
Genetic Diagnosis in Sudden Cardiac Death: The Crucial Role of Multidisciplinary Care

 and
and 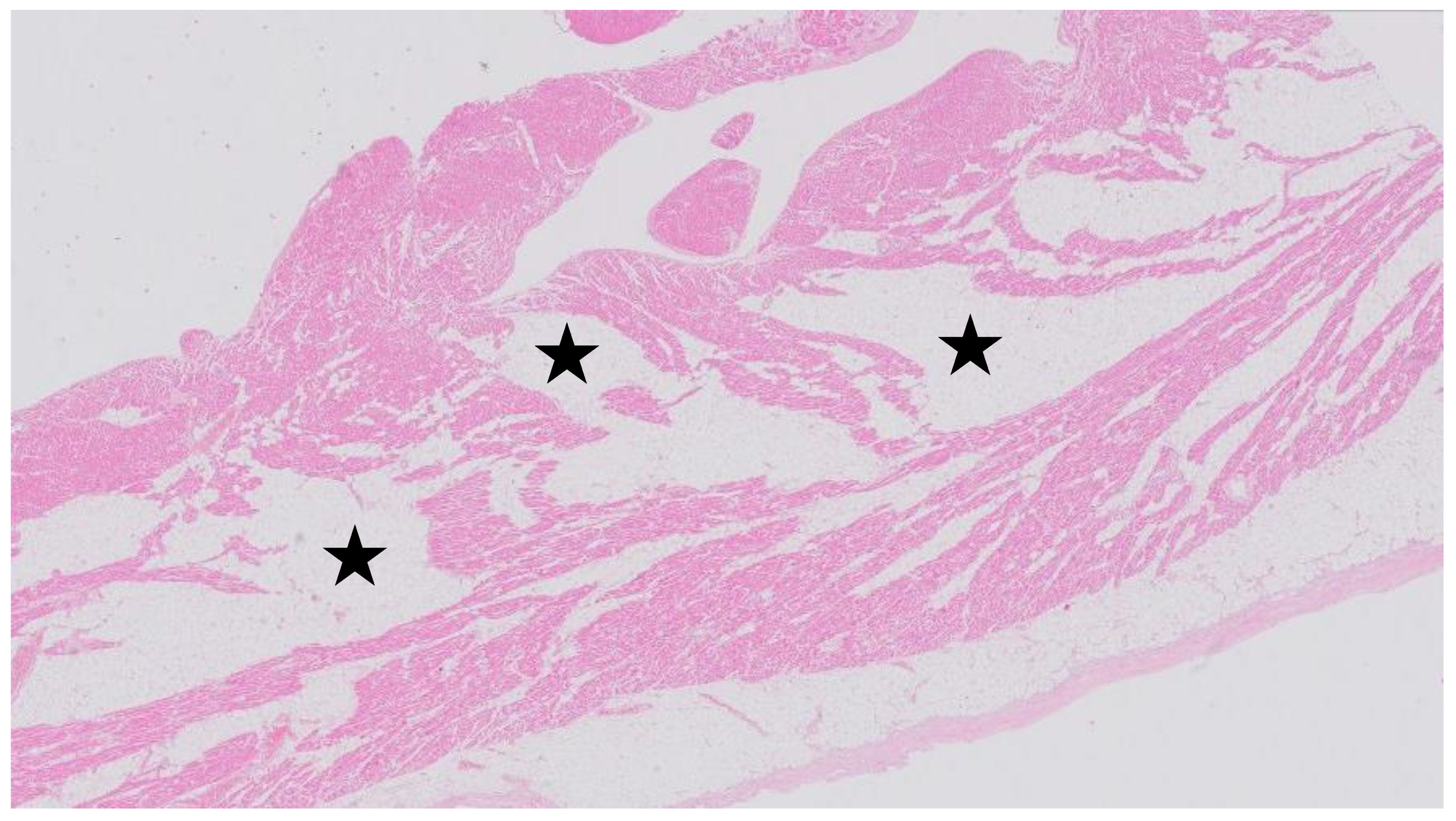{kind=link}
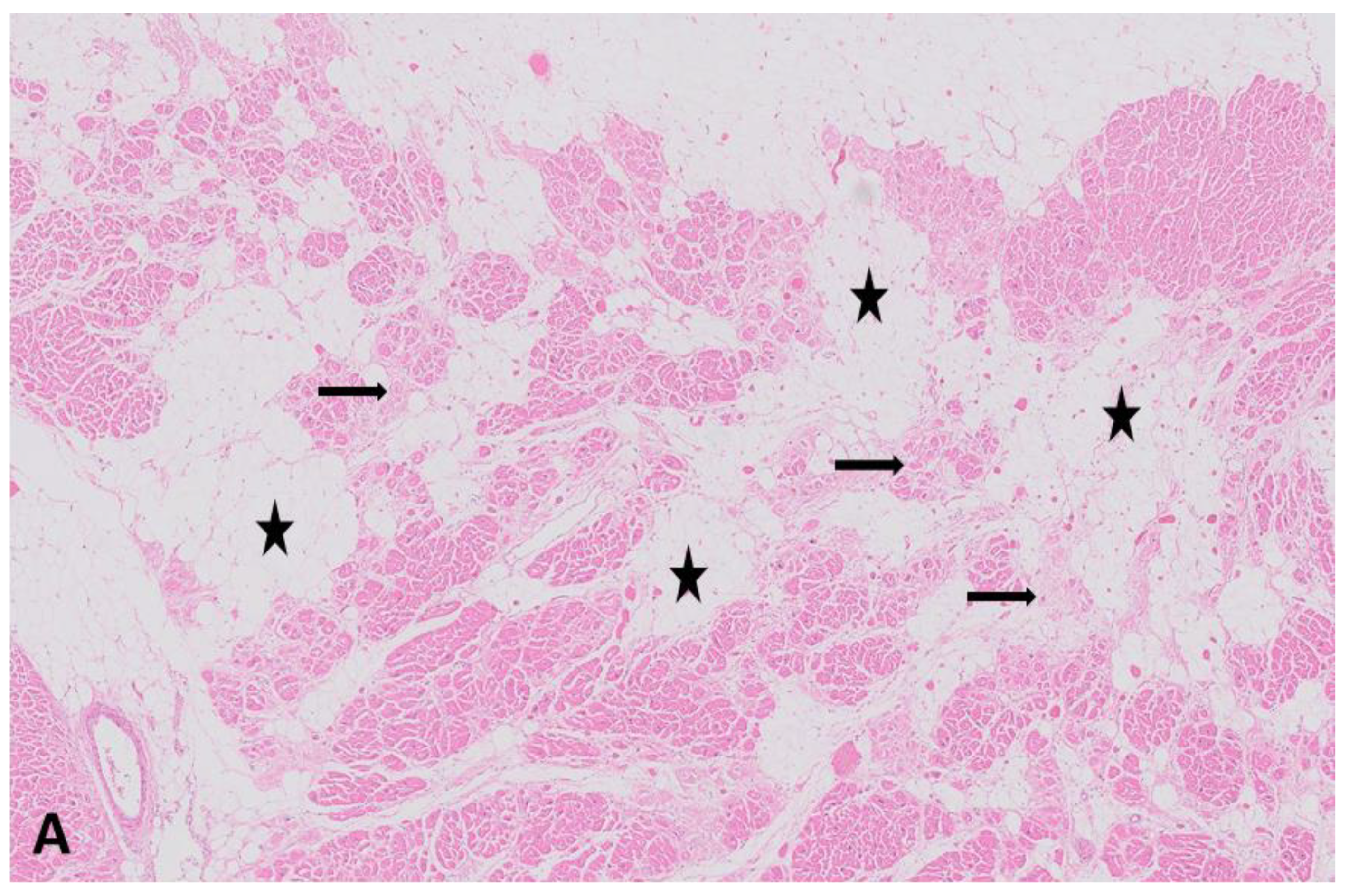{kind=link}
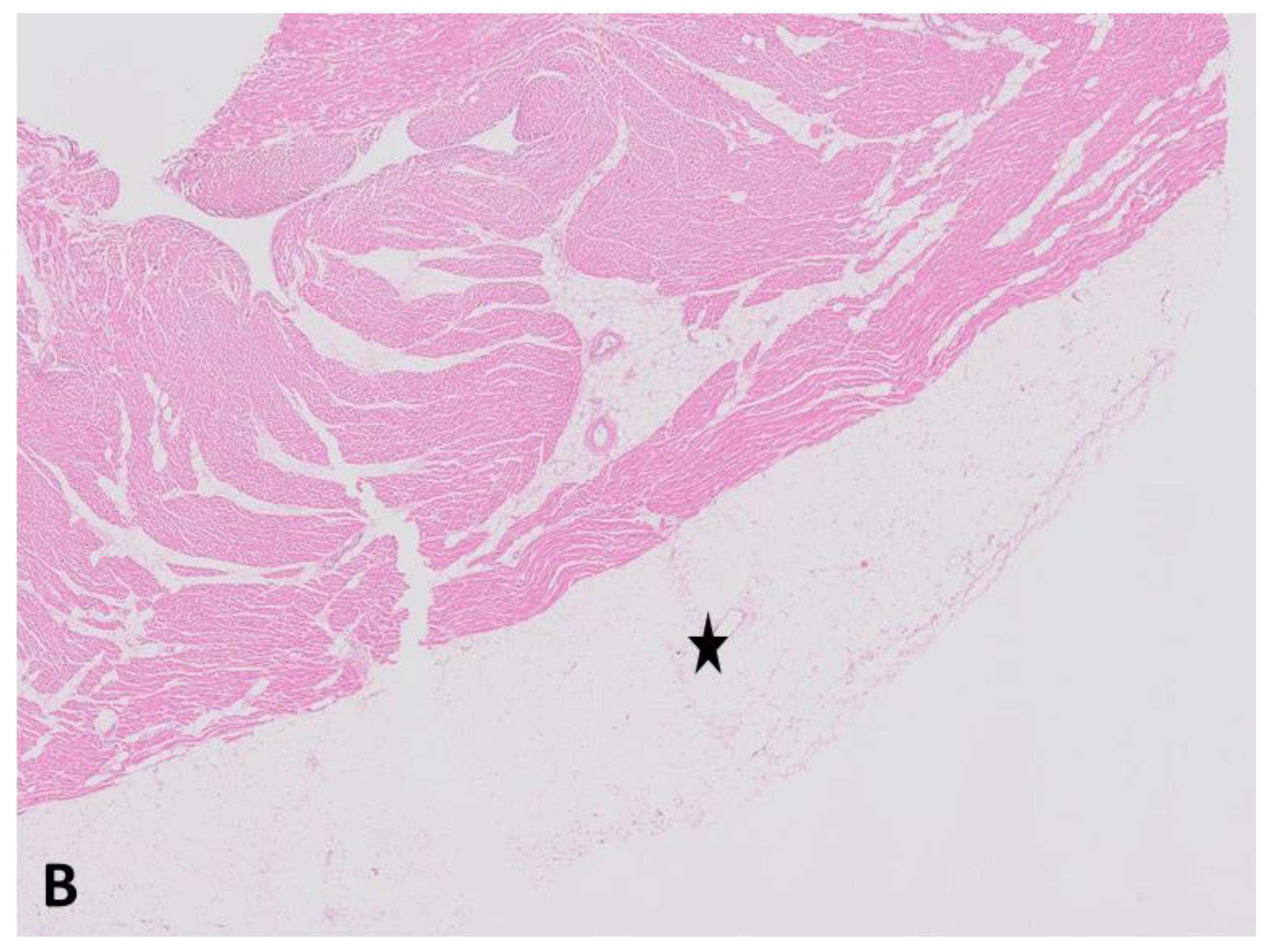{kind=link}
Abstract
1. Introduction
2. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Priori, S.G.; Blomström-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 2015, 36, 2793–2867. [Google Scholar] [PubMed]
- Bagnall, R.D.; Ingles, J.; Dinger, M.E.; Cowley, M.J.; Ross, S.B.; Minoche, A.E.; Lal, S.; Turner, C.; Colley, A.; Rajagopalan, S.; et al. Whole Genome Sequencing Improves Outcomes of Genetic Testing in Patients with Hypertrophic Cardiomyopathy. J. Am. Coll Cardiol. 2018, 72, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Overwater, E.; Marsili, L.; Baars, M.J.; Baas, A.F.; van de Beek, I.; Dulfer, E.; van Hagen, J.M.; Hilhorst-Hofstee, Y.; Kempers, M.; Krapels, I.P.; et al. Results of next-generation sequencing gene panel diagnostics including copy-number variation analysis in 810 patients suspected of heritable thoracic aortic disorders. Hum. Mutat. 2018, 39, 1173–1192. [Google Scholar] [CrossRef] [PubMed]
- Lahrouchi, N.; Raju, H.; Lodder, E.M.; Papatheodorou, E.; Ware, J.S.; Papadakis, M.; Tadros, R.; Cole, D.; Skinner, J.R.; Crawford, J.; et al. Utility of Post-Mortem Genetic Testing in Cases of Sudden Arrhythmic Death Syndrome. J. Am. Coll Cardiol. 2017, 69, 2134–2145. [Google Scholar] [CrossRef] [PubMed]
- Stiles, M.K.; Wilde, A.A.; Abrams, D.J.; Ackerman, M.J.; Albert, C.M.; Behr, E.R.; Chugh, S.S.; Cornel, M.C.; Gardner, K.; Ingles, J.; et al. 2020 APHRS/HRS expert consensus statement on the investigation of decedents with sudden unexplained death and patients with sudden cardiac arrest, and of their families. Heart Rhythm 2020, 18, e1–e50. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.; Daubert, J.P.; de Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy: Executive summary. Heart Rhythm 2019, 16, e373–e407. [Google Scholar] [CrossRef] [PubMed]
- Verdonschot, J.A.J.; Vanhoutte, E.K.; Claes, G.R.; Helderman-van den Enden, A.T.; Hoeijmakers, J.G.; Hellebrekers, D.M.; de Haan, A.; Christiaans, I.; Lekanne Deprez, R.H.; Boen, H.M.; et al. A mutation update for the FLNC gene in myopathies and cardiomyopathies. Hum. Mutat. 2020, 41, 1091–1111. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.L.; Akhtar, M.M.; Sabater-Molina, M.; Futema, M.; Asimaki, A.; Protonotarios, A.; Dalageorgou, C.; Pittman, A.M.; Suarez, M.P.; Aguilera, B.; et al. Filamin C variants are associated with a distinctive clinical and immunohistochemical arrhythmogenic cardiomyopathy phenotype. Int. J. Cardiol. 2020, 307, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Hefner, G.; Hahn, M.; Hohner, M.; Roll, S.C.; Klimke, A.; Hiemke, C. QTc Time Correlates with Amitriptyline and Venlafaxine Serum Levels in Elderly Psychiatric Inpatients. Pharmacopsychiatry 2019, 52, 38–43. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van der Crabben, S.N.; Komdeur, F.L.; Nossent, E.J.; Deprez, R.H.L.; Broekhuizen, E.A.; van der Werf, C.; Vermeer, A.M.C.; Niessen, H.W.M.; Houweling, A.C. Genetic Diagnosis in Sudden Cardiac Death: The Crucial Role of Multidisciplinary Care. Cardiogenetics 2021, 11, 68-72. https://doi.org/10.3390/cardiogenetics11020008
van der Crabben SN, Komdeur FL, Nossent EJ, Deprez RHL, Broekhuizen EA, van der Werf C, Vermeer AMC, Niessen HWM, Houweling AC. Genetic Diagnosis in Sudden Cardiac Death: The Crucial Role of Multidisciplinary Care. Cardiogenetics. 2021; 11(2):68-72. https://doi.org/10.3390/cardiogenetics11020008
Chicago/Turabian Stylevan der Crabben, S. N., F. L. Komdeur, E. J. Nossent, R. H. Lekanne Deprez, E. A. Broekhuizen, C. van der Werf, A. M. C. Vermeer, H. W. M. Niessen, and A. C. Houweling. 2021. "Genetic Diagnosis in Sudden Cardiac Death: The Crucial Role of Multidisciplinary Care" Cardiogenetics 11, no. 2: 68-72. https://doi.org/10.3390/cardiogenetics11020008
APA Stylevan der Crabben, S. N., Komdeur, F. L., Nossent, E. J., Deprez, R. H. L., Broekhuizen, E. A., van der Werf, C., Vermeer, A. M. C., Niessen, H. W. M., & Houweling, A. C. (2021). Genetic Diagnosis in Sudden Cardiac Death: The Crucial Role of Multidisciplinary Care. Cardiogenetics, 11(2), 68-72. https://doi.org/10.3390/cardiogenetics11020008