
A Family with a Single LMNA Mutation Illustrates Diversity in Cardiac Phenotypes Associated with Laminopathic Progeroid Syndromes

 ,
,
Abstract
:1. Introduction
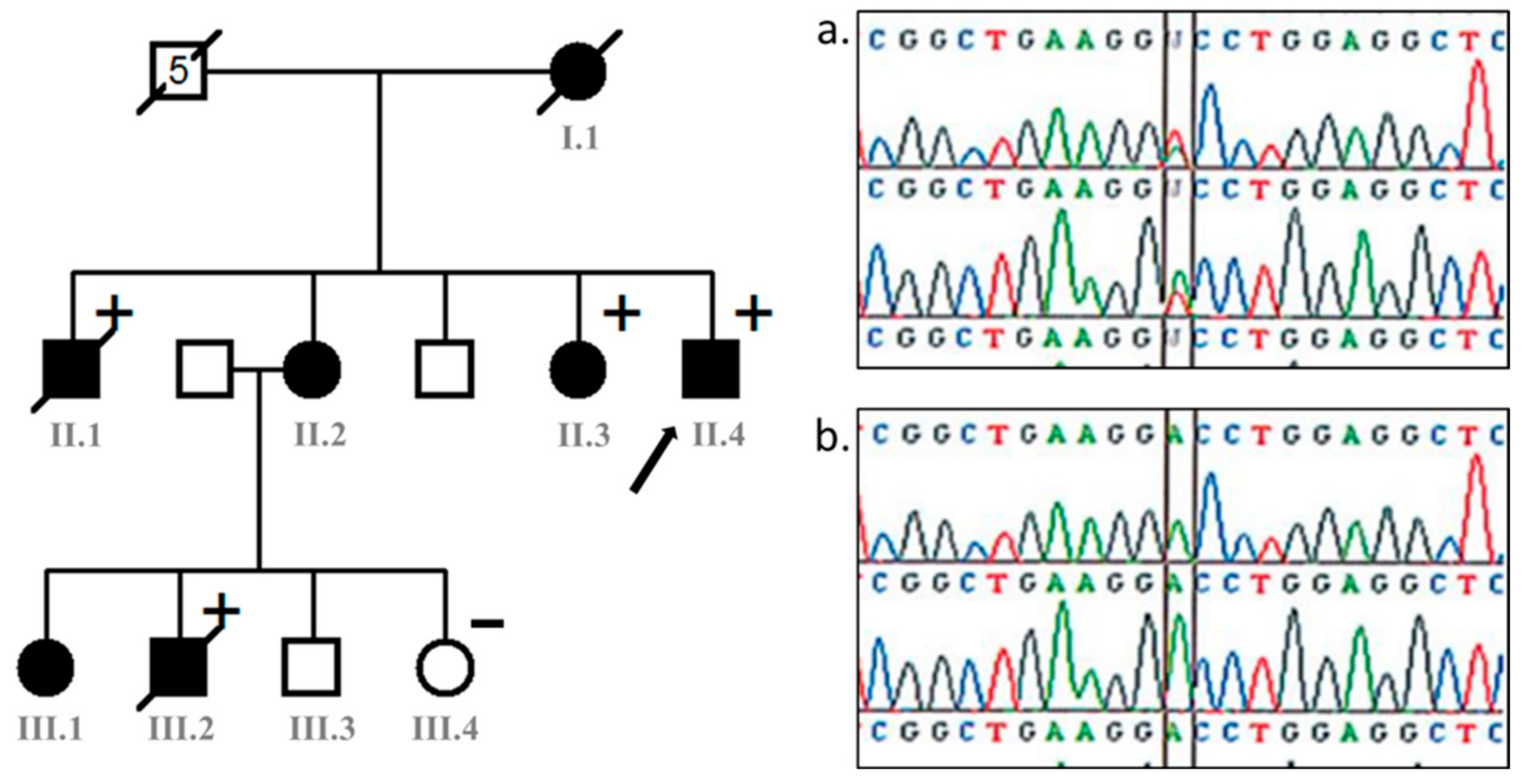
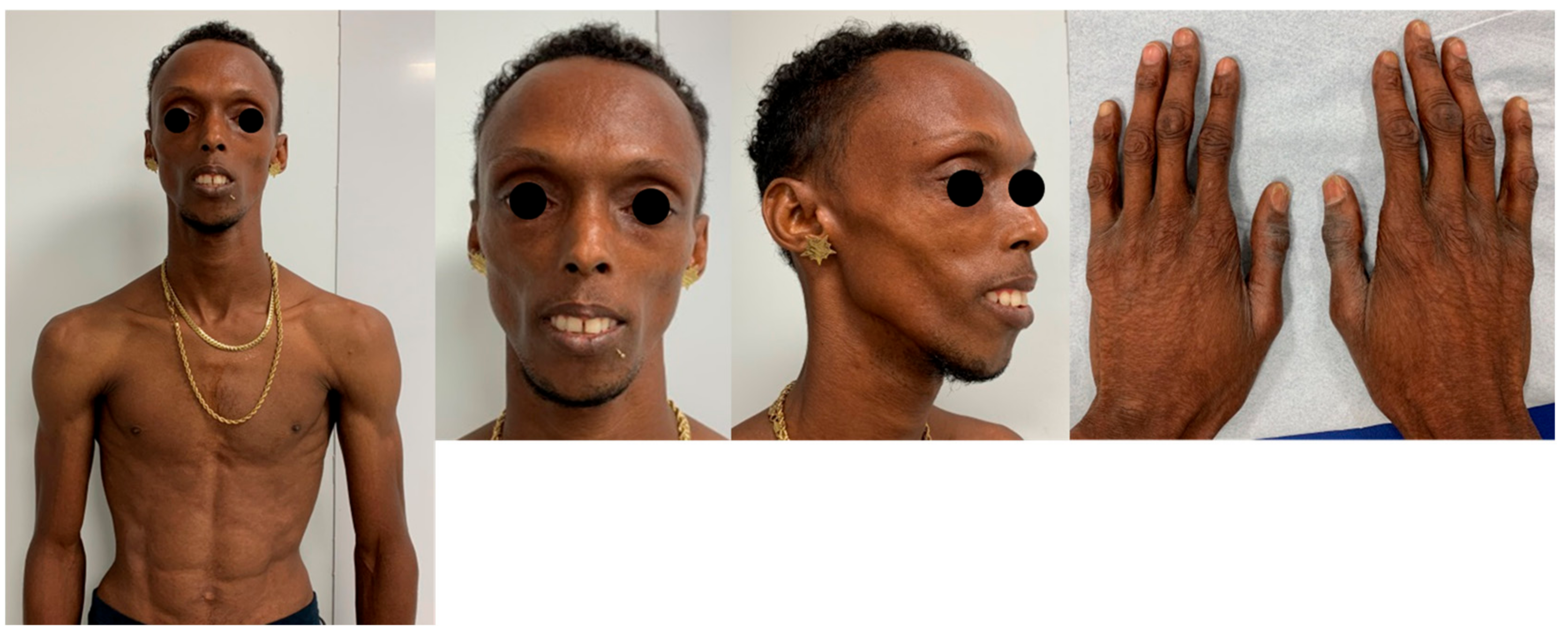
2. Case Presentation
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schreiber, K.H.; Kennedy, B.K. When Lamins Go Bad: Nuclear Structure and Disease. Cell 2013, 152, 1365–1375. [Google Scholar] [CrossRef] [PubMed]
- Bonne, G.; Barletta, M.R.D.; Varnous, S.; Bécane, H.-M.; Hammouda, E.-H.; Merlini, L.; Muntoni, F.; Greenberg, C.R.; Gary, F.; Urtizberea, J.-A.; et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat. Genet. 1999, 21, 285–288. [Google Scholar] [CrossRef] [PubMed]
- De Sandre-Giovannoli, A.; Chaouch, M.; Kozlov, S.; Vallat, J.-M.; Tazir, M.; Kassouri, N.; Szepetowski, P.; Hammadouche, T.; Vandenberghe, A.; Stewart, C.L.; et al. Homozygous Defects in LMNA, Encoding Lamin A/C Nuclear-Envelope Proteins, Cause Autosomal Recessive Axonal Neuropathy in Human (Charcot-Marie-Tooth Disorder Type 2) and Mouse. Am. J. Hum. Genet. 2002, 70, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Fatkin, D.; MacRae, C.; Sasaki, T.; Wolff, M.R.; Porcu, M.; Frenneaux, M.; Atherton, J.; Vidaillet, H.J.; Spudich, S.; De Girolami, U.; et al. Missense Mutations in the Rod Domain of the Lamin A/C Gene as Causes of Dilated Cardiomyopathy and Conduction-System Disease. N. Engl. J. Med. 1999, 341, 1715–1724. [Google Scholar] [CrossRef] [PubMed]
- Muchir, A. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum. Mol. Genet. 2000, 9, 1453–1459. [Google Scholar] [CrossRef]
- Raffaele di Barletta, M.; Ricci, E.; Galluzzi, G.; Tonali, P.; Mora, M.; Morandi, L.; Romorini, A.; Voit, T.; Orstavik, K.H.; Merlini, L.; et al. Different Mutations in the LMNA Gene Cause Autosomal Dominant and Autosomal Recessive Emery-Dreifuss Muscular Dystrophy. Am. J. Hum. Genet. 2000, 66, 1407–1412. [Google Scholar] [CrossRef]
- Cao, H. Nuclear lamin A/C R482Q mutation in Canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum. Mol. Genet. 2000, 9, 109–112. [Google Scholar] [CrossRef]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin A cause Hutchinson–Gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef]
- Novelli, G.; Muchir, A.; Sangiuolo, F.; Helbling-Leclerc, A.; D’Apice, M.R.; Massart, C.; Capon, F.; Sbraccia, P.; Federici, M.; Lauro, R.; et al. Mandibuloacral Dysplasia Is Caused by a Mutation in LMNA-Encoding Lamin A/C. Am. J. Hum. Genet. 2002, 71, 426–431. [Google Scholar] [CrossRef]
- De Sandre-Giovannoli, A.; Bernard, R.; Cau, P.; Navarro, C.; Amiel, J.; Boccaccio, I.; Lyonnet, S.; Stewart, C.L.; Munnich, A.; Le Merrer, M.; et al. Lamin a truncation in Hutchinson-Gilford progeria. Science 2003, 300, 2055. [Google Scholar] [CrossRef]
- Brodehl, A.; Gerull, B. Genetic Insights into Primary Restrictive Cardiomyopathy. JCM 2022, 11, 2094. [Google Scholar] [CrossRef] [PubMed]
- Caux, F.; Dubosclard, E.; Lascols, O.; Buendia, B.; Chazouillères, O.; Cohen, A.; Courvalin, J.-C.; Laroche, L.; Capeau, J.; Vigouroux, C.; et al. A new clinical condition linked to a novel mutation in lamins A and C with generalized lipoatrophy, insulin-resistant diabetes, disseminated leukomelanodermic papules, liver steatosis, and cardiomyopathy. J. Clin. Endocrinol. Metab. 2003, 88, 1006–1013. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Subramanyam, L.; Agarwal, A.K.; Simha, V.; Levine, B.; D’Apice, M.R.; Novelli, G.; Crow, Y. Atypical Progeroid Syndrome due to Heterozygous Missense LMNA Mutations. J. Clin. Endocrinol. Metab. 2009, 94, 4971–4983. [Google Scholar] [CrossRef]
- Hennekam, R.C.M. Hutchinson-Gilford progeria syndrome: Review of the phenotype. Am. J. Med. Genet. A 2006, 140, 2603–2624. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, F.; Evangelisti, C.; Cenni, V.; Fazio, A.; Paganelli, F.; Martelli, A.M.; Lattanzi, G. The Cutting Edge: The Role of mTOR Signaling in Laminopathies. Int. J. Mol. Sci. 2019, 20, 847. [Google Scholar] [CrossRef] [PubMed]
- Mosbah, H.; Vatier, C.; Boccara, F.; Jéru, I.; Lascols, O.; Vantyghem, M.-C.; Fève, B.; Donadille, B.; Sarrazin, E.; Benabbou, S.; et al. Looking at New Unexpected Disease Targets in LMNA-Linked Lipodystrophies in the Light of Complex Cardiovascular Phenotypes: Implications for Clinical Practice. Cells 2020, 9, 765. [Google Scholar] [CrossRef]
- Lee, S.; Park, S.M.; Kim, H.J.; Kim, J.-W.; Yu, D.S.; Lee, Y.B. Genomic diagnosis by whole genome sequencing in a Korean family with atypical progeroid syndrome. J. Dermatol. 2015, 42, 1149–1152. [Google Scholar] [CrossRef]
- Masson, E.; Zou, W.-B.; Génin, E.; Cooper, D.N.; Le Gac, G.; Fichou, Y.; Pu, N.; Rebours, V.; Férec, C.; Liao, Z.; et al. Expanding ACMG variant classification guidelines into a general framework. Hum. Genom. 2022, 16, 31. [Google Scholar] [CrossRef]
- Simm, D.; Hatje, K.; Kollmar, M. Waggawagga: Comparative visualization of coiled-coil predictions and detection of stable single α-helices (SAH domains). Bioinformatics 2015, 31, 767–769. [Google Scholar] [CrossRef]
- Bera, M.; Ainavarapu, S.R.K.; Sengupta, K. Significance of 1B and 2B domains in modulating elastic properties of lamin A. Sci. Rep. 2016, 6, 27879. [Google Scholar] [CrossRef]
- Wahbi, K.; Ben Yaou, R.; Gandjbakhch, E.; Anselme, F.; Gossios, T.; Lakdawala, N.K.; Stalens, C.; Sacher, F.; Babuty, D.; Trochu, J.-N.; et al. Development and Validation of a New Risk Prediction Score for Life-Threatening Ventricular Tachyarrhythmias in Laminopathies. Circulation 2019, 140, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Muchir, A.; Wu, W.; Sera, F.; Homma, S.; Worman, H.J. Mitogen-activated protein kinase kinase 1/2 inhibition and angiotensin II converting inhibition in mice with cardiomyopathy caused by lamin A/C gene mutation. Biochem. Biophys. Res. Commun. 2014, 452, 958–961. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, T.A.; Sahni, N.; Kubben, N.; Hill, D.E.; Vidal, M.; Burgess, R.C.; Roukos, V.; Misteli, T. Systematic identification of pathological lamin A interactors. Mol. Biol. Cell 2014, 25, 1493–1510. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.S.; Gordon, D.M.; Majumdar, U.; Lawrence, P.J.; Matos-Nieves, A.; Myers, K.; Kamp, A.N.; Leonard, J.C.; McBride, K.L.; White, P.; et al. Use of machine learning to classify high-risk variants of uncertain significance in lamin A/C cardiac disease. Heart Rhythm. 2022, 19, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, H.A.; Akimenko, M.-A.; Tesson, F. Cellular and Animal Models of Striated Muscle Laminopathies. Cells 2019, 8, 291. [Google Scholar] [CrossRef]
- Treiber, G.; Flaus Furmaniuk, A.; Guilleux, A.; Medjane, S.; Bonfanti, O.; Schneebeli, S.; Bernard, C.; Le-Moullec, N.; Bakiri, F.; Pholsena, M.; et al. A recurrent familial partial lipodystrophy due to a monoallelic or biallelic LMNA founder variant highlights the multifaceted cardiac manifestations of metabolic laminopathies. Eur. J. Endocrinol. 2021, 185, 453–462. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pedigree Number | I.1 | II.1 | II.2 | II.3 | II.4 | III.2 | III.1 |
|---|---|---|---|---|---|---|---|
| Sex | F | M | F | F | M | M | F |
| Age of diagnostic (years) | 44 | 44 | 34 | 25 | 18 | 10 | 11 |
| Age of death and age (years) | 48 | 44 | Alive (46) | Alive (37) | Alive (28) | 15 | Alive (21) |
| Height/Weight (cm/kg) | 167/41 | 166/40 | 175/32 | 160/30 | 170/53 | 170/41 | 168/40 |
| BMI (kg/m2) | 14.7 | 14.5 | 10.4 | 11.7 | 18.3 | 14.2 | 14.2 |
| Dysmorphism pattern | + | + | + | + | + | + | + |
| Lipodystrophy | + | + | + | + | + | + | + |
| High-pitched voice | + | - | + | + | + | - | + |
| Bird’s face | + | + | + | + | + | + | + |
| Alopecia | - | - | + | - | + | - | - |
| Cutaneous atrophy | nd | nd | + | - | + | - | - |
| Scoliosis | - | - | - | + | - | + | nd |
| ECG Conduction defects | LBBB | Unspecific delayed ventricular conduction | Incomplete RBBB | RBBB | Incomplete RBBB | LBBB | Incomplete RBBB |
| Ventricular arrythmias | Frequent VPB | Frequent VPBs | nd | Sustained VT | Frequent VPBs | - | nd |
| Valve Thickening | + | + | + | - | + | - | - |
| Aortic Stenosis | + | + | + | - | - | - | - |
| Congestive Heart Failure | + | - | - | - | - | + | - |
| Coronary angiogram | nd | Three-vessel disease | nd | Three-vessel disease | nd | Normal | nd |
| Hepatic cell insufficiency/Type 2 diabetis | - | - | - | - | - | - | - |
| LMNA c.407A>T (p.Asp136Val) | nd | + | nd | + | + | + | nd |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giguet-Valard, A.-G.; Monfort, A.; Lucron, H.; Mosbah, H.; Boccara, F.; Vatier, C.; Vigouroux, C.; Richard, P.; Wahbi, K.; Bellance, R.; et al. A Family with a Single LMNA Mutation Illustrates Diversity in Cardiac Phenotypes Associated with Laminopathic Progeroid Syndromes. Cardiogenetics 2023, 13, 135-144. https://doi.org/10.3390/cardiogenetics13040013
Giguet-Valard A-G, Monfort A, Lucron H, Mosbah H, Boccara F, Vatier C, Vigouroux C, Richard P, Wahbi K, Bellance R, et al. A Family with a Single LMNA Mutation Illustrates Diversity in Cardiac Phenotypes Associated with Laminopathic Progeroid Syndromes. Cardiogenetics. 2023; 13(4):135-144. https://doi.org/10.3390/cardiogenetics13040013
Chicago/Turabian StyleGiguet-Valard, Anna-Gaëlle, Astrid Monfort, Hugues Lucron, Helena Mosbah, Franck Boccara, Camille Vatier, Corinne Vigouroux, Pascale Richard, Karim Wahbi, Remi Bellance, and et al. 2023. "A Family with a Single LMNA Mutation Illustrates Diversity in Cardiac Phenotypes Associated with Laminopathic Progeroid Syndromes" Cardiogenetics 13, no. 4: 135-144. https://doi.org/10.3390/cardiogenetics13040013
APA StyleGiguet-Valard, A.-G., Monfort, A., Lucron, H., Mosbah, H., Boccara, F., Vatier, C., Vigouroux, C., Richard, P., Wahbi, K., Bellance, R., Sarrazin, E., & Inamo, J. (2023). A Family with a Single LMNA Mutation Illustrates Diversity in Cardiac Phenotypes Associated with Laminopathic Progeroid Syndromes. Cardiogenetics, 13(4), 135-144. https://doi.org/10.3390/cardiogenetics13040013