
Functional Characterization of the A414G Loss-of-Function Mutation in HCN4 Associated with Sinus Bradycardia



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmid Construction and Transfection
2.2. Patch-Clamp Experiments
2.3. Computer Simulations
2.4. Statistics
3. Results
3.1. Voltage-Clamp Experiments
3.1.1. Current Density
3.1.2. Activation Kinetics
3.1.3. Deactivation Kinetics and Reversal Potentials
3.2. Computer Simulations
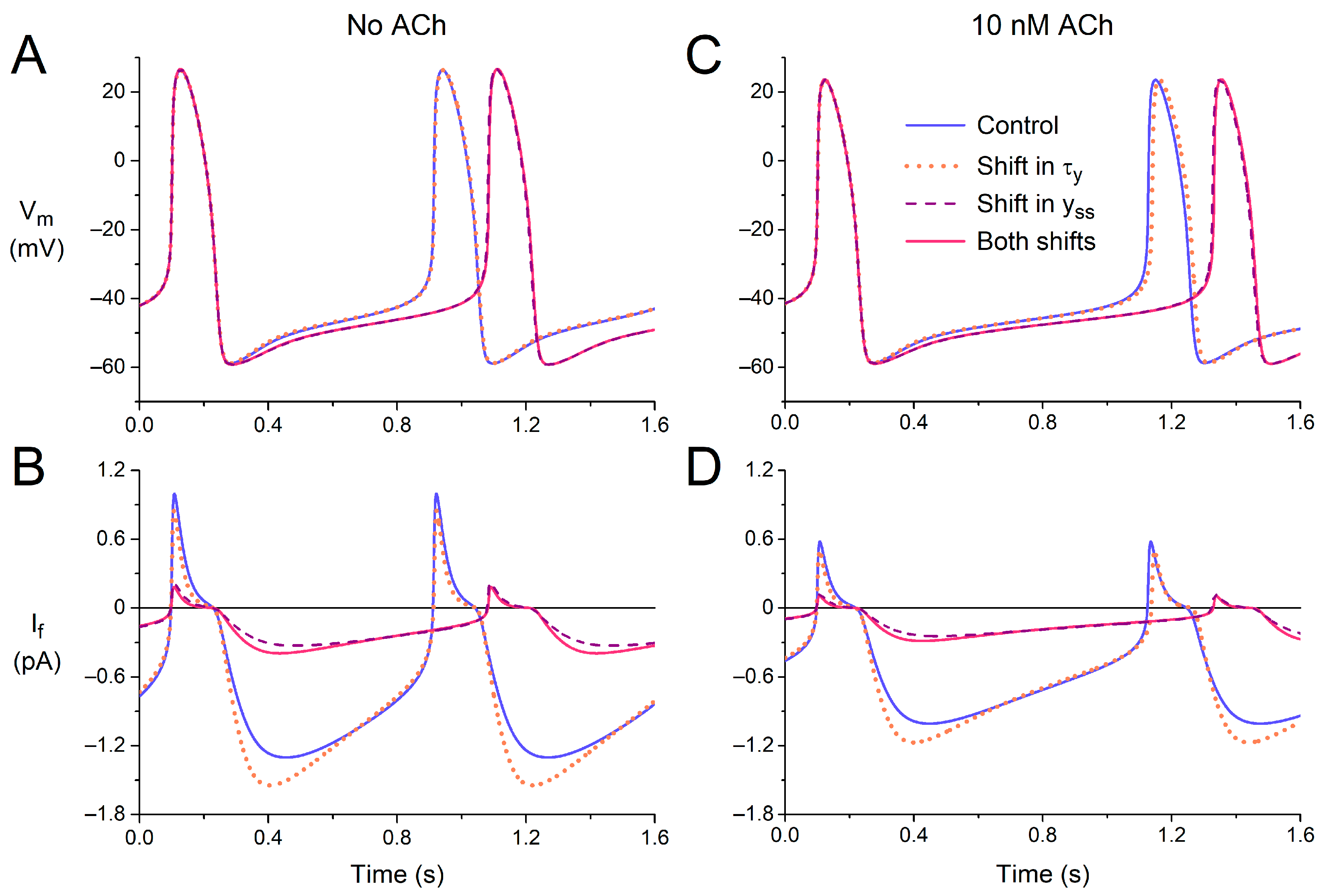
3.2.1. Effects of Shifts in Voltage Dependence
3.2.2. Effects of Atrial Load
3.2.3. Combined Effects of Atrial Load and Vagal Tone
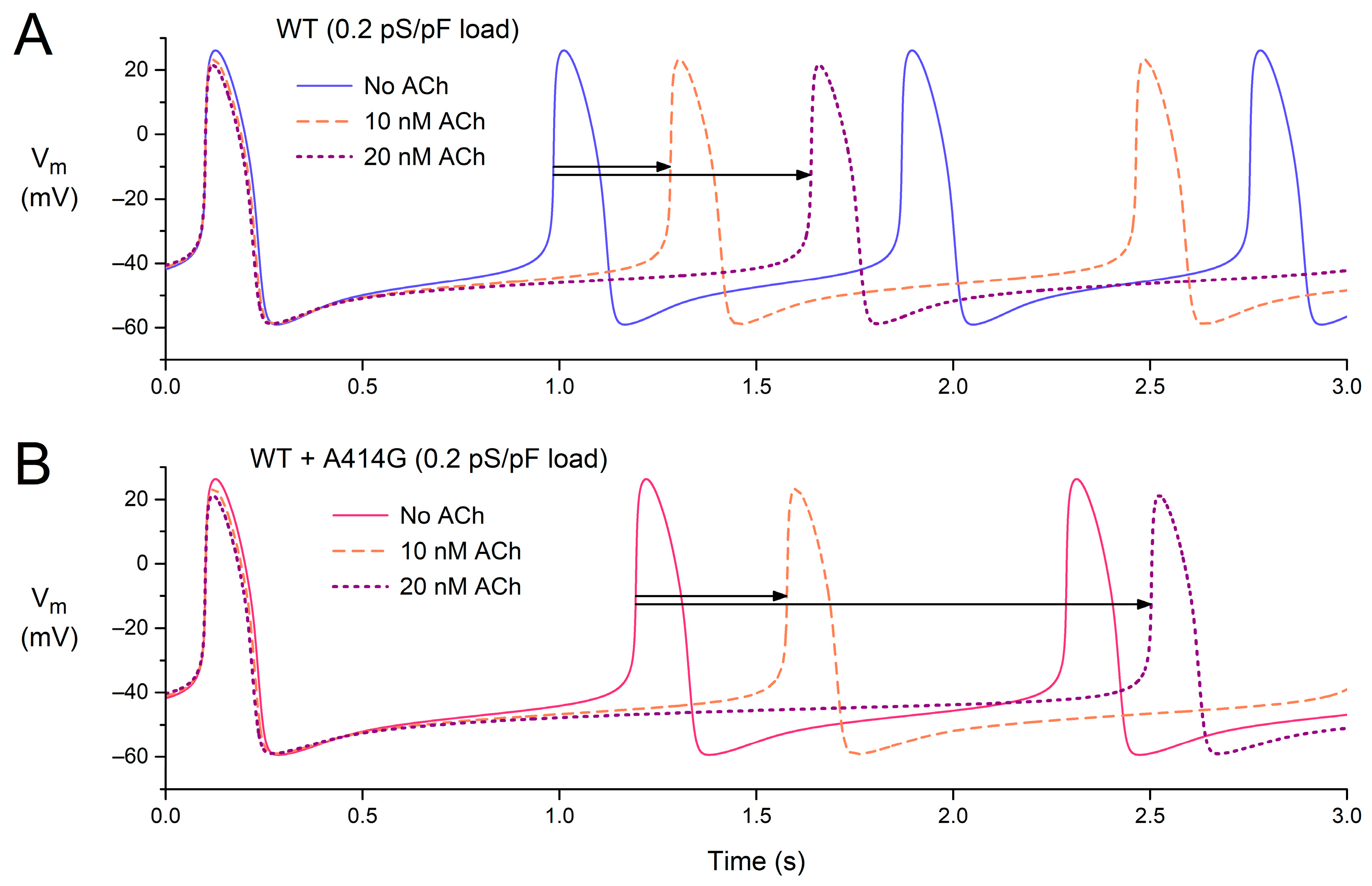
3.2.4. Beating Rate in the Presence of Atrial Load and Vagal Tone
4. Discussion
4.1. Overview
4.2. Bradycardia and LVNC
4.3. Limitations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Verkerk, A.O.; Van Ginneken, A.C.G.; Wilders, R. Pacemaker activity of the human sinoatrial node: Role of the hyperpolarization-activated current, If. Int. J. Cardiol. 2009, 132, 318–336. [Google Scholar] [CrossRef]
- Nof, E.; Antzelevitch, C.; Glikson, M. The contribution of HCN4 to normal sinus node function in humans and animal models. Pacing Clin. Electrophysiol. 2010, 33, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Baruscotti, M.; Barbuti, A.; Bucchi, A. The cardiac pacemaker current. J. Mol. Cell. Cardiol. 2010, 48, 55–64. [Google Scholar] [CrossRef] [PubMed]
- DiFrancesco, D. The role of the funny current in pacemaker activity. Circ. Res. 2010, 106, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Hennis, K.; Biel, M.; Wahl-Schott, C.; Fenske, S. Beyond pacemaking: HCN channels in sinoatrial node function. Prog. Biophys. Mol. Biol. 2021, 166, 51–60. [Google Scholar] [CrossRef]
- Hoekstra, M.; Van Ginneken, A.C.G.; Wilders, R.; Verkerk, A.O. HCN4 current during human sinoatrial node-like action potentials. Prog. Biophys. Mol. Biol. 2021, 166, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Wymore, R.; Yu, H.; Wu, J.; Wymore, R.T.; Pan, Z.; Robinson, R.B.; Dixon, J.E.; McKinnon, D.; Cohen, I.S. Distribution and prevalence of hyperpolarization-activated cation channel (HCN) mRNA expression in cardiac tissues. Circ. Res. 1999, 85, e1–e6. [Google Scholar] [CrossRef]
- Tellez, J.O.; Dobrzynski, H.; Greener, I.D.; Graham, G.M.; Laing, E.; Honjo, H.; Hubbard, S.J.; Boyett, M.R.; Billeter, R. Differential expression of ion channel transcripts in atrial muscle and sinoatrial node in rabbit. Circ. Res. 2006, 99, 1384–1393. [Google Scholar] [CrossRef]
- Brioschi, C.; Micheloni, S.; Tellez, J.O.; Pisoni, G.; Longhi, R.; Moroni, P.; Billeter, R.; Barbuti, A.; Dobrzynski, H.; Boyett, M.R.; et al. Distribution of the pacemaker HCN4 channel mRNA and protein in the rabbit sinoatrial node. J. Mol. Cell. Cardiol. 2009, 47, 221–227. [Google Scholar] [CrossRef]
- Chandler, N.J.; Greener, I.D.; Tellez, J.O.; Inada, S.; Musa, H.; Molenaar, P.; DiFrancesco, D.; Baruscotti, M.; Longhi, R.; Anderson, R.H.; et al. Molecular architecture of the human sinus node: Insights into the function of the cardiac pacemaker. Circulation 2009, 119, 1562–1575. [Google Scholar] [CrossRef]
- Schulze-Bahr, E.; Neu, A.; Friederich, P.; Kaupp, U.B.; Breithardt, G.; Pongs, O.; Isbrandt, D. Pacemaker channel dysfunction in a patient with sinus node disease. J. Clin. Investig. 2003, 111, 1537–1545. [Google Scholar] [CrossRef] [PubMed]
- Baruscotti, M.; Bottelli, G.; Milanesi, R.; DiFrancesco, J.C.; DiFrancesco, D. HCN-related channelopathies. Pflügers Arch. 2010, 460, 405–415. [Google Scholar] [CrossRef] [PubMed]
- DiFrancesco, D. Funny channel gene mutations associated with arrhythmias. J. Physiol. 2013, 591, 4117–4124. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.O.; Wilders, R. Pacemaker activity of the human sinoatrial node: Effects of HCN4 mutations on the hyperpolarization-activated current. Europace 2014, 16, 384–395. [Google Scholar] [CrossRef]
- Verkerk, A.O.; Wilders, R. Pacemaker activity of the human sinoatrial node: An update on the effects of mutations in HCN4 on the hyperpolarization-activated current. Int. J. Mol. Sci. 2015, 16, 3071–3094. [Google Scholar] [CrossRef] [PubMed]
- DiFrancesco, D. HCN4, sinus bradycardia and atrial fibrillation. Arrhythmia Electrophysiol. Rev. 2015, 4, 9–13. [Google Scholar] [CrossRef]
- Milano, A.; Vermeer, A.M.C.; Lodder, E.M.; Barc, J.; Verkerk, A.O.; Postma, A.V.; Van der Bilt, I.A.C.; Baars, M.J.H.; Van Haelst, P.L.; Caliskan, K.; et al. HCN4 mutations in multiple families with bradycardia and left ventricular noncompaction cardiomyopathy. J. Am. Coll. Cardiol. 2014, 64, 745–756. [Google Scholar] [CrossRef]
- Schweizer, P.A.; Schröter, J.; Greiner, S.; Haas, J.; Yampolsky, P.; Mereles, D.; Buss, S.J.; Seyler, C.; Bruehl, C.; Draguhn, A.; et al. The symptom complex of familial sinus node dysfunction and myocardial noncompaction is associated with mutations in the HCN4 channel. J. Am. Coll. Cardiol. 2014, 64, 757–767. [Google Scholar] [CrossRef]
- Millat, G.; Janin, A.; De Tauriac, O.; Roux, A.; Dauphin, C. HCN4 mutation as a molecular explanation on patients with bradycardia and non-compaction cardiomyopathy. Eur. J. Med. Genet. 2015, 58, 439–442. [Google Scholar] [CrossRef]
- Vermeer, A.M.C.; Lodder, E.M.; Thomas, D.; Duijkers, F.A.M.; Marcelis, C.; Van Gorselen, E.O.F.; Fortner, P.; Buss, S.J.; Mereles, D.; Katus, H.A.; et al. Dilation of the aorta ascendens forms part of the clinical spectrum of HCN4 mutations. J. Am. Coll. Cardiol. 2016, 67, 2313–2315. [Google Scholar] [CrossRef]
- Servatius, H.; Porro, A.; Pless, S.A.; Schaller, A.; Asatryan, B.; Tanner, H.; De Marchi, S.F.; Roten, L.; Seiler, J.; Haeberlin, A.; et al. Phenotypic spectrum of HCN4 mutations: A clinical case. Circ. Genom. Precis. Med. 2018, 11, e002033. [Google Scholar] [CrossRef]
- Hanania, H.L.; Regalado, E.S.; Guo, D.-C.; Xu, L.; Demo, E.; Sallee, D.; Milewicz, D.M. Do HCN4 variants predispose to thoracic aortic aneurysms and dissections? Circ. Genom. Precis. Med. 2019, 12, e002626. [Google Scholar] [CrossRef]
- Alonso-Fernández-Gatta, M.; Gallego-Delgado, M.; Caballero, R.; Villacorta, E.; Díaz-Peláez, E.; García-Berrocal, B.; Crespo-García, T.; Plata-Izquierdo, B.; Marcos-Vadillo, E.; García-Cuenllas, L.; et al. A rare HCN4 variant with combined sinus bradycardia, left atrial dilatation, and hypertrabeculation/left ventricular noncompaction phenotype. Rev. Esp. Cardiol. 2021, 74, 781–789. [Google Scholar] [CrossRef]
- Brunet-Garcia, L.; Odori, A.; Fell, H.; Field, E.; Roberts, A.M.; Starling, L.; Kaski, J.P.; Cervi, E. Noncompaction cardiomyopathy, sick sinus disease, and aortic dilatation: Too much for a single diagnosis? JACC Case Rep. 2022, 4, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Chanavat, V.; Janin, A.; Millat, G. A fast and cost-effective molecular diagnostic tool for genetic diseases involved in sudden cardiac death. Clin. Chim. Acta 2016, 453, 80–85. [Google Scholar] [CrossRef]
- Richard, P.; Ader, F.; Roux, M.; Donal, E.; Eicher, J.C.; Aoutil, N.; Huttin, O.; Selton-Suty, C.; Coisne, D.; Jondeau, G.; et al. Targeted panel sequencing in adult patients with left ventricular non-compaction reveals a large genetic heterogeneity. Clin. Genet. 2019, 95, 356–367. [Google Scholar] [CrossRef]
- Paszkowska, A.; Piekutowska-Abramczuk, D.; Ciara, E.; Mirecka-Rola, A.; Brzezinska, M.; Wicher, D.; Kostrzewa, G.; Sarnecki, J.; Ziółkowska, L. Clinical presentation of left ventricular noncompaction cardiomyopathy and bradycardia in three families carrying HCN4 pathogenic variants. Genes 2022, 13, 477. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Ohno, S.; Murakami, T.; Yoshida, K.; Mishima, H.; Fukuoka, T.; Kimoto, H.; Sakamoto, R.; Ohkusa, T.; Aiba, T.; et al. Sick sinus syndrome with HCN4 mutations shows early onset and frequent association with atrial fibrillation and left ventricular noncompaction. Heart Rhythm 2017, 14, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, R.; Kinoshita, K.; Hata, Y.; Abe, M.; Matsuoka, K.; Hirono, K.; Kano, M.; Nakazawa, M.; Ichida, F.; Nishida, N.; et al. A mutant HCN4 channel in a family with bradycardia, left bundle branch block, and left ventricular noncompaction. Heart Vessel. 2018, 33, 802–819. [Google Scholar] [CrossRef]
- Ponińska, J.; Michalak, E.; Śpiewak, M.; Lutyńska, A.; Płoski, R.; Bilińska, Z.T. A novel HCN4 variant related to familial sinus bradycardia, left ventricular noncompaction, and thoracic aortic aneurysm. Pol. Arch. Intern. Med. 2021, 131, 70–72. [Google Scholar] [CrossRef]
- Weigl, I.; Geschwill, P.; Reiss, M.; Bruehl, C.; Draguhn, A.; Koenen, M.; Sedaghat-Hamedani, F.; Meder, B.; Thomas, D.; Katus, H.A.; et al. The C-terminal HCN4 variant P883R alters channel properties and acts as genetic modifier of atrial fibrillation and structural heart disease. Biochem. Biophys. Res. Commun. 2019, 519, 141–147. [Google Scholar] [CrossRef]
- Fabbri, A.; Fantini, M.; Wilders, R.; Severi, S. Computational analysis of the human sinus node action potential: Model development and effects of mutations. J. Physiol. 2017, 595, 2365–2396. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.O.; Wilders, R. Mechanism of sinus bradycardia in carriers of the A414G mutation in the HCN4 gene. Comp. Cardiol. 2019, 46, 147. [Google Scholar] [CrossRef]
- DiFrancesco, D.; Ferroni, A.; Mazzanti, M.; Tromba, C. Properties of the hyperpolarizing-activated current (if) in cells isolated from the rabbit sino-atrial node. J. Physiol. 1986, 377, 61–88. [Google Scholar] [CrossRef] [PubMed]
- Van Ginneken, A.C.G.; Giles, W. Voltage clamp measurements of the hyperpolarization-activated inward current If in single cells from rabbit sino-atrial node. J. Physiol. 1991, 434, 57–83. [Google Scholar] [CrossRef]
- Lloyd, C.M.; Lawson, J.R.; Hunter, P.J.; Nielsen, P.F. The CellML model repository. Bioinformatics 2008, 24, 2122–2123. [Google Scholar] [CrossRef]
- Cuellar, A.A.; Lloyd, C.M.; Nielsen, P.F.; Bullivant, D.P.; Nickerson, D.P.; Hunter, P.J. An overview of CellML 1.1, a biological model description language. Simulation 2003, 79, 740–747. [Google Scholar] [CrossRef]
- Garny, A.; Kohl, P.; Noble, D. Cellular open resource (COR): A public CellML based environment for modelling biological function. Int. J. Bifurc. Chaos 2003, 13, 3579–3590. [Google Scholar] [CrossRef]
- Verkerk, A.O.; Den Ruijter, H.M.; Bourier, J.; Boukens, B.J.; Brouwer, I.A.; Wilders, R.; Coronel, R. Dietary fish oil reduces pacemaker current and heart rate in rabbit. Heart Rhythm 2009, 6, 1485–1492. [Google Scholar] [CrossRef]
- Kodama, I.; Boyett, M.R. Regional differences in the electrical activity of the rabbit sinus node. Pflügers Arch. 1985, 404, 214–226. [Google Scholar] [CrossRef]
- Boyett, M.R.; Honjo, H.; Kodama, I. The sinoatrial node, a heterogeneous pacemaker structure. Cardiovasc. Res. 2000, 47, 658–687. [Google Scholar] [CrossRef]
- Kirchhof, C.J.H.J.; Bonke, F.I.M.; Allessie, M.A.; Lammers, W.J.E.P. The influence of the atrial myocardium on impulse formation in the rabbit sinus node. Pflügers Arch. 1987, 410, 198–203. [Google Scholar] [CrossRef]
- Towbin, J.A.; Lorts, A.; Jefferies, J.L. Left ventricular non-compaction cardiomyopathy. Lancet 2015, 386, 813–825. [Google Scholar] [CrossRef]
- Cannie, D.; Elliott, P. The genetics of left ventricular noncompaction. Curr. Opin. Cardiol. 2021, 36, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J.; Stöllberger, C. Left ventricular noncompaction syndrome: Genetic insights and therapeutic perspectives. Curr. Cardiol. Rep. 2020, 22, 84. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Makita, N.; Xing, Y.; Watanabe, S.; Futatani, T.; Ye, F.; Saito, K.; Ibuki, K.; Watanabe, K.; Hirono, K.; et al. SCN5A variants in Japanese patients with left ventricular noncompaction and arrhythmia. Mol. Genet. Metab. 2008, 93, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Nakamura, Y.; Terano, K.; Ando, T.; Hishitani, T.; Hoshino, K. Isolated non-compaction of the ventricular myocardium associated with long QT syndrome: A report of 2 cases. Circ. J. 2009, 73, 2169–2172. [Google Scholar] [CrossRef]
- Jongbloed, R.; Marcelis, C.; Velter, C.; Doevendans, P.; Geraedts, J.; Smeets, H. DHPLC analysis of potassium ion channel genes in congenital long QT syndrome. Hum. Mutat. 2002, 20, 382–391. [Google Scholar] [CrossRef]
- Bellocq, C.; Wilders, R.; Schott, J.-J.; Louérat-Oriou, B.; Boisseau, P.; Le Marec, H.; Escande, D.; Baró, I. A common antitussive drug, clobutinol, precipitates the long QT syndrome 2. Mol. Pharmacol. 2004, 66, 1093–1102. [Google Scholar] [CrossRef]
- Xu, B.; Li, K.; Liu, F.; Kong, L.; Yang, J.; Zhou, B.; Lv, T.; Liu, Y.; She, F.; He, R.; et al. Mexiletine shortened QT interval and reduced ventricular arrhythmias in a pedigree of type 2 long QT syndrome combined with left ventricular non-compaction. Int. Heart J. 2021, 62, 427–431. [Google Scholar] [CrossRef]
- Caiffa, T.; Tessitore, A.; Leoni, L.; Reffo, E.; Chicco, D.; D’Agata Mottolese, B.; Rubinato, E.; Girotto, G.; Lenarduzzi, S.; Barbi, E.; et al. Long QT syndrome and left ventricular non-compaction in a family with KCNH2 mutation: A case report. Front. Pediatr. 2022, 10, 970240. [Google Scholar] [CrossRef] [PubMed]
- Maddali, M.M.; Thomas, E.; Al Abri, I.A.; Patel, M.H.; Al Maskari, S.N.; Al Yamani, M.I. Dilated cardiomyopathy phenotype-associated left ventricular noncompaction and congenital long QT syndrome type-2 in infants with KCNH2 gene mutation: Anesthetic considerations. J. Cardiothorac. Vasc. Anesth. 2022, 36, 3662–3667. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Liu, X.; Hao, X.; Zhou, X.; Wang, J.; Han, J.; Liang, M.; Zhang, H.; He, Y. Case report: Biventricular noncompaction cardiomyopathy with pulmonary stenosis and bradycardia in a fetus with KCNH2 mutation. Front. Genet. 2022, 13, 821226. [Google Scholar] [CrossRef]
- Nakashima, K.; Kusakawa, I.; Yamamoto, T.; Hirabayashi, S.; Hosoya, R.; Shimizu, W.; Sumitomo, N. A left ventricular noncompaction in a patient with long QT syndrome caused by a KCNQ1 mutation: A case report. Heart Vessel. 2013, 28, 126–129. [Google Scholar] [CrossRef]
- Kharbanda, M.; Hunter, A.; Tennant, S.; Moore, D.; Curtis, S.; Hancox, J.C.; Murday, V. Long QT syndrome and left ventricular noncompaction in 4 family members across 2 generations with KCNQ1 mutation. Eur. J. Med. Genet. 2017, 60, 233–238. [Google Scholar] [CrossRef]
- Verkerk, A.O.; Wilders, R. Human sinoatrial node pacemaker activity: Role of the slow component of the delayed rectifier K+ current, IKs. Int. J. Mol. Sci. 2023, 24, 7264. [Google Scholar] [CrossRef]
- Wilders, R.; Verkerk, A.O. Long QT syndrome and sinus bradycardia–A mini review. Front. Cardiovasc. Med. 2018, 5, 106. [Google Scholar] [CrossRef]
- Hodgkin, A.L.; Huxley, A.F. A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 1952, 117, 500–544. [Google Scholar] [CrossRef] [PubMed]
- DiFrancesco, D. The cardiac hyperpolarizing-activated current, if. Origins and developments. Prog. Biophys. Mol. Biol. 1985, 46, 163–183. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.C.; Joyner, R.W. Electrotonic influences on action potentials from isolated ventricular cells. Circ. Res. 1990, 67, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, E.I.; Honjo, H.; Anno, T.; Boyett, M.R.; Kodama, I.; Toyama, J. Modulation of pacemaker activity of sinoatrial node cells by electrical load imposed by an atrial cell model. Am. J. Physiol. 1995, 269, H1735–H1742. [Google Scholar] [CrossRef] [PubMed]
- Kohl, P.; Noble, D.; Winslow, R.L.; Hunter, P.J. Computational modelling of biological systems: Tools and visions. Philos. Trans. A Math. Phys. Eng. Sci. 2000, 358, 579–610. [Google Scholar] [CrossRef]
- Clayton, R.H.; Bernus, O.; Cherry, E.M.; Dierckx, H.; Fenton, F.H.; Mirabella, L.; Panfilov, A.V.; Sachse, F.B.; Seemann, G.; Zhang, H. Models of cardiac tissue electrophysiology: Progress, challenges and open questions. Prog. Biophys. Mol. Biol. 2011, 104, 22–48. [Google Scholar] [CrossRef]
- Maleckar, M.M.; Greenstein, J.L.; Trayanova, N.A.; Giles, W.R. Mathematical simulations of ligand-gated and cell-type specific effects on the action potential of human atrium. Prog. Biophys. Mol. Biol. 2008, 98, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Peischard, S.; Möller, M.; Disse, P.; Ho, H.T.; Verkerk, A.O.; Strutz-Seebohm, N.; Budde, T.; Meuth, S.G.; Schweizer, P.A.; Morris, S.; et al. Virus-induced inhibition of cardiac pacemaker channel HCN4 triggers bradycardia in human-induced stem cell system. Cell. Mol. Life Sci. 2022, 79, 440. [Google Scholar] [CrossRef]
- Wiesinger, A.; Li, J.; Fokkert, L.; Bakker, P.; Verkerk, A.O.; Christoffels, V.M.; Boink, G.J.J.; Devalla, H.D. A single cell transcriptional roadmap of human pacemaker cell differentiation. Elife 2022, 11, e76781. [Google Scholar] [CrossRef]
- Li, J.; Wiesinger, A.; Fokkert, L.; Boukens, B.J.; Verkerk, A.O.; Christoffels, V.M.; Boink, G.J.J.; Devalla, H.D. Molecular and electrophysiological evaluation of human cardiomyocyte subtypes to facilitate generation of composite cardiac models. J. Tissue Eng. 2022, 13, 20417314221127908. [Google Scholar] [CrossRef]
- Li, J.; Wiesinger, A.; Fokkert, L.; Boukens, B.; Verkerk, A.; Christoffels, V.; Boink, G.; Devalla, H. Comparative characterization of single cells and engineered heart tissues from hiPSC-derived cardiomyocyte subtypes. Cardiovasc. Res. 2022, 118 (Suppl. S1), cvac066.024. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verkerk, A.O.; Wilders, R. Functional Characterization of the A414G Loss-of-Function Mutation in HCN4 Associated with Sinus Bradycardia. Cardiogenetics 2023, 13, 117-134. https://doi.org/10.3390/cardiogenetics13030012
Verkerk AO, Wilders R. Functional Characterization of the A414G Loss-of-Function Mutation in HCN4 Associated with Sinus Bradycardia. Cardiogenetics. 2023; 13(3):117-134. https://doi.org/10.3390/cardiogenetics13030012
Chicago/Turabian StyleVerkerk, Arie O., and Ronald Wilders. 2023. "Functional Characterization of the A414G Loss-of-Function Mutation in HCN4 Associated with Sinus Bradycardia" Cardiogenetics 13, no. 3: 117-134. https://doi.org/10.3390/cardiogenetics13030012
APA StyleVerkerk, A. O., & Wilders, R. (2023). Functional Characterization of the A414G Loss-of-Function Mutation in HCN4 Associated with Sinus Bradycardia. Cardiogenetics, 13(3), 117-134. https://doi.org/10.3390/cardiogenetics13030012