
Spontaneous Coronary Artery Dissection as Presenting Feature of Vascular Ehlers-Danlos Syndrome


{kind=link}
Abstract
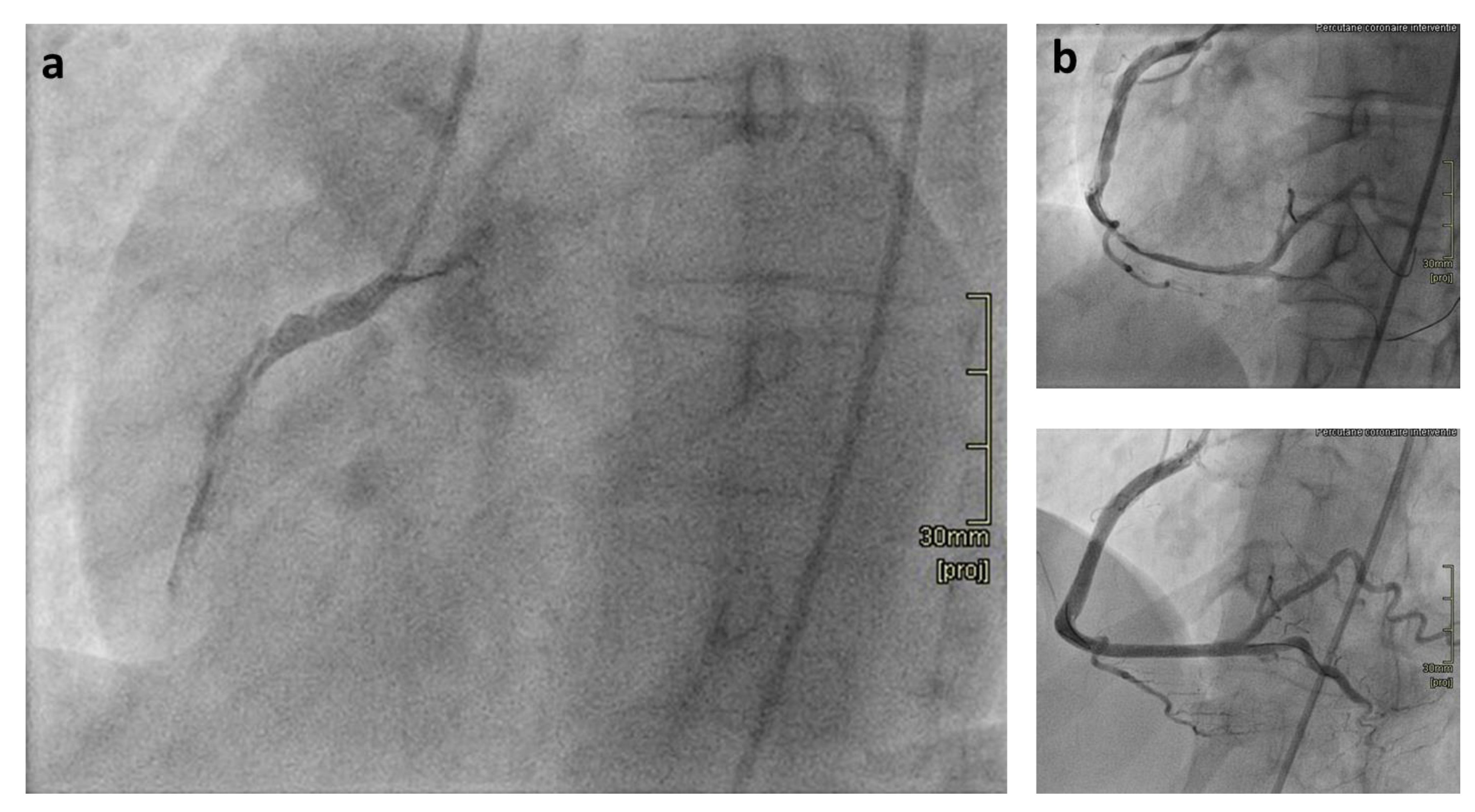
:1. Introduction
2. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vrints, C.J. Spontaneous Coronary Artery Dissection. Heart 2010, 96, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Carss, K.J.; Baranowska, A.A.; Armisen, J.; Webb, T.R.; Hamby, S.E.; Premawardhana, D.; Al-Hussaini, A.; Wood, A.; Wang, Q.; Deevi, S.V.V.; et al. Spontaneous Coronary Artery Dissection: Insights on Rare Genetic Variation from Genome Sequencing. Circ. Genom. Precis. Med. 2020, 13, e003030. [Google Scholar] [CrossRef] [PubMed]
- Byers, P.H.; Belmont, J.; Black, J.; De Backer, J.; Frank, M.; Jeunemaitre, X.; Johnson, D.; Pepin, M.; Robert, L.; Sanders, L.; et al. Diagnosis, natural history, and management in vascular Ehlers-Danlos syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 40–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 8–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, K.T.; Perdu, J.; De Backer, J.; Bozec, E.; Collignon, P.; Emmerich, J.; Fauret, A.L.; Fiessinger, J.N.; Germain, D.P.; Georgesco, G.; et al. Effect of celiprolol on prevention of cardiovascular events in vascular Ehlers-Danlos syndrome: A prospective randomised, open, blinded-endpoints trial. Lancet 2010, 376, 1476–1484. [Google Scholar] [CrossRef]
- Henkin, S.; Negrotto, S.M.; Tweet, M.S.; Kirmani, S.; Deyle, D.R.; Gulati, R.; Olson, T.M.; Hayes, S.N. Spontaneous coronary artery dissection and its association with heritable connective tissue disorders. Heart 2016, 102, 876–881. [Google Scholar] [CrossRef] [PubMed]
- Overwater, E.; Marsili, L.; Baars, M.J.H.; Baas, A.F.; van de Beek, I.; Dulfer, E.; van Hagen, J.M.; Hilhorst-Hofstee, Y.; Kempers, M.; Krapels, I.P.; et al. Results of next-generation sequencing gene panel diagnostics including copy-number variation analysis in 810 patients suspected of heritable thoracic aortic disorders. Hum. Mutat. 2018, 39, 1173–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beitsch, P.D.; Whitworth, P.W.; Hughes, K.; Patel, R.; Rosen, B.; Compagnoni, G.; Baron, P.; Simmons, R.; Smith, L.A.; Grady, I.; et al. Underdiagnosis of Hereditary Breast Cancer: Are Genetic Testing Guidelines a Tool or an Obstacle? J. Clin. Oncol. 2019, 37, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Copur, M.S. Universal Genetic Testing for All Breast Cancer Patients. Oncology 2019, 33, 68371. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bos, J.; Overwater, E.; Dirksen, M.T.; Simsek, S.; Demirdas, S.; Houweling, A.C. Spontaneous Coronary Artery Dissection as Presenting Feature of Vascular Ehlers-Danlos Syndrome. Cardiogenetics 2021, 11, 129-131. https://doi.org/10.3390/cardiogenetics11030014
Bos J, Overwater E, Dirksen MT, Simsek S, Demirdas S, Houweling AC. Spontaneous Coronary Artery Dissection as Presenting Feature of Vascular Ehlers-Danlos Syndrome. Cardiogenetics. 2021; 11(3):129-131. https://doi.org/10.3390/cardiogenetics11030014
Chicago/Turabian StyleBos, J., E. Overwater, M.T. Dirksen, S. Simsek, S. Demirdas, and A.C. Houweling. 2021. "Spontaneous Coronary Artery Dissection as Presenting Feature of Vascular Ehlers-Danlos Syndrome" Cardiogenetics 11, no. 3: 129-131. https://doi.org/10.3390/cardiogenetics11030014
APA StyleBos, J., Overwater, E., Dirksen, M. T., Simsek, S., Demirdas, S., & Houweling, A. C. (2021). Spontaneous Coronary Artery Dissection as Presenting Feature of Vascular Ehlers-Danlos Syndrome. Cardiogenetics, 11(3), 129-131. https://doi.org/10.3390/cardiogenetics11030014