicted Deleterious Variants in Cardiomyopathy Genes Prognosticate Mortality and Composite Outcomes in the UK Biobank. JACC Heart Fail. 2024, 12, 918–932. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Song, S.; Shi, A.; Hu, S. Identification of Potential lncRNA-miRNA-mRNA Regulatory Network Contributing to Arrhythmogenic Right Ventricular Cardiomyopathy. J. Cardiovasc. Dev. Dis. 2024, 11, 168. [Google Scholar] [CrossRef] [PubMed]
- Braunwald, E. Cardiac cell therapy: A call for action. Eur. Heart J. 2022, 43, 2352–2353. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| ESC | AHA/ACC/HRS | HRS | ETF/ITF | |
|---|---|---|---|---|
| Definition of ACM | ARVC: presence of predominantly RV dilatation and/or dysfunction in the presence of histological involvement and/or ECG abnormalities in accordance with published criteria [19]. There is progressive myocardial atrophy with fibro-fatty replacement of the RV myocardium, but lesions can also be present in the LV myocardium. | Inherited cardiomyopathy that predominantly affects the RV but can affect the LV, causing areas of myocardial replacement with fibrosis and adipose tissue that frequently causes VA and SCD. | Arrhythmogenic heart muscle disorder not explained by ischemic, hypertensive, or valvular heart disease. | Heart muscle disease characterized by prominent non-ischemic myocardial scarring predisposing to ventricular electrical instability, that may affect both ventricles, with variants being RV-dominant, Biv-, or LV-dominant. |
| ARVC | ARVC diagnosis should be suspected in adolescents or young adults with palpitations, syncope, or aborted sudden death; frequent VEs or VT of LBBB morphology; right precordial TWI (V1–V3) in routine ECG testing; low QRS voltages in the peripheral leads and terminal activation delay in the right precordial leads; RV dilatation on 2D echo. Revised Task Force Criteria for the diagnosis of ARVC: [3]
No major or minor morpho-functional and/or structural LV criteria +
| Presence of clinical symptoms along with the presence of Revised Task Force Criteria for the diagnosis of ARVC) [3]:
| The diagnosis of ARVC should be considered in the following: patients with exercise-related palpitations and/or syncope; survivors of SCA (particularly during exercise); and individuals with frequent VEs (>500 in 24 h) and/or VT of LBBB morphology in the absence of other heart disease. Revised Task Force Criteria for the diagnosis of ARVC [3]:
| European Task Force Proposed Diagnostic Criteria for the diagnosis of ACM [21]: No major or minor morpho-functional or structural (tissue characterization) LV criteria +
|
| ALVC | The NDLVC phenotype includes ALVC, left-dominant ARVC, or arrhythmogenic DCM. The term NDLVC defined by the presence of non-ischemic LV scarring or fatty replacement regardless of the presence of global or regional wall motion abnormalities, or isolated global LV hypokinesia without scarring. General endorsement of Padua criteria: [20] No major or minor morpho-functional and/or structural RV criteria + ≥1 major structural LV criteria + pathogenic or likely pathogenic ACM-causing gene mutation. | Not reported | Not reported | European Task Force Proposed Diagnostic Criteria for the diagnosis of ACM [21]: No major or minor morpho-functional or structural (tissue characterization) RV criteria + the following:
|
| Biventricular ACM | General endorsement of Padua criteria: [20] Presence of ≥1 major or minor morpho-functional and/or structural RV criteria + ≥1 major or minor morpho-functional or structural (tissue characterization) LV criteria +
| Not reported | Not reported | European Task Force Proposed Diagnostic Criteria for the diagnosis of ACM: [21] Presence of ≥1 major or minor morpho-functional or structural (tissue characterization) RV criteria + Presence of ≥1 major or minor morpho-functional or structural (tissue characterization) LV criteria
|
| Desmosomal ACM | ||
|---|---|---|
| Gene Variant | Cell Damage | Phenotype |
| Plakoglobin |
| ARVC (mainly) ALVC Naxos disease (hair and skin) |
| Plakophilin C |
| ARVC |
| Desmocollin 2 |
| ARVC |
| Desmoglein 2 |
| ARVC (mainly) ALVC Biv-ACM |
| Desmoplakin |
| ALVC (mainly) Biv-ACM ARVC |
| Non-desmosomal ACM | ||
| Gene variant | Cell damage | Phenotype |
| Phospholamban | ALVC (mainly) Biv-ACM ARVC | |
| Transmembrane protein 43 |
| ALVC (mainly) Biv-ACM (fast deterioration) |
| Filamin C |
| ALVC (mainly) Biv-ACM (fast deterioration) Skeletal myofibrillar myopathy |
| Desmin |
| ALVC (mainly) Biv-ACM Skeletal myofibrillar myopathy Conduction system abnormalities |
| Lamin A/C |
| ALVC (mainly) ARVC BivACM (fast deterioration) Emery–Dreifuss muscular dystrophy Limb–girdle muscular dystrophy 1B Familial lipodystrophy Hutchinson–Gliford progeria [59] |
| SCN5A |
| ARVC ALVC |
| Clinical Trials | Description |
|---|---|
| NCT05885412 Phase 1—dose escalation | Intravenous injected recombinant AAV vector containing PKP2 (RP-A601) in subjects with high-risk PKP2-ACM |
| NCT06109181 Phase 1/2—open label, dose escalating, multicentric trial | Safety and tolerability of LX2020 (AAV vector encoding PKP2 gene) in 10 adult patients with PKP2-ACM |
| NCT06228924—RIDGE-1 open label, phase 1 | Fifteen patients across two designated dose groups who are experiencing symptomatic PKP2-ACM, each cohort receiving a single endovenous dose of TN-401 (AAV9 containing PKP2 transgene) |
| NCT06311708 multicentric, observational | Prevalence of pre-existing antibodies to AAV9 in a population of PKP2-ACM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galanti, K.; Iezzi, L.; Rizzuto, M.L.; Falco, D.; Negri, G.; Pham, H.N.; Mansour, D.; Giansante, R.; Stuppia, L.; Mazzocchetti, L.; et al. Desmosomal Versus Non-Desmosomal Arrhythmogenic Cardiomyopathies: A State-of-the-Art Review. Cardiogenetics 2025, 15, 22. https://doi.org/10.3390/cardiogenetics15030022
Galanti K, Iezzi L, Rizzuto ML, Falco D, Negri G, Pham HN, Mansour D, Giansante R, Stuppia L, Mazzocchetti L, et al. Desmosomal Versus Non-Desmosomal Arrhythmogenic Cardiomyopathies: A State-of-the-Art Review. Cardiogenetics. 2025; 15(3):22. https://doi.org/10.3390/cardiogenetics15030022
Chicago/Turabian StyleGalanti, Kristian, Lorena Iezzi, Maria Luana Rizzuto, Daniele Falco, Giada Negri, Hoang Nhat Pham, Davide Mansour, Roberta Giansante, Liborio Stuppia, Lorenzo Mazzocchetti, and et al. 2025. "Desmosomal Versus Non-Desmosomal Arrhythmogenic Cardiomyopathies: A State-of-the-Art Review" Cardiogenetics 15, no. 3: 22. https://doi.org/10.3390/cardiogenetics15030022
APA StyleGalanti, K., Iezzi, L., Rizzuto, M. L., Falco, D., Negri, G., Pham, H. N., Mansour, D., Giansante, R., Stuppia, L., Mazzocchetti, L., Gallina, S., Mantini, C., Khanji, M. Y., Chahal, C. A. A., & Ricci, F. (2025). Desmosomal Versus Non-Desmosomal Arrhythmogenic Cardiomyopathies: A State-of-the-Art Review. Cardiogenetics, 15(3), 22. https://doi.org/10.3390/cardiogenetics15030022