

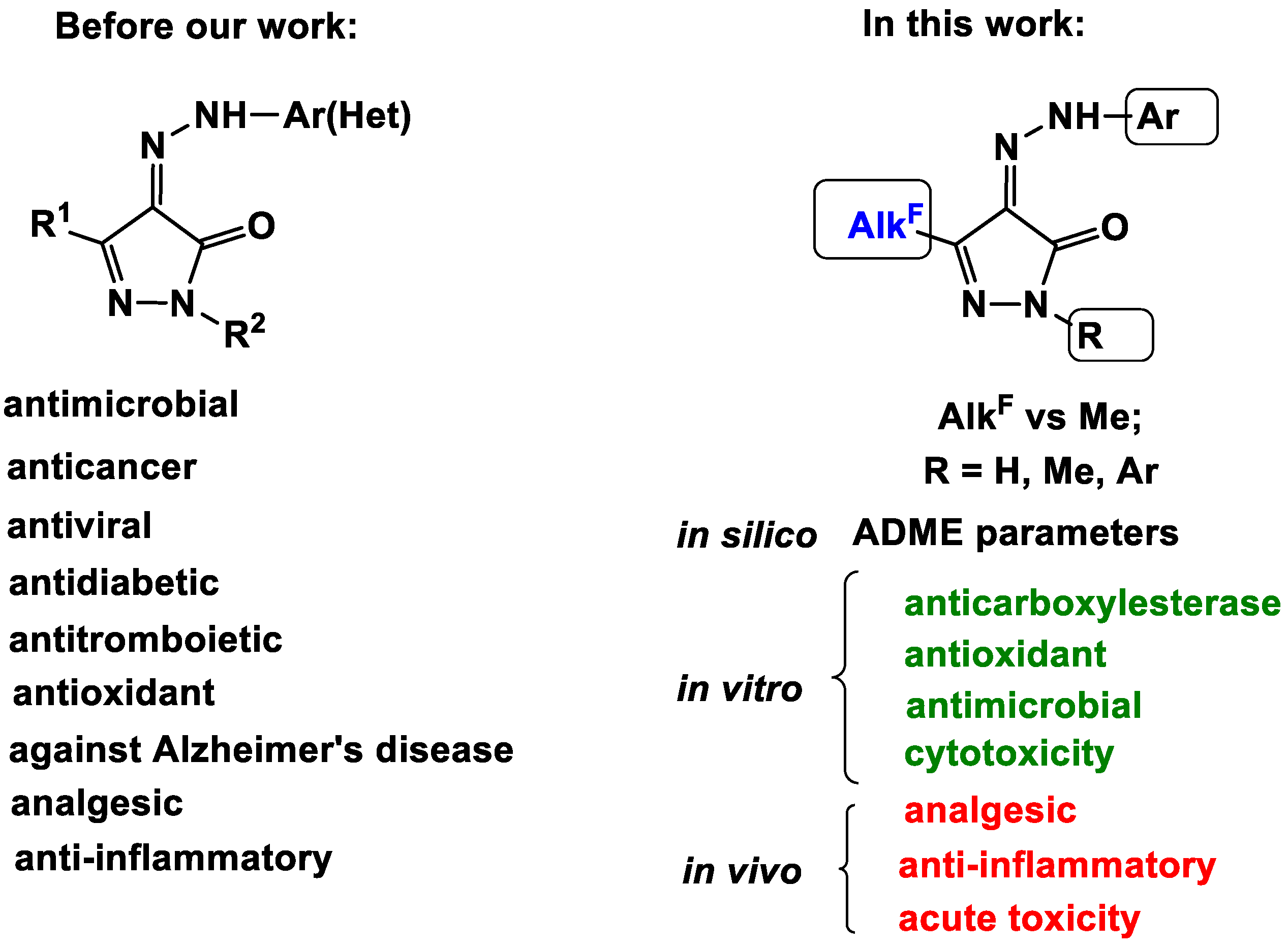
Powerful Potential of Polyfluoroalkyl-Containing 4-Arylhydrazinylidenepyrazol-3-ones for Pharmaceuticals
 , , , , , , , , , , , and add
Show full author list
, , , , , , , , , , , and add
Show full author list
Abstract
:1. Introduction
2. Results
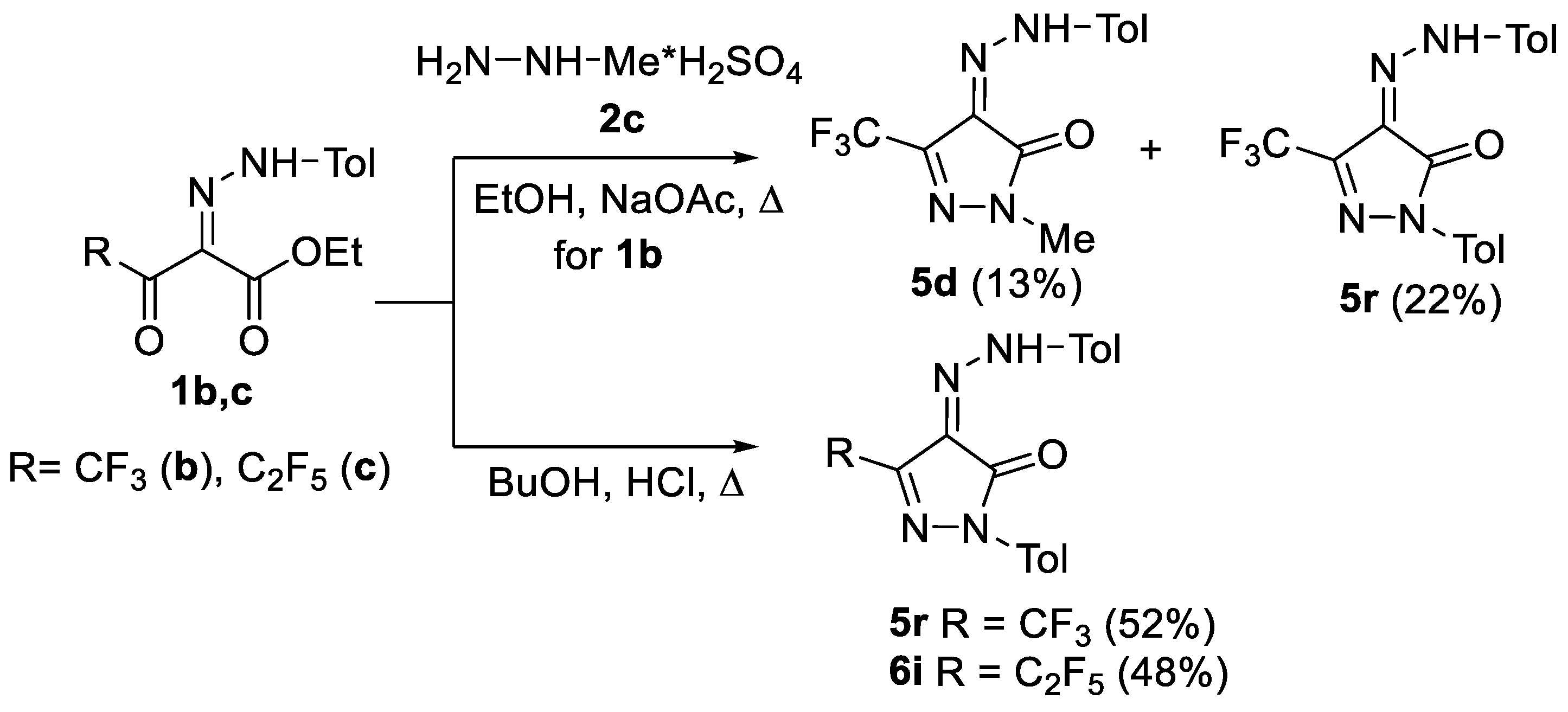
2.1. Chemistry
2.2. In Silico ADME Studies
2.3. Biological Evaluation
2.3.1. Esterase Profile of 4-AHPs 5–9
2.3.2. Antioxidant Activity
2.3.3. Antimicrobial Activity
2.3.4. Cytotoxicity
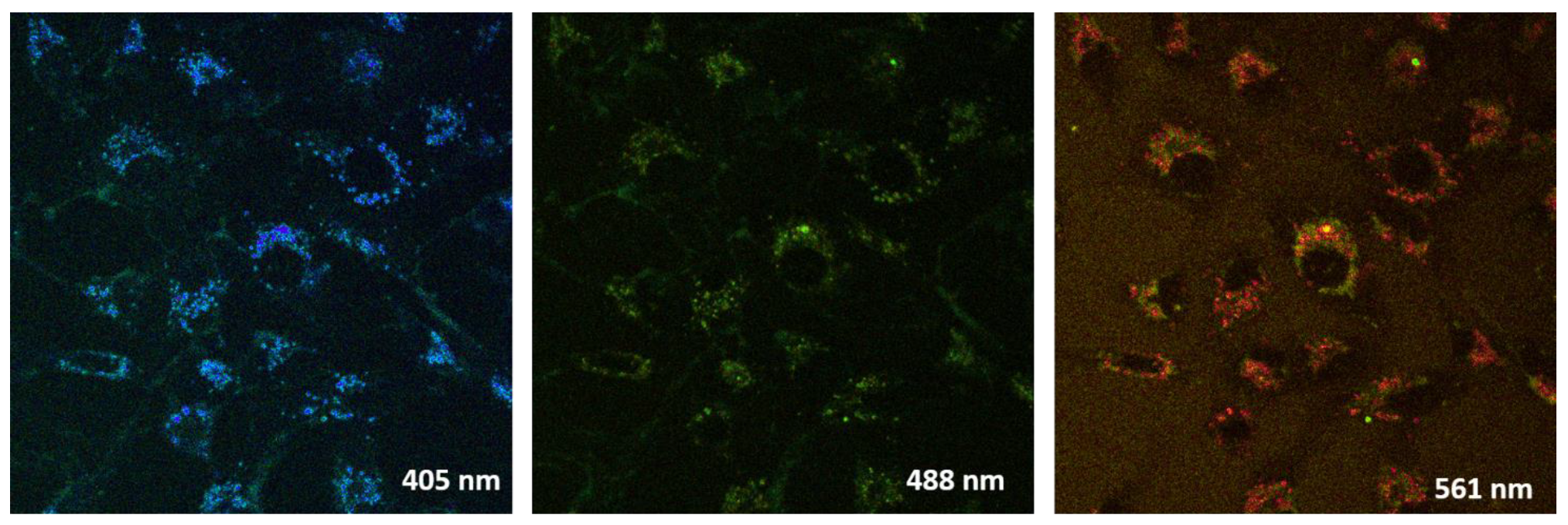
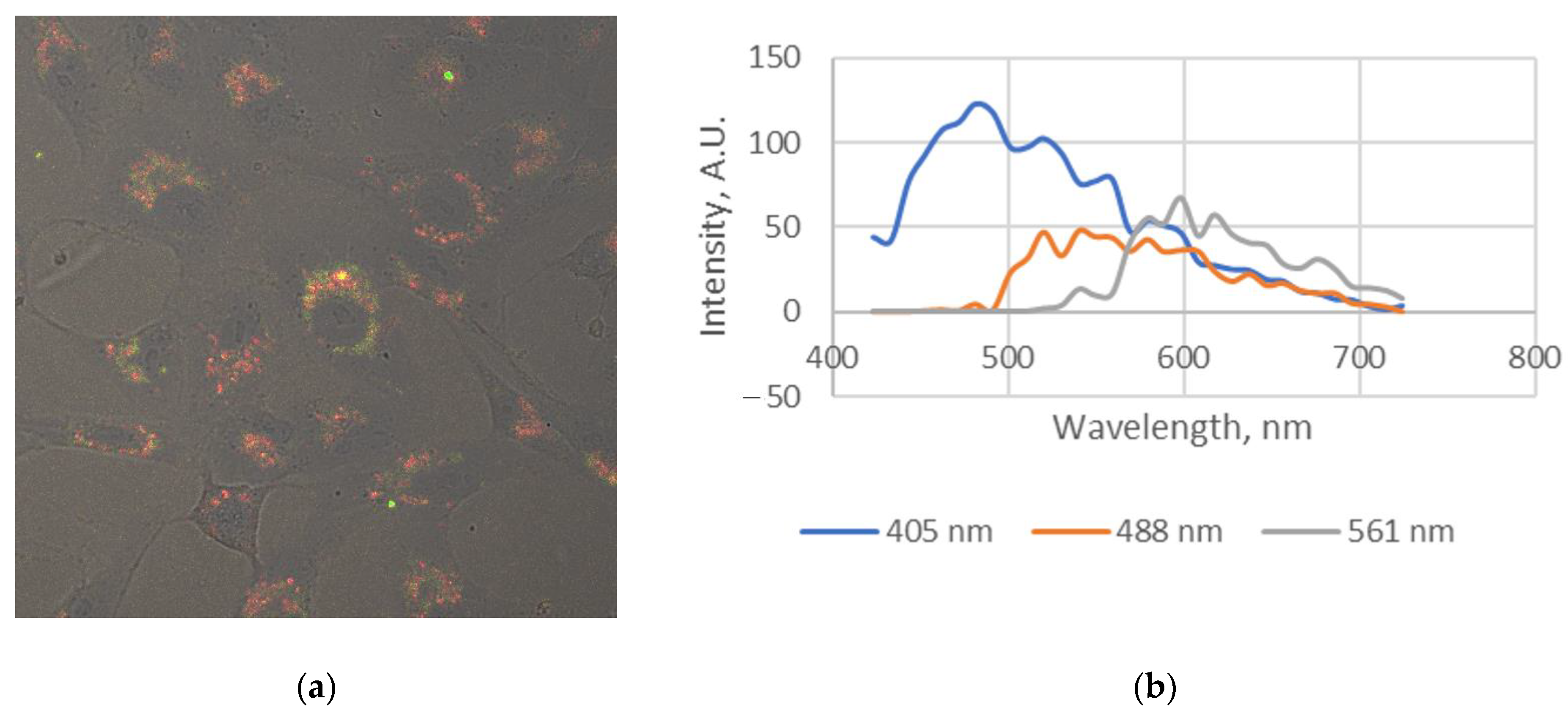
2.3.5. Live Cell Visualization
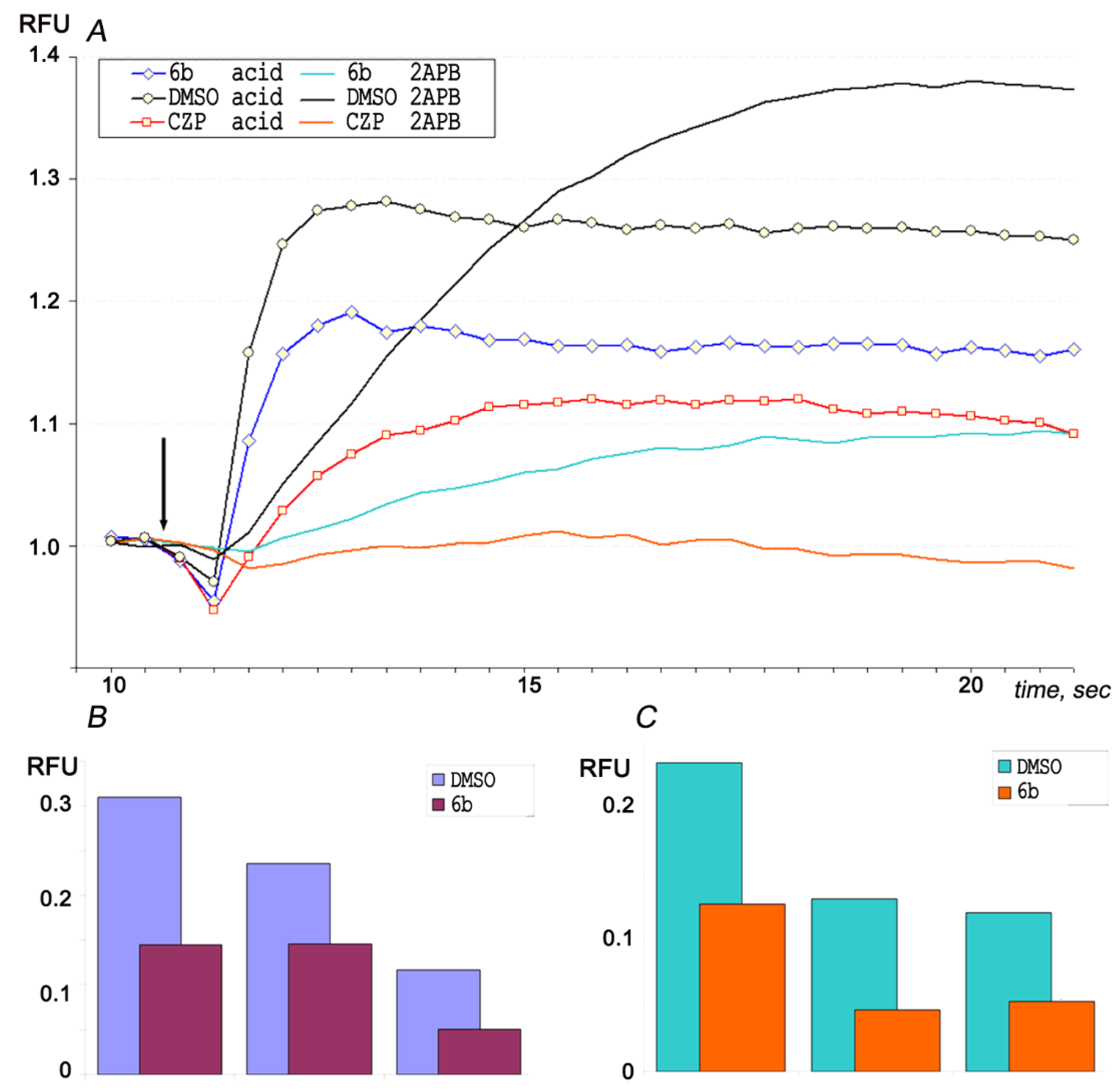
2.3.6. Analgesic and Anti-Inflammatory Activity
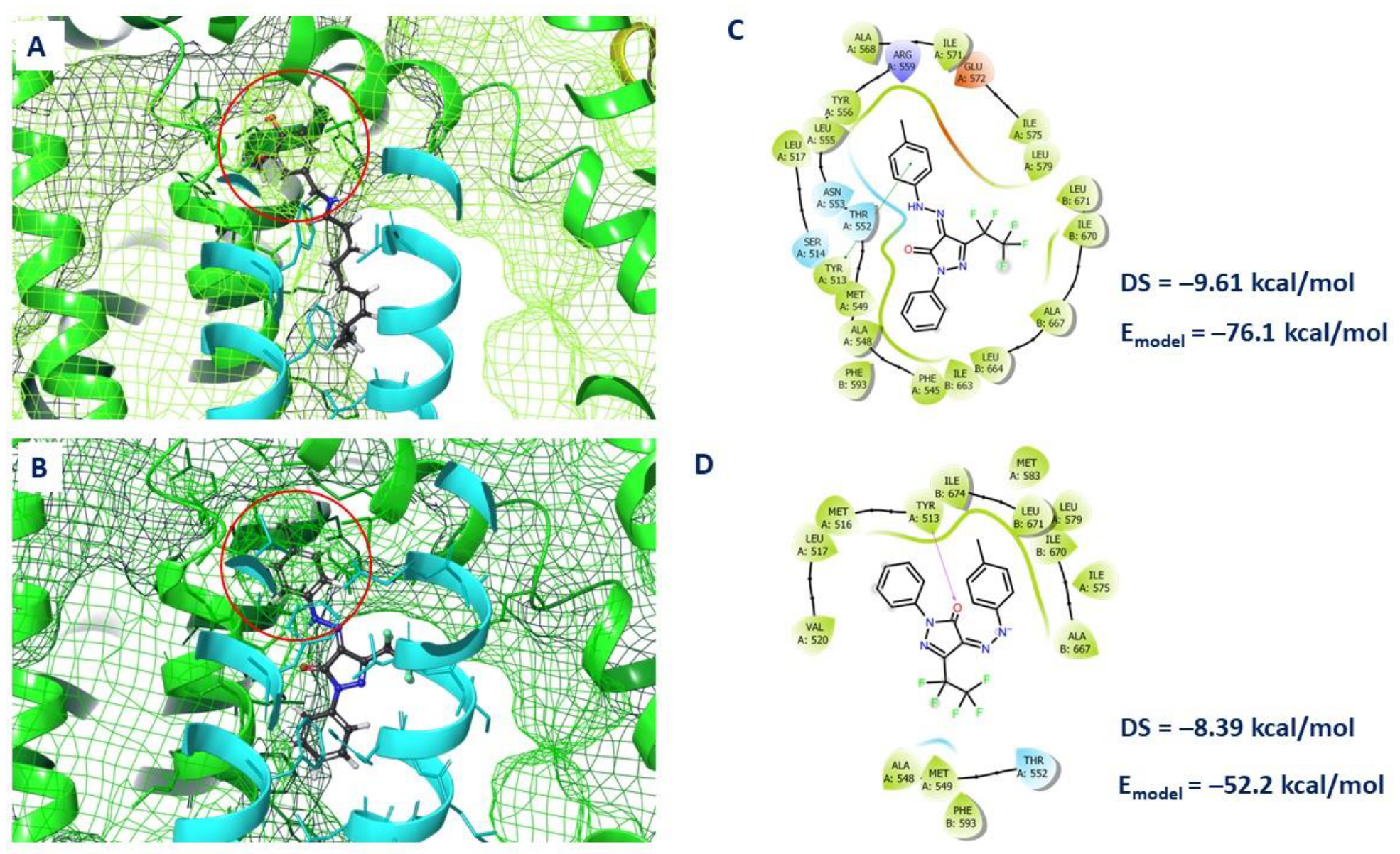
2.4. Molecular Docking
3. Material and Methods
3.1. Chemistry
3.1.1. Synthesis of Compounds 5a–s, 6a–i, 7a,b 8a,b, 9a,b (General Procedure)
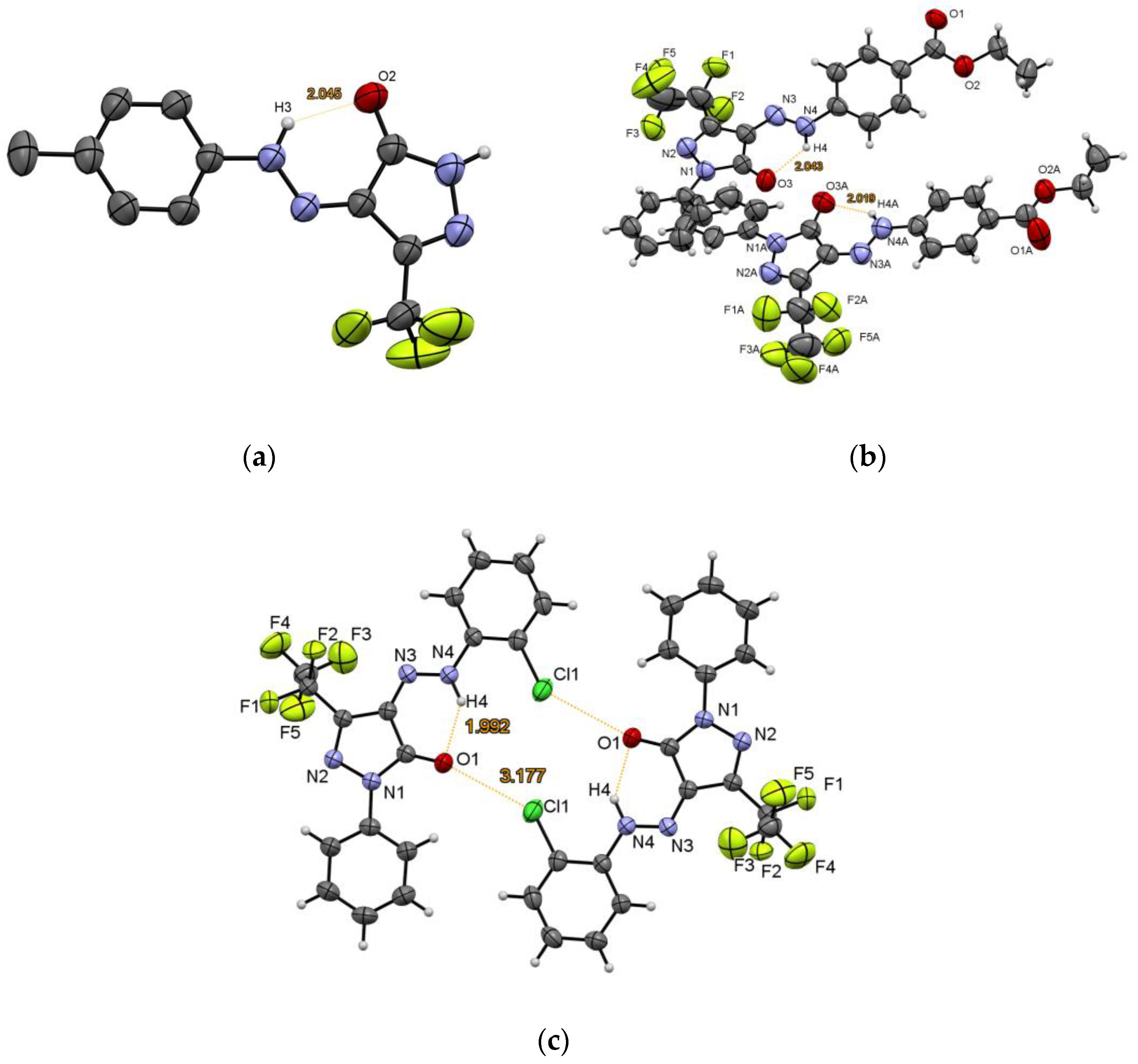
3.1.2. XRD Experiments
3.2. Biological Studies
3.2.1. Inhibition of Porcine Liver CES, Human AChE and Equine BChE
3.2.2. ABTS Radical Cation Scavenging Activity Assay
3.2.3. Ferric Reducing Antioxidant Power (FRAP) Assay
3.2.4. Oxygen Radical Absorbance Capacity (ORAC) Assay
3.2.5. Antimicrobial Activity
3.2.6. In Vitro Experiments
3.2.7. In Vivo Experiments
3.3. Quantum-Mechanical Calculations
3.3.1. Ligand Preparation
3.3.2. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Zhao, Z.; Dai, X.; Li, C.; Wang, X.; Tian, J.; Feng, Y.; Xie, J.; Ma, C.; Nie, Z.; Fan, P.; et al. Pyrazolone structural motif in medicinal chemistry: Retrospect and prospect. Eur. J. Med. Chem. 2020, 186, 111893. [Google Scholar] [CrossRef] [PubMed]
- Havrylyuk, D.; Roman, O.; Lesyk, R. Synthetic approaches, structure activity relationship and biological applications for pharmacologically attractive pyrazole/pyrazoline–thiazolidine-based hybrids. Eur. J. Med. Chem. 2016, 113, 145–166. [Google Scholar] [CrossRef] [PubMed]
- Küçükgüzel, Ş.G.; Şenkardeş, S. Recent advances in bioactive pyrazoles. Eur. J. Med. Chem. 2015, 97, 786–815. [Google Scholar] [CrossRef] [PubMed]
- Keri, R.S.; Chand, K.; Ramakrishnappa, T.; Nagaraja, B.M. Recent Progress on Pyrazole Scaffold-Based Antimycobacterial Agents. Arch. Pharm. 2015, 348, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Pizzuti, L.; Barschak, A.; Stefanello, F.; Farias, M.; Lencina, C.; Roesch-Ely, M.; Cunico, W.; Moura, S.; Pereira, C. Environment-friendly Synthesis of Bioactive Pyrazoles. Curr. Org. Chem. 2014, 18, 115–126. [Google Scholar] [CrossRef]
- Liu, J.-J.; Zhao, M.-y.; Zhang, X.; Zhao, X.; Zhu, H.-L. Pyrazole Derivatives as Antitumor, Anti-Inflammatory and Antibacterial Agents. Mini-Rev. Med. Chem. 2013, 13, 1957–1966. [Google Scholar] [CrossRef]
- Sahoo, J.; Sahoo, C.R.; Nandini Sarangi, P.K.; Prusty, S.K.; Padhy, R.N.; Paidesetty, S.K. Molecules with versatile biological activities bearing antipyrinyl nucleus as pharmacophore. Eur. J. Med. Chem. 2020, 186, 111911. [Google Scholar] [CrossRef]
- Uramaru, N.; Shigematsu, H.; Toda, A.; Eyanagi, R.; Kitamura, S.; Ohta, S. Design, Synthesis, and Pharmacological Activity of Nonallergenic Pyrazolone-Type Antipyretic Analgesics. J. Med. Chem. 2010, 53, 8727–8733. [Google Scholar] [CrossRef]
- Bailly, C.; Hecquet, P.-E.; Kouach, M.; Thuru, X.; Goossens, J.-F. Chemical reactivity and uses of 1-phenyl-3-methyl-5-pyrazolone (PMP), also known as edaravone. Biorg. Med. Chem. 2020, 28, 115463. [Google Scholar] [CrossRef]
- Horrocks, P.; Pickard, M.R.; Parekh, H.H.; Patel, S.P.; Pathak, R.B. Synthesis and biological evaluation of 3-(4-chlorophenyl)-4-substituted pyrazole derivatives. Org. Biomol. Chem. 2013, 11, 4891. [Google Scholar] [CrossRef]
- Chikkula, K.V.; Sundararajan, R. Analgesic, anti-inflammatory, and antimicrobial activities of novel isoxazole/pyrimidine/pyrazole substituted benzimidazole analogs. Med. Chem. Res. 2017, 26, 3026–3037. [Google Scholar] [CrossRef]
- Manojkumar, P.; Ravi, T.K.; Gopalakrishnan, S. Antioxidant and antibacterial studies of arylazopyrazoles and arylhydrazonopyrazolones containing coumarin moiety. Eur. J. Med. Chem. 2009, 44, 4690–4694. [Google Scholar] [CrossRef] [PubMed]
- Bekhit, A.A.; Ashour, H.M.A.; Guemei, A.A. Novel Pyrazole Derivatives as Potential Promising Anti-inflammatory Antimicrobial Agents. Arch. Pharm. 2005, 338, 167–174. [Google Scholar] [CrossRef]
- Oraby, A.K.; Abdellatif, K.R.A.; Abdelgawad, M.A.; Attia, K.M.; Dawe, L.N.; Georghiou, P.E. 2,4-Disubstituted Phenylhydrazonopyrazolone and Isoxazolone Derivatives as Antibacterial Agents: Synthesis, Preliminary Biological Evaluation and Docking Studies. ChemistrySelect 2018, 3, 3295–3301. [Google Scholar] [CrossRef]
- Grosskopf, S.; Eckert, C.; Arkona, C.; Radetzki, S.; Böhm, K.; Heinemann, U.; Wolber, G.; von Kries, J.-P.; Birchmeier, W.; Rademann, J. Selective Inhibitors of the Protein Tyrosine Phosphatase SHP2 Block Cellular Motility and Growth of Cancer Cells in vitro and in vivo. ChemMedChem 2015, 10, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Stornaiuolo, M.; La Regina, G.; Passacantilli, S.; Grassia, G.; Coluccia, A.; La Pietra, V.; Giustiniano, M.; Cassese, H.; Di Maro, S.; Brancaccio, D.; et al. Structure-Based Lead Optimization and Biological Evaluation of BAX Direct Activators as Novel Potential Anticancer Agents. J. Med. Chem. 2015, 58, 2135–2148. [Google Scholar] [CrossRef] [PubMed]
- Sayed, A.R.; Gomha, S.M.; Abdelrazek, F.M.; Farghaly, M.S.; Hassan, S.A.; Metz, P. Design, efficient synthesis and molecular docking of some novel thiazolyl-pyrazole derivatives as anticancer agents. BMC Chem. 2019, 13, 116. [Google Scholar] [CrossRef] [Green Version]
- Abdelgawad, M.A.; Bakr, R.B.; Omar, H.A. Design, synthesis and biological evaluation of some novel benzothiazole/benzoxazole and/or benzimidazole derivatives incorporating a pyrazole scaffold as antiproliferative agents. Bioorg. Chem. 2017, 74, 82–90. [Google Scholar] [CrossRef]
- Mahadevan, D.; Powis, G.; Mash, E.A.; George, B.; Gokhale, V.M.; Zhang, S.; Shakalya, K.; Du-Cuny, L.; Berggren, M.; Ali, M.A.; et al. Discovery of a novel class of AKT pleckstrin homology domain inhibitors. Mol. Cancer Ther. 2008, 7, 2621–2632. [Google Scholar] [CrossRef] [Green Version]
- Erickson-Miller, C.L.; Jenkins, J.; Theodore, D. Novel combinations. WO2008073864. 2008. Available online: https://patents.google.com/patent/WO2008073864A1 (accessed on 13 December 2022).
- Dogo-Isonagie, C.; Lee, S.-L.; Lohith, K.; Liu, H.; Mandadapu, S.R.; Lusvarghi, S.; O’Connor, R.D.; Bewley, C.A. Design and synthesis of small molecule-sulfotyrosine mimetics that inhibit HIV-1 entry. Biorg. Med. Chem. 2016, 24, 1718–1728. [Google Scholar] [CrossRef]
- Acharya, P.; Dogo-Isonagie, C.; LaLonde, J.M.; Lam, S.N.; Leslie, G.J.; Louder, M.K.; Frye, L.L.; Debnath, A.K.; Greenwood, J.R.; Luongo, T.S.; et al. Structure-Based Identification and Neutralization Mechanism of Tyrosine Sulfate Mimetics That Inhibit HIV-1 Entry. ACS Chem. Biol. 2011, 6, 1069–1077. [Google Scholar] [CrossRef]
- Singh, U.P.; Bhat, H.R.; Verma, A.; Kumawat, M.K.; Kaur, R.; Gupta, S.K.; Singh, R.K. Phenyl hydrazone bearing pyrazole and pyrimidine scaffolds: Design and discovery of a novel class of non-nucleoside reverse transcriptase inhibitors (NNRTIs) against HIV-1 and their antibacterial properties. RSC Adv. 2013, 3, 17335. [Google Scholar] [CrossRef]
- Lyer, P.C.; Zhao, J.; Emert-Sedlak, L.A.; Moore, K.K.; Smithgall, T.E.; Day, B.W. Synthesis and structure–activity analysis of diphenylpyrazolodiazene inhibitors of the HIV-1 Nef virulence factor. Bioorg. Med. Chem. Lett. 2014, 24, 1702–1706. [Google Scholar] [CrossRef] [Green Version]
- Arnost, M.; Pierce, A.; Haar, E.t.; Lauffer, D.; Madden, J.; Tanner, K.; Green, J. 3-Aryl-4-(arylhydrazono)-1H-pyrazol-5-ones: Highly ligand efficient and potent inhibitors of GSK3β. Bioorg. Med. Chem. Lett. 2010, 20, 1661–1664. [Google Scholar] [CrossRef] [PubMed]
- Duffy, K.J.; Darcy, M.G.; Delorme, E.; Dillon, S.B.; Eppley, D.F.; Erickson-Miller, C.; Giampa, L.; Hopson, C.B.; Huang, Y.; Keenan, R.M.; et al. Hydrazinonaphthalene and Azonaphthalene Thrombopoietin Mimics Are Nonpeptidyl Promoters of Megakaryocytopoiesis. J. Med. Chem. 2001, 44, 3730–3745. [Google Scholar] [CrossRef]
- Farrukh, S.U.B.; Javed, I.; Ather, A.Q.; Emwas, A.-H.; Alazmi, M.; Gao, X.; Chotana, G.A.; Davis, T.P.; Ke, P.C.; Saleem, R.S.Z. Synthesis and identification of novel pyridazinylpyrazolone based diazo compounds as inhibitors of human islet amyloid polypeptide aggregation. Bioorg. Chem. 2019, 84, 339–346. [Google Scholar] [CrossRef]
- Turkan, F.; Cetin, A.; Taslimi, P.; Karaman, H.S.; Gulçin, İ. Synthesis, characterization, molecular docking and biological activities of novel pyrazoline derivatives. Arch. Pharm. 2019, 352, 1800359. [Google Scholar] [CrossRef]
- Shetty, S.; Kalluraya, B.; Nithinchandra; Peethambar, S.K.; Telkar, S.B. Type II diabetes-related enzyme inhibition and molecular modeling study of a novel series of pyrazolone derivatives. Med. Chem. Res. 2013, 23, 2834–2846. [Google Scholar] [CrossRef]
- Abdelgawad, M.A.; Labib, M.B.; Ali, W.A.M.; Kamel, G.; Azouz, A.A.; El-Nahass, E.L.S. Design, synthesis, analgesic, anti-inflammatory activity of novel pyrazolones possessing aminosulfonyl pharmacophore as inhibitors of COX-2/5-LOX enzymes: Histopathological and docking studies. Bioorg. Chem. 2018, 78, 103–114. [Google Scholar] [CrossRef]
- Anwar, T.; Nadeem, H.; Sarwar, S.; Naureen, H.; Ahmed, S.; Khan, A.; Arif, M. Investigation of antioxidant and anti-nociceptive potential of isoxazolone, pyrazolone derivatives, and their molecular docking studies. Drug Dev. Res. 2020, 81, 893–903. [Google Scholar] [CrossRef]
- Gaffer, H.E.; Abdel-Fattah, S.; Etman, H.A.; Abdel-Latif, E. Synthesis and Antioxidant Activity of Some New Thiazolyl-Pyrazolone Derivatives. J. Heterocycl. Chem. 2017, 54, 331–340. [Google Scholar] [CrossRef]
- Jenkins, J.M.; Williams, D.; Deng, Y.; Uhl, J.; Kitchen, V.; Collins, D.; Erickson-Miller, C.L. Phase 1 clinical study of eltrombopag, an oral, nonpeptide thrombopoietin receptor agonist. Blood 2007, 109, 4739–4741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussel, J.B.; Cheng, G.; Saleh, M.N.; Psaila, B.; Kovaleva, L.; Meddeb, B.; Kloczko, J.; Hassani, H.; Mayer, B.; Stone, N.L.; et al. Eltrombopag for the Treatment of Chronic Idiopathic Thrombocytopenic Purpura. N. Engl. J. Med. 2007, 357, 2237–2247. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, Y.; Liu, J.; Han, S.; Chen, L.; Wang, H.; Peng, Y. A systematic review and meta-analysis of the safety and efficacy of anti-thymocyte globulin combined with eltrombopag in the treatment of severe aplastic anemia. Ann. Palliat. Med. 2021, 10, 5549–5560. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.; Bhawana, P.; Fulekar, M.H. Microbial decolorization and degradation of synthetic dyes: A review. Rev. Environ. Sci. Bio/Technol. 2012, 12, 75–97. [Google Scholar] [CrossRef]
- Yamjala, K.; Nainar, M.S.; Ramisetti, N.R. Methods for the analysis of azo dyes employed in food industry—A review. Food Chem. 2016, 192, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Metwally, M.A.; Abdel-Galil, E.; Metwally, A.; Amer, F.A. New azodisperse dyes with thiazole, thiophene, pyridone and pyrazolone moiety for dyeing polyester fabrics. Dye. Pigm. 2012, 92, 902–908. [Google Scholar] [CrossRef]
- Shams, H.Z.; Helal, M.H.; Mohamed, F.A. Synthesis of a new series of polyfunctionally substituted pyrazole azo dye systems for dyeing of synthetic and modified cellulose fibers. Pigm. Resin Technol. 2001, 30, 99–108. [Google Scholar] [CrossRef]
- Helal, M.H. Synthesis of a new series of thiazole and pyrazole azo dye systems for dyeing synthetic fibers. Pigm. Resin Technol. 2001, 30, 296–300. [Google Scholar] [CrossRef]
- Ben Mansour, H.; Corroler, D.; Barillier, D.; Ghedira, K.; Chekir, L.; Mosrati, R. Evaluation of genotoxicity and pro-oxidant effect of the azo dyes: Acids yellow 17, violet 7 and orange 52, and of their degradation products by Pseudomonas putida mt-2. Food Chem. Toxicol. 2007, 45, 1670–1677. [Google Scholar] [CrossRef]
- Sener, N.; Sener, I.; Yavuz, S.; Karci, F. Synthesis, Absorption Properties and Biological Evaluation of Some Novel Disazo Dyes Derived from Pyrazole Derivatives. Asian J. Chem. 2015, 27, 3003–3012. [Google Scholar] [CrossRef]
- Meanwell, N.A. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J.L.; Soloshonok, V.A.; Izawa, K.; Liu, H. Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev. 2016, 116, 422–518. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Sumii, Y.; Shibata, N. Contribution of Organofluorine Compounds to Pharmaceuticals. ACS Omega 2020, 5, 10633–10640. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Remete, A.M.; Dobson, L.S.; Kiss, L.; Izawa, K.; Moriwaki, H.; Soloshonok, V.A.; O’Hagan, D. Next generation organofluorine containing blockbuster drugs. J. Fluor. Chem. 2020, 239, 109639. [Google Scholar] [CrossRef]
- Nair, A.S.; Singh, A.K.; Kumar, A.; Kumar, S.; Sukumaran, S.; Koyiparambath, V.P.; Pappachen, L.K.; Rangarajan, T.M.; Kim, H.; Mathew, B. FDA-Approved Trifluoromethyl Group-Containing Drugs: A Review of 20 Years. Processes 2022, 10, 2054. [Google Scholar] [CrossRef]
- Saleh, A.M.; Taha, M.O.; Aziz, M.A.; Al-Qudah, M.A.; AbuTayeh, R.F.; Rizvi, S.A. Novel anticancer compound [trifluoromethyl-substituted pyrazole N-nucleoside] inhibits FLT3 activity to induce differentiation in acute myeloid leukemia cells. Cancer Lett. 2016, 375, 199–208. [Google Scholar] [CrossRef]
- Abdou, I.; Saleh, A.; Zohdi, H. Synthesis and Antitumor Activity of 5-Trifluoromethyl-2,4- dihydropyrazol-3-one Nucleosides. Molecules 2004, 9, 109–116. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, A.M.S.; Abou-Elkhair, R.A.I.; El-Torky, A.M.; Hassan, A.E.A. 3-Trifluoromethylpyrazolones derived nucleosides: Synthesis and antiviral evaluation. Nucl. Nucl. Nucleic Acids 2019, 38, 590–603. [Google Scholar] [CrossRef]
- Khalil, N.S.A.M. A facile synthesis, structure, and antimicrobial evaluation of novel 4-arylhydrazono-5-trifluoromethyl-2,4-dihydropyrazol-3-ones, their N- and N,O-bis-β-d-glucosides. Carbohydr. Res. 2009, 344, 1654–1659. [Google Scholar] [CrossRef]
- Narayana Rao, D.V.; Raghavendra Guru Prasad, A.; Spoorthy, Y.N.; Raghunatha Rao, D.; Ravindranath, L.K. In vitro microbiological evaluation of novel bis pyrazolones. Ann. Pharm. Fr. 2014, 72, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, B.F.; El-Ahl, A.-A.S.; Badria, F.A. Synthesis of New 2-Naphthyl Ethers and Their Protective Activities against DNA Damage Induced by Bleomycin-Iron. Chem. Pharm. Bull. 2009, 57, 1348–1351. [Google Scholar] [CrossRef] [Green Version]
- Lyčka, A.; Liptaj, T.; Jirman, J. 13C and 15N NMR spectra of 3-methyl-1-phenylpyrazole-4,5-dione 4-(4′-substituted phenyl)hydrazones. Collect. Czech. Chem. Commun. 1987, 52, 727–735. [Google Scholar] [CrossRef]
- Khalil, A.; Hassan, M.; Mohamed, M.; Elsayed, A. Metal salt-catalyzed diazocoupling of 3-substituted-1-pyrazol-2-in-5-ones in aqueous medium. Dye. Pigm. 2005, 66, 241–245. [Google Scholar] [CrossRef]
- El-Metwally, S.; Khalil, A.K. Reactions of 1,3-Diphenyl-2-pyrazolin-5-one and 4-Amino1,5-dimethyl-2-phenyl-1H-pyrazol-3(2H)-one. Synthesis of Some New Pyrazoles and Pyrazolones. Acta Chim. Slov. 2010, 57, 941–947. [Google Scholar]
- Kedar, N.A.; Shirodkar, S.G. Synthesis and characterization of monomeric complexes of Cu(II) and Ni(II) with 3-methyl-4-(2-arylhydrazono)-1H-pyrazol-5(4H)-one. J. Chem. Pharm. Res 2014, 6, 801–807. [Google Scholar]
- Zhang, Y.; Hu, H.; Liu, C.-J.; Cao, D.; Wang, B.; Sun, Y.; Abdukader, A. Highly Efficient Brønsted Acidic Ionic Liquid Promoted Direct Diazenylation of Pyrazolones with Aryltriazenes under Mild Conditions. Asian J. Org. Chem. 2017, 6, 102–107. [Google Scholar] [CrossRef]
- Pandit, R.P.; Kim, S.H.; Lee, Y.R. Iodine-mediated construction of polyfunctionalized arylazopyrazoles from β-ketoesters or 2-arylpyrazol-3-ones and arylhydrazines. Org. Biomol. Chem. 2016, 14, 6996–7000. [Google Scholar] [CrossRef]
- Pandit, R.P.; Lee, Y.R. Construction of Multifunctionalized Azopyrazoles by Silver-Catalyzed Cascade Reaction of Diazo Compounds. Adv. Synth. Catal. 2015, 357, 2657–2664. [Google Scholar] [CrossRef]
- Kuo, W.-F.; Chiu, C.-Y.; Lin, I.T.; Sheu, S.-Y.; Lm, K.-H.; Yeh, M.-Y. The Decomposition Reaction of 4-Acetylsydnones Arylhydrazones. J. Chin. Chem. Soc. 2000, 47, 1171–1176. [Google Scholar] [CrossRef]
- Kuzueva, O.G.; Burgart, Y.V.; Saloutin, V.I. Synthesis of 2-arylhydrazones of aliphatic fluorine-containing 1,2,3-tricarbonyl compounds and their reactions with dinucleophiles. Russ. Chem. Bull. 1998, 47, 673–678. [Google Scholar] [CrossRef]
- Burgart, Y.V.; Fokin, A.S.; Kuzueva, O.G.; Chupakhin, O.N.; Saloutin, V.I. Synthesis of fluorinated 2(3)-arylhydrazones of 1,2,3-tri(1,2,3,4-tetra)carbonyl compounds and their heterocyclization reactions. J. Fluor. Chem. 1998, 92, 101–108. [Google Scholar] [CrossRef]
- Boltneva, N.P.; Makhaeva, G.F.; Shchegol’kov, E.V.; Burgart, Y.V.; Saloutin, V.I. Selective Carboxylesterase Inhibitors for Improving Efficacy, Safety and Rational use of Ester-Containing Drugs. Biomed. Chem. Res. Methods 2018, 1, e00026. [Google Scholar] [CrossRef] [Green Version]
- Makhaeva, G.F.; Rudakova, E.V.; Kovaleva, N.V.; Lushchekina, S.V.; Boltneva, N.P.; Proshin, A.N.; Shchegolkov, E.V.; Burgart, Y.V.; Saloutin, V.I. Cholinesterase and carboxylesterase inhibitors as pharmacological agents. Russ. Chem. Bull. 2019, 68, 967–984. [Google Scholar] [CrossRef]
- Boltneva, N.P.; Makhaeva, G.F.; Kovaleva, N.V.; Lushchekina, S.V.; Burgart, Y.V.; Shchegol’kov, E.V.; Saloutin, V.I.; Chupakhin, O.N. Alkyl 2-arylhydrazinylidene-3-oxo-3-polyfluoroalkylpropionates as new effective and selective inhibitors of carboxylesterase. Dokl. Biochem. Biophys. 2016, 465, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Makhaeva, G.F.; Lushchekina, S.V.; Boltneva, N.P.; Serebryakova, O.G.; Kovaleva, N.V.; Rudakova, E.V.; Elkina, N.A.; Shchegolkov, E.V.; Burgart, Y.V.; Stupina, T.S.; et al. Novel potent bifunctional carboxylesterase inhibitors based on a polyfluoroalkyl-2-imino-1,3-dione scaffold. Eur. J. Med. Chem. 2021, 218, 113385. [Google Scholar] [CrossRef]
- Bernardin, J.; Pechmeze, J. Wasserunloesliche Azofarbstoffe, Verfahren zu Ihrer Herstellung und Ihre Verwendung. DE2657110. 1977. Available online: https://patents.google.com/patent/DE2657110A1 (accessed on 13 December 2022).
- Hassan, A.E.A.; Moustafa, A.H.; Tolbah, M.M.; Zohdy, H.F.; Haikal, A.Z. Synthesis and Antimicrobial Evaluation of Novel Pyrazolones and Pyrazolone Nucleosides. Nucl. Nucl. Nucleic Acids 2012, 31, 783–800. [Google Scholar] [CrossRef]
- Burgart, Y.V.; Agafonova, N.A.; Shchegolkov, E.V.; Borisevich, S.S.; Khursan, S.L.; Maslova, V.V.; Triandafilova, G.A.; Solodnikov, S.Y.; Krasnykh, O.P.; Saloutin, V.I. The competitive N1-, N2-, O- and C-methylation of 3-trifluoromethyl-1H-pyrazol-5-ol for synthesis of analgesic compounds. J. Fluor. Chem. 2019, 218, 1–10. [Google Scholar] [CrossRef]
- Nemytova, N.A.; Shchegol’kov, E.V.; Burgart, Y.V.; Slepukhin, P.A.; Borisevich, S.S.; Khursan, S.L.; Saloutin, V.I. Regiocontrolled N-, O- and C-methylation of 1-phenyl-3-polyfluoroalkyl-1H-pyrazol-5-ols. J. Fluor. Chem. 2018, 206, 72–81. [Google Scholar] [CrossRef]
- Balani, S.; Miwa, G.; Gan, L.-S.; Wu, J.-T.; Lee, F. Strategy of Utilizing In Vitro and In Vivo ADME Tools for Lead Optimization and Drug Candidate Selection. Curr. Top. Med. Chem. 2005, 5, 1033–1038. [Google Scholar] [CrossRef]
- Schrödinger Release 2020-4: QikProp, Schrödinger; LLC: New York, NY, USA, 2020.
- Makhaeva, G.F.; Serebryakova, O.G.; Boltneva, N.P.; Galenko, T.G.; Aksinenko, A.Y.; Sokolov, V.B.; Martynov, I.V. Esterase profile and analysis of structure-inhibitor selectivity relationships for homologous phosphorylated 1-hydroperfluoroisopropanols. Dokl. Biochem. Biophys. 2008, 423, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Makhaeva, G.F.; Radchenko, E.V.; Baskin, I.I.; Palyulin, V.A.; Richardson, R.J.; Zefirov, N.S. Combined QSAR studies of inhibitor properties ofO-phosphorylated oximes toward serine esterases involved in neurotoxicity, drug metabolism and Alzheimer’s disease. SAR QSAR Environ. Res. 2012, 23, 627–647. [Google Scholar] [CrossRef] [PubMed]
- Potter, P.; Wadkins, R. Carboxylesterases—Detoxifying Enzymes and Targets for Drug Therapy. Curr. Med. Chem. 2006, 13, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Di, L. The Impact of Carboxylesterases in Drug Metabolism and Pharmacokinetics. Curr. Drug Metab. 2019, 20, 91–102. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, C.; He, W.; Liu, D. Regulations of Xenobiotics and Endobiotics on Carboxylesterases: A Comprehensive Review. Eur. J. Drug Metab. Pharmacokinet. 2016, 41, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, M.J.; Potter, P.M. Carboxylesterase inhibitors. Expert Opin. Ther. Pat. 2011, 21, 1159–1171. [Google Scholar] [CrossRef]
- Zou, L.-W.; Jin, Q.; Wang, D.-D.; Qian, Q.-K.; Hao, D.-C.; Ge, G.-B.; Yang, L. Carboxylesterase Inhibitors: An Update. Curr. Med. Chem. 2018, 25, 1627–1649. [Google Scholar] [CrossRef]
- Casey Laizure, S.; Herring, V.; Hu, Z.; Witbrodt, K.; Parker, R.B. The Role of Human Carboxylesterases in Drug Metabolism: Have We Overlooked Their Importance? Pharmacotherapy 2013, 33, 210–222. [Google Scholar] [CrossRef] [Green Version]
- Lian, J.; Nelson, R.; Lehner, R. Carboxylesterases in lipid metabolism: From mouse to human. Protein Cell 2017, 9, 178–195. [Google Scholar] [CrossRef]
- Merali, Z.; Ross, S.; Paré, G. The pharmacogenetics of carboxylesterases: CES1 and CES2 genetic variants and their clinical effect. Drug Metabol. Drug Interact. 2014, 29, 143–151. [Google Scholar] [CrossRef]
- Radchenko, E.V.; Mel’nikov, A.A.; Makhaeva, G.F.; Palyulin, V.A.; Zefirov, N.S. Molecular design of O-phosphorylated oximes—Selective inhibitors of butyrylcholinesterase. Dokl. Biochem. Biophys. 2012, 443, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Radchenko, E.V.; Makhaeva, G.F.; Boltneva, N.P.; Serebryakova, O.G.; Serkov, I.V.; Proshin, A.N.; Palyulin, V.A.; Zefirov, N.S. Molecular design of N,N-disubstituted 2-aminothiazolines as selective carboxylesterase inhibitors. Russ. Chem. Bull. 2016, 65, 570–575. [Google Scholar] [CrossRef]
- Tsurkan, L.G.; Hatfield, M.J.; Edwards, C.C.; Hyatt, J.L.; Potter, P.M. Inhibition of human carboxylesterases hCE1 and hiCE by cholinesterase inhibitors. Chem. Biol. Interact. 2013, 203, 226–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makhaeva, G.F.; Elkina, N.A.; Shchegolkov, E.V.; Boltneva, N.P.; Lushchekina, S.V.; Serebryakova, O.G.; Rudakova, E.V.; Kovaleva, N.V.; Radchenko, E.V.; Palyulin, V.A.; et al. Synthesis, molecular docking, and biological evaluation of 3-oxo-2-tolylhydrazinylidene-4,4,4-trifluorobutanoates bearing higher and natural alcohol moieties as new selective carboxylesterase inhibitors. Bioorg. Chem. 2019, 91, 103097. [Google Scholar] [CrossRef]
- Burgart, Y.V.; Elkina, N.A.; Shchegolkov, E.V.; Krasnykh, O.P.; Maslova, V.V.; Triandafilova, G.A.; Solodnikov, S.Y.; Makhaeva, G.F.; Serebryakova, O.G.; Rudakova, E.V.; et al. Synthesis of Biologically Active 6-(Tolylhydrazinylidene)Pyrazolo[1,5-a]Pyrimidinones. Chem. Heterocycl. Comp. 2020, 56, 199–207. [Google Scholar] [CrossRef]
- Khudina, O.G.; Shchegol’kov, E.V.; Burgart, Y.V.; Boltneva, N.P.; Rudakova, E.V.; Makhaeva, G.F.; Saloutin, V.I. Intramolecular cyclization of polyfluoroalkyl-containing 2-(arylhydrazinylidene)-1,3-diketones. J. Fluor. Chem. 2018, 210, 117–125. [Google Scholar] [CrossRef]
- Elkina, N.A.; Burgart, Y.V.; Shchegolkov, E.V.; Krasnykh, O.P.; Maslova, V.V.; Triandafilova, G.A.; Solodnikov, S.S.; Muryleva, A.A.; Misiurina, M.A.; Slita, A.V.; et al. Competitive routes to cyclizations of polyfluoroalkyl-containing 2-tolylhydrazinylidene-1,3-diketones with 3-aminopyrazoles into bioactive pyrazoloazines. J. Fluor. Chem. 2020, 240, 109648. [Google Scholar] [CrossRef]
- Messaad, M.; Dhouib, I.; Abdelhedi, M.; Khemakhem, B. Synthesis, bioassay and molecular docking of novel pyrazole and pyrazolone derivatives as acetylcholinesterase inhibitors. J. Mol. Struct. 2022, 1263, 133105. [Google Scholar] [CrossRef]
- Munteanu, I.G.; Apetrei, C. Analytical Methods Used in Determining Antioxidant Activity: A Review. Int. J. Mol. Sci. 2021, 22, 3380. [Google Scholar] [CrossRef]
- Hunger, K.; Herbst, W. Pigments, Organic. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2000. [Google Scholar] [CrossRef]
- Geng, J.; Xu, D.; Chang, F.-F.; Tao, T.; Huang, W. From heterocyclic hydrazone to hydrazone-azomethine dyes: Solvent and pH induced hydrazone and azo-keto transformation for a family of pyrazolone-based heterocyclic dyes. Dye. Pigm. 2017, 137, 101–110. [Google Scholar] [CrossRef]
- Gao, P.; Jiang, H.; Chen, W.; Cui, Z. An intramolecular Mannich type reaction of ortho-amino aromatic azo dye and its detection effect for formaldehyde. Dye. Pigm. 2020, 179, 108376. [Google Scholar] [CrossRef]
- Szadkowski, B.; Marzec, A.; Rogowski, J.; Maniukiewicz, W.; Zaborski, M. Insight into the formation mechanism of azo dye-based hybrid colorant: Physico-chemical properties and potential applications. Dye. Pigm. 2019, 167, 236–244. [Google Scholar] [CrossRef]
- OECD. Guideline 423: Acute Oral toxicity; OECD: Paris, France, 1996. [Google Scholar]
- Gein, V.L.; Popov, A.V.; Kolla, V.É.; Popova, N.A.; Potemkin, K.D. Synthesis and biological activity of 1,5-diaryl-3-arylamino-4-carboxymethyl-2,5-dihydro-2-pyrrolones and 1,5-diaryl-4-carboxymethyltetrahydropyrrole-2, 3-diones. Pharm. Chem. J. 1993, 27, 343–346. [Google Scholar] [CrossRef]
- Nassini, R.; Fusi, C.; Materazzi, S.; Coppi, E.; Tuccinardi, T.; Marone, I.M.; De Logu, F.; Preti, D.; Tonello, R.; Chiarugi, A.; et al. The TRPA1 channel mediates the analgesic action of dipyrone and pyrazolone derivatives. Br. J. Pharmacol. 2015, 172, 3397–3411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malvar, D.d.C.; Soares, D.M.; Fabrício, A.S.C.; Kanashiro, A.; Machado, R.R.; Figueiredo, M.J.; Rae, G.A.; de Souza, G.E.P. The antipyretic effect of dipyrone is unrelated to inhibition of PGE2 synthesis in the hypothalamus. Br. J. Pharmacol. 2011, 162, 1401–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulsen, C.E.; Armache, J.-P.; Gao, Y.; Cheng, Y.; Julius, D. Structure of the TRPA1 ion channel suggests regulatory mechanisms. Nature 2015, 520, 511–517. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Lin King, J.V.; Paulsen, C.E.; Cheng, Y.; Julius, D. Irritant-evoked activation and calcium modulation of the TRPA1 receptor. Nature 2020, 585, 141–145. [Google Scholar] [CrossRef]
- Basbaum, A.I.; Bautista, D.M.; Scherrer, G.; Julius, D. Cellular and Molecular Mechanisms of Pain. Cell 2009, 139, 267–284. [Google Scholar] [CrossRef] [Green Version]
- Gladkikh, I.N.; Sintsova, O.V.; Leychenko, E.V.; Kozlov, S.A. TRPV1 Ion Channel: Structural Features, Activity Modulators, and Therapeutic Potential. Biochemistry 2021, 86, S50–S70. [Google Scholar] [CrossRef]
- Khudina, O.G.; Burgart, Y.V.; Shchegol’kov, E.V.; Saloutin, V.I.; Kazheva, O.N.; Chekhlov, A.N.; D’yachenko, O.A. Steric structure of alkyl 2-aryl(hetaryl)hydrazono-3-fluoroalkyl-3-oxopropionates. Russ. J. Org. Chem. 2009, 45, 801–809. [Google Scholar] [CrossRef]
- Saloutin, V.I.; Fomin, A.N.; Pashkevich, K.I. Reaction of fluorine-containing beta-ketoesters with bifunctional N-nucleophiles. Bull. Acad. Sci. USSR Div. Chem. Sci. 1985, 34, 135–141. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Palatinus, L.; Chapuis, G. SUPERFLIP– a computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J. Appl. Crystallogr. 2007, 40, 786–790. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. A short history ofSHELX. Acta Crystallogr. Sect. A 2007, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterri, S.H.; Johnsen, B.A.; Fonnum, F. A radiochemical assay method for carboxylesterase, and comparison of enzyme activity towards the substrates methyl [1-14C] butyrate and 4-nitrophenyl butyrate. Biochem. Pharmacol. 1985, 34, 2779–2785. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef]
- Benzie, I.F.F.; Strain, J.J. The Ferric Reducing Ability of Plasma (FRAP) as a Measure of “Antioxidant Power”: The FRAP Assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef] [Green Version]
- Benzie, I.F.F.; Strain, J.J. [2] Ferric reducing/antioxidant power assay: Direct measure of total antioxidant activity of biological fluids and modified version for simultaneous measurement of total antioxidant power and ascorbic acid concentration. Methods Enzymol. 1999, 299, 15–27. [Google Scholar] [CrossRef]
- Meir, S.; Kanner, J.; Akiri, B.; Philosoph-Hadas, S. Determination and Involvement of Aqueous Reducing Compounds in Oxidative Defense Systems of Various Senescing Leaves. J. Agric. Food Chem. 2002, 43, 1813–1819. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Kovaleva, N.V.; Rudakova, E.V.; Boltneva, N.P.; Lushchekina, S.V.; Faingold, I.I.; Poletaeva, D.A.; Soldatova, Y.V.; Kotelnikova, R.A.; Serkov, I.V.; et al. New Multifunctional Agents Based on Conjugates of 4-Amino-2,3-polymethylenequinoline and Butylated Hydroxytoluene for Alzheimer’s Disease Treatment. Molecules 2020, 25, 5891. [Google Scholar] [CrossRef] [PubMed]
- Ou, B.; Hampsch-Woodill, M.; Prior, R.L. Development and Validation of an Improved Oxygen Radical Absorbance Capacity Assay Using Fluorescein as the Fluorescent Probe. J. Agric. Food Chem. 2001, 49, 4619–4626. [Google Scholar] [CrossRef] [PubMed]
- Burgart, Y.V.; Makhaeva, G.F.; Krasnykh, O.P.; Borisevich, S.S.; Agafonova, N.A.; Kovaleva, N.V.; Boltneva, N.P.; Rudakova, E.V.; Shchegolkov, E.V.; Triandafilova, G.A.; et al. Synthesis of 4-Aminopyrazol-5-ols as Edaravone Analogs and Their Antioxidant Activity. Molecules 2022, 27, 7722. [Google Scholar] [CrossRef] [PubMed]
- Arendrup, M.C.; Kahlmeter, G.; Guinea, J.; Meletiadis, J. How to: Perform antifungal susceptibility testing of microconidia-forming dermatophytes following the new reference EUCAST method E.Def 11.0, exemplified by Trichophyton. Clin. Microbiol. Infect. 2021, 27, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Tudela, J.L.; Arendrup, M.C.; Barchiesi, F.; Bille, J.; Chryssanthou, E.; Cuenca-Estrella, M.; Dannaoui, E.; Denning, D.W.; Donnelly, J.P.; Dromer, F.; et al. EUCAST Definitive Document EDef 7.1: Method for the determination of broth dilution MICs of antifungal agents for fermentative yeasts. Clin. Microbiol. Infect. 2008, 14, 398–405. [Google Scholar] [CrossRef]
- Arendrup, M.C.; Cuenca-Estrella, M.; Lass-Flörl, C.; Hope, W. EUCAST technical note on the EUCAST definitive document EDef 7.2: Method for the determination of broth dilution minimum inhibitory concentrations of antifungal agents for yeasts EDef 7.2 (EUCAST-AFST)*. Clin. Microbiol. Infect. 2012, 18, E246–E247. [Google Scholar] [CrossRef] [Green Version]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Guzii, A.G.; Makarieva, T.N.; Korolkova, Y.V.; Andreev, Y.A.; Mosharova, I.V.; Tabakmaher, K.M.; Denisenko, V.A.; Dmitrenok, P.S.; Ogurtsova, E.K.; Antonov, A.S.; et al. Pulchranin A, isolated from the Far-Eastern marine sponge, Monanchora pulchra: The first marine non-peptide inhibitor of TRPV-1 channels. Tetrahedron Lett. 2013, 54, 1247–1250. [Google Scholar] [CrossRef]
- Korolkova, Y.; Makarieva, T.; Tabakmakher, K.; Shubina, L.; Kudryashova, E.; Andreev, Y.; Mosharova, I.; Lee, H.-S.; Lee, Y.-J.; Kozlov, S. Marine Cyclic Guanidine Alkaloids Monanchomycalin B and Urupocidin A Act as Inhibitors of TRPV1, TRPV2 and TRPV3, but not TRPA1 Receptors. Mar. Drugs 2017, 15, 87. [Google Scholar] [CrossRef] [Green Version]
- Mironov, A.N. Guidelines for Preclinical Study of Medicinal Products. Part One (In Russian); Grif&Ko: Moscow, Russia, 2012. [Google Scholar]
- Vogel, H.G. Drug Discovery and Evaluation; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 2008. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Petersson, G.A.; Al-Laham, M.A. A complete basis set model chemistry. II. Open-shell systems and the total energies of the first-row atoms. J. Chem. Phys. 1991, 94, 6081–6090. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadezhdin, K.D.; Neuberger, A.; Nikolaev, Y.A.; Murphy, L.A.; Gracheva, E.O.; Bagriantsev, S.N.; Sobolevsky, A.I. Extracellular cap domain is an essential component of the TRPV1 gating mechanism. Nat. Comm. 2021, 12, 2154. [Google Scholar] [CrossRef]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | RF | R1 | R2 | Yield (Method), % |
|---|---|---|---|---|
| 5a | CF3 | H | H | 58 (A) |
| 5b | CF3 | H | Me-4 | 59 (A) |
| 5c | CF3 | H | Br-4 | 78 (B) |
| 5d | CF3 | Me | Me-4 | 45 (A) |
| 5e | CF3 | Ph | H | 61 (A) |
| 5f | CF3 | Ph | Me-4 | 62 (A) |
| 5g | CF3 | Ph | OMe-4 | 74 (B) |
| 5h | CF3 | Ph | F-4 | 84 (B) |
| 5i | CF3 | Ph | Cl-2 | 86 (B) |
| 5j | CF3 | Ph | Cl2-2,6 | 79 (B) |
| 5k | CF3 | Ph | Br-4 | 83 (B) |
| 5l | CF3 | Ph | CO2Et-4 | 73 (B) |
| 5m | CF3 | Ph | SO2Me-4 | 82 (B) |
| 5n | CF3 | Ph | SO2NH2-4 | 78 (B) |
| 5o | CF3 | Ph | SO3H-4 | 67 (B) |
| 5p | CF3 | C6H4SO2NH2-4 | Me-4 | 70 (B) |
| 5q | CF3 | C6H4SO2Me-4 | Me-4 | 61 (B) |
| 6a | C2F5 | H | Me-4 | 59 (A) |
| 6b | C2F5 | Ph | Me-4 | 80 (B) |
| 6c | C2F5 | Ph | Cl-2 | 71 (B) |
| 6d | C2F5 | Ph | Cl-3 | 94 (B) |
| 6e | C2F5 | Ph | Cl2-2,6 | 64 (B) |
| 6f | C2F5 | Ph | Cl2-2,4 | 73 (B) |
| 6g | C2F5 | Ph | CO2Et-4 | 83 (B) |
| 6h | C2F5 | Ph | SO2Me-4 | 68 (B) |
| 7a | C3F7 | H | Me-4 | 65 (B) |
| 7b | C3F7 | Ph | Me-4 | 75 (B) |
| 8a | C4F9 | H | Me-4 | 78 (B) |
| 8b | C4F9 | Ph | Me-4 | 95 (B) |
| 9a | Me | H | Me-4 | 61 (A) |
| 9b | Me | Ph | Me-4 | 63 (A) |
| Com-pound | # Stars a | Lipinski’s Rule of Five | Jorgensen’s Rule of Three | QPlog BB k | QPlog Khsa l | HOA, % m | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| mol MW b | Donor HB c | Accpt HB d | QPlog Po/w e | # Rot f | PSA g | Qplog S h | QPP Caco i | # Met j | |||||
| min | 0 | 130.0 | 0.0 | 2.0 | −2.0 | 0 | 7.0 | −6.5 | <25 poor | 1.0 | −3.0 | −1.5 | <25 is poor |
| max | 5 | 725.0 | 6.0 | 20.0 | 6.5 | 15 | 200.0 | 0.5 | >500 great | 8.0 | 1.2 | 1.5 | >80 is high |
| 5a | 0 | 256.2 | 1.0 | 4.5 | 1.9 | 3 | 81.3 | −3.1 | 606 | 2 | −0.5 | −0.3 | 88 |
| 5b | 0 | 270.2 | 1.0 | 4.5 | 2.2 | 3 | 81.3 | −3.6 | 606 | 2 | −0.5 | −0.2 | 90 |
| 5c | 2 | 335.1 | 1.0 | 4.5 | 2.4 | 3 | 81.3 | −3.9 | 606 | 1 | −0.3 | −0.2 | 91 |
| 5d | 0 | 284.2 | 0.0 | 4.5 | 2.9 | 3 | 64.5 | −3.8 | 1991.9 | 2 | 0.0 | −0.2 | 100 |
| 5e | 0 | 332.3 | 0.0 | 4.5 | 4.1 | 3 | 65.5 | −5.0 | 2458 | 2 | 0.0 | 0.2 | 100 |
| 5f | 0 | 346.3 | 0.0 | 4.5 | 4.4 | 3 | 65.5 | −5.6 | 2457 | 2 | 0.0 | 0.3 | 100 |
| 5g | 0 | 362.3 | 0.0 | 5.3 | 4.1 | 4 | 737 | −5.2 | 2456 | 2 | −0.1 | 0.2 | 100 |
| 5h | 0 | 350.3 | 0.0 | 4.5 | 4.3 | 3 | 65.5 | −5.4 | 2458 | 1 | 0.1 | 0.2 | 100 |
| 5i | 1 | 366.7 | 0.0 | 4.5 | 4.5 | 3 | 64.4 | −5.6 | 2868 | 2 | 0.2 | 0.3 | 100 |
| 5j | 1 | 401.2 | 0.0 | 4.5 | 4.8 | 3 | 62.4 | −5.8 | 3043 | 2 | 0.4 | 0.4 | 100 |
| 5k | 1 | 411.2 | 0.0 | 4.5 | 4.6 | 3 | 65.5 | −5.9 | 2458 | 1 | 0.2 | 0.3 | 100 |
| 5l | 1 | 404.3 | 0.0 | 6.5 | 4.1 | 5 | 98.3 | −6.0 | 752 | 1 | −0.8 | 0.2 | 100 |
| 5m | 1 | 410.4 | 0.0 | 8.5 | 2.6 | 4 | 103.2 | −4.4 | 441 | 1 | −0.9 | −0.5 | 89 |
| 5n | 1 | 411.4 | 2.0 | 9.0 | 2.0 | 5 | 131.1 | −4.8 | 114 | 1 | −1.6 | −0.3 | 75 |
| 5o | 1 | 412.3 | 1.0 | 8.5 | 2.7 | 5 | 127.5 | −4.8 | 31 | 1 | −1.6 | −0.4 | 70 |
| 5p | 1 | 425.4 | 2.0 | 9.0 | 2.2 | 5 | 129.2 | −5.3 | 119 | 2 | −1.6 | −0.1 | 77 |
| 5q | 1 | 424.4 | 0.0 | 8.5 | 2.9 | 4 | 101.5 | −4.9 | 467 | 2 | −0.9 | −0.3 | 92 |
| 5r | 0 | 360.3 | 0.0 | 4.5 | 4.7 | 3 | 65.5 | −6.2 | 2456 | 3 | 0.0 | 0.5 | 100 |
| 6a | 0 | 320.2 | 1.0 | 4.5 | 3.0 | 4 | 81.0 | −4.3 | 695 | 2 | −0.4 | 0.0 | 96 |
| 6b | 0 | 396.3 | 0.0 | 4.5 | 5.0 | 4 | 62.4 | −6.2 | 2906 | 2 | 0.1 | 0.5 | 100 |
| 6c | 1 | 416.7 | 0,0 | 4.5 | 5.2 | 4 | 61.3 | −6.1 | 3334 | 2 | 0.4 | 0.4 | 100 |
| 6d | 2 | 416.7 | 0.0 | 4.5 | 5.2 | 4 | 62.4 | −6.4 | 2902 | 2 | 0.3 | 0.5 | 100 |
| 6e | 2 | 451.2 | 0.0 | 4.5 | 5.6 | 4 | 61.1 | −6.7 | 3689 | 2 | 0.5 | 0.6 | 100 |
| 6f | 3 | 451.2 | 0.0 | 4.5 | 5.7 | 4 | 61.4 | −6.9 | 3337 | 1 | 0.5 | 0.6 | 100 |
| 6g | 2 | 454.4 | 0.0 | 6.5 | 4.8 | 6 | 98.0 | −6.7 | 854 | 1 | −0.7 | 0.4 | 100 |
| 6h | 1 | 460.4 | 0.0 | 8.5 | 3.2 | 5 | 100.2 | −5.0 | 519 | 1 | −0.8 | −0.3 | 94 |
| 7a | 2 | 370.2 | 1.0 | 4.5 | 3.4 | 5 | 77.3 | −4.8 | 756 | 2 | −0.4 | 0.1 | 100 |
| 7b | 2 | 446.3 | 0.0 | 4.5 | 5.7 | 5 | 62.3 | −6.9 | 2931 | 2 | 0.2 | 0.7 | 100 |
| 8a | 1 | 420.2 | 1.0 | 4.5 | 4.1 | 6 | 81.1 | −5.6 | 700 | 2 | −0.4 | 0.3 | 100 |
| 8b | 2 | 496.3 | 0.0 | 4.5 | 6.5 | 6 | 63.2 | −7.9 | 3115 | 2 | 0.2 | 0.9 | 100 |
| 9a | 0 | 216.2 | 1.0 | 4.5 | 1.5 | 3 | 81.8 | −2.8 | 527 | 3 | −0.8 | −0.3 | 84 |
| 9b | 0 | 292.3 | 0.0 | 4.5 | 3.7 | 3 | 66.0 | −4.0 | 2136 | 3 | −0.3 | 0.2 | 100 |
| No | Structure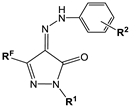 | Inhibitory Activity IC50 ± SEM (µM) or Inhibition % at 20 µM | ||||
|---|---|---|---|---|---|---|
| RF | R1 | R2 | Porcine CES | Human AChE | Equine BChE | |
| 5a | CF3 | H | H | 0.248 ± 0.022 | 12.8 ± 2.3% | 10.7 ± 1.8% |
| 5b | CF3 | H | Me-4 | 10.1 ± 1.7% | n.a. | n.a. |
| 5c | CF3 | H | Br-4 | 5.5 ± 0.9% | 3.4 ± 0.6% | 5.9 ± 1.0% |
| 5d | CF3 | Me | Me-4 | 2.56 ± 0.16 | 4.5 ± 1.1% | n.a. |
| 5e | CF3 | Ph | H | 0.481 ± 0.043 | n.a. | 5.1 ± 0.9% |
| 5f | CF3 | Ph | Me-4 | 1.01 ± 0.09 | n.a. | n.a. |
| 5g | CF3 | Ph | OMe-4 | 2.16 ± 0.20 | 12.3 ± 2.3% | n.a. |
| 5h | CF3 | Ph | F-4 | 1.90 ± 0.15 | 7.0 ± 1.2% | 3.2 ± 0.6% |
| 5i | CF3 | Ph | Cl-2 | 23.2 ± 1.8% | 6.3 ± 1.0% | 6.4 ± 1.3% |
| 5j | CF3 | Ph | Cl2-2,6 | 2.26 ± 0.18 | n.a. | 7.1 ± 1.2% |
| 5k | CF3 | Ph | Br-4 | 23.2 ± 2.0% | 5.4 ± 0.9% | 6.0 ± 1.1% |
| 5l | CF3 | Ph | CO2Et-4 | 0.871 ± 0.069 | 16.7 ± 1.5% | 3.2 ± 0.8% |
| 5m | CF3 | Ph | SO2Me-4 | 20.4 ± 1.6% | 5.5 ± 0.5% | 4.0 ± 1.0% |
| 5n | CF3 | Ph | SO2NH2-4 | n.a. | n.a. | 3.5 ± 0.6% |
| 5o | CF3 | Ph | SO3H-4 | 5.2 ± 1.0% | n.a. | 7.4 ± 1.2% |
| 5p | CF3 | C6H4SO2NH2-4 | Me-4 | 5.7 ± 1.0% | n.a. | 3.6 ± 0.9% |
| 5q | CF3 | C6H4SO2Me-4 | Me-4 | n.a. | 4.3 ± 0.8% | n.a. |
| 5r | CF3 | C6H4Me-4 | Me-4 | 11.9 ± 0.9 | n.a. | 4.0 ± 1.0% |
| 6a | C2F5 | H | Me-4 | 7.8 ± 1.4% | n.a. | 3.3 ± 0.9% |
| 6b | C2F5 | Ph | Me-4 | 0.954 ± 0.62 | 8.3 ± 1.4% | 5.7 ± 1.3% |
| 6c | C2F5 | Ph | Cl-2 | 7.34 ± 0.66 | 3.3 ± 0.6% | 6.5 ± 1.0% |
| 6d | C2F5 | Ph | Cl-3 | 1.68 ± 0.13 | n.a. | 7.8 ± 1.2% |
| 6e | C2F5 | Ph | Cl2-2,6 | n.a. | 6.0 ± 1.0% | 6.2 ± 1.2% |
| 6f | C2F5 | Ph | Cl2-2,4 | 6.6 ± 0.9% | 3.4 ± 0.7% | 4.3 ± 0.8% |
| 6g | C2F5 | Ph | CO2Et-4 | 1.81 ± 0.14 | 6.2 ± 0.9% | 4.2 ± 0.8% |
| 6h | C2F5 | Ph | SO2Me-4 | 30.8 ± 2.7% | 4.1 ± 0.8% | 5.2 ± 0.9% |
| 7a | C3F7 | H | Me-4 | 9.9 ± 0.7% | 35.6 ± 0.8% | 10.5 ± 1.1% |
| 7b | C3F7 | Ph | Me-4 | 2.44 ± 0.18 | 12.6 ± 1.8% | 4.5 ± 1.2% |
| 8a | C4F9 | H | Me-4 | 15.2 ± 1.1 | n.a. | n.a. |
| 8b | C4F9 | Ph | Me-4 | 12.1 ± 1.9% | 9.0 ± 1.4% | n.a. |
| 9a | Me | H | Me-4 | n.a. | n.a. | 4.3 ± 1.0% |
| 9b | Me | Ph | Me-4 | 10.3 ± 2.4% | n.a. | n.a. |
| BNPP | 1.80 ± 0.11 | n.a. | n.a. | |||
| Tacrine | n.a. | 0.601 ± 0.047 | 0.0295 ± 0.0002 | |||
| No | Compounds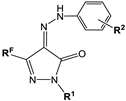 | Ki (µM) | αKi (µM) | ||
|---|---|---|---|---|---|
| RF | R1 | R2 | |||
| 5a | CF3 | H | H | 0.116 ± 0.008 | 0.255 ± 0.013 |
| 5d | CF3 | Me | Me-4 | 2.75 ± 0.22 | 3.98 ± 0.31 |
| 5f | CF3 | Ph | Me-4 | 0.309 ± 0.014 | 0.735 ± 0.007 |
| No | Structure | Antioxidant Activity | |||||
|---|---|---|---|---|---|---|---|
| ABTS.+-Scavenging Activity, (Mean ± SEM ) | FRAP (* TE) | ORAC-FL (* TE) | |||||
| RF | R1 | R2 | * TEAC | IC50, μM | |||
| 5a | CF3 | H | H | 0.50 ± 0.03 | 42.8 ± 2.3 | 0.09 ± 0.01 | 2.30 ± 0.20 |
| 5b | CF3 | H | Me-4 | 0.42 ± 0.03 | 46.2 ± 2.4 | 0.05 ± 0.02 | 2.37 ± 0.32 |
| 5c | CF3 | H | Br-4 | 0.40 ± 0.02 | 41.5 ± 2.4 | n.a. | 1.73 ± 0.21 |
| 5d | CF3 | Me | Me-4 | 0.15 ± 0.01 | 160.8 ± 3.2 | n.a. | 1.27 ± 0.15 |
| 5e | CF3 | Ph | H | 0.02 | n.d. | n.a. | i.t.s. |
| 5f | CF3 | Ph | Me-4 | 0.1 ± 0.006 | n.d. | n.a. | i.t.s. |
| 5g | CF3 | Ph | OMe-4 | 0.35 ± 0.03 | 55.6 ± 2.4 | n.a. | 2.20 ± 0.36 |
| 5h | CF3 | Ph | F-4 | 0.05 ± 0.004 | n.d. | n.a. | n.a. |
| 5i | CF3 | Ph | Cl-2 | n.a. | n.d. | n.a. | i.t.s. |
| 5j | CF3 | Ph | Cl2-2,6 | n.a. | n.d. | n.a. | n.d. |
| 5k | CF3 | Ph | Br-4 | n.a. | n.d. | n.a. | i.t.s. |
| 5l | CF3 | Ph | CO2Et-4 | n.a. | n.d. | n.a. | i.t.s. |
| 5m | CF3 | Ph | SO2Me-4 | n.a. | n.d. | n.a. | i.t.s. |
| 5n | CF3 | Ph | SO2NH2-4 | n.a. | n.d. | n.a. | i.t.s. |
| 5o | CF3 | Ph | SO3H-4 | n.a. | n.d. | n.a. | i.t.s. |
| 5p | CF3 | C6H4SO2NH2-4 | Me-4 | 0.03 ± 0.002 | n.d. | n.a. | i.t.s. |
| 5q | CF3 | C6H4SO2Me-4 | Me-4 | 0.02 ± 0.001 | n.d. | n.a. | i.t.s. |
| 5r | CF3 | C6H4Me-4 | Me-4 | 0.13 ± 0.007 | n.d. | n.a. | n.a. |
| 6a | C2F5 | H | Me-4 | 0.45 ± 0.03 | 46.8 ± 2.1 | 0.15 ± 0.02 | 2.67 ± 0.29 |
| 6b | C2F5 | Ph | Me-4 | 0.04 ± 0.002 | n.d. | n.a. | n.a. |
| 6c | C2F5 | Ph | Cl-2 | n.a. | n.d. | n.a. | n.d. |
| 6d | C2F5 | Ph | Cl-3 | n.a. | n.d. | n.a. | n.d. |
| 6e | C2F5 | Ph | Cl2-2,6 | n.a. | n.d. | n.a. | n.d. |
| 6f | C2F5 | Ph | Cl2-2,4 | n.a. | n.d. | n.a. | n.d. |
| 6g | C2F5 | Ph | CO2Et-4 | n.a. | n.d. | n.a. | n.d. |
| 6h | C2F5 | Ph | SO2Me-4 | n.a. | n.d. | n.a. | i.t.s. |
| 7a | C3F7 | H | Me-4 | 0.54 ± 0.03 | 41.5 ± 1.8 | n.a. | 2.80 ± 0.01 |
| 7b | C3F7 | Ph | Me-4 | 0.03 ± 0.002 | n.d. | n.a. | n.a. |
| 8a | C4F9 | H | Me-4 | 0.43 ± 0.03 | 48.3 ± 2.2 | n.a. | 2.33 ± 0.15 |
| 8b | C4F9 | Ph | Me-4 | 0.05 ± 0.03 | >400 | n.a. | n.a. |
| 9a | Me | H | Me-4 | 0.86 ± 0.04 | 25.4 ± 1.1 | 0.51 ± 0.01 | 3.23 ± 0.50 |
| 9b | Me | Ph | Me-4 | 0.80 ± 0.05 | 28.3 ± 1.2 | 0.11 ± 0.01 | 2.67 ± 0.58 |
| Trolox | 1.0 | 19.7 ± 0.8 | 1.0 | 1.0 | |||
| Edaravone | 0.96 ± 0.04 | 21.4 ± 1.8 | 0.80 ± 0.01 | 3.71 ± 0.06 | |||
| No | RF | R1 | R2 | MIC (μg/mL) | No | RF | R1 | R2 | MIC (μg/mL) |
|---|---|---|---|---|---|---|---|---|---|
| 5b | CF3 | H | Me-4 | 62.5 | 6a | C2F5 | H | Me-4 | 62.5 |
| 5c | CF3 | H | Br-4 | 125 | 6b | C2F5 | Ph | Me-4 | 0.9 |
| 5d | CF3 | Me | Me-4 | 15.6 | 6c | C2F5 | Ph | Cl-2 | >250 |
| 5e | CF3 | Ph | H | >250 | 6d | C2F5 | Ph | Cl-3 | 15.6 |
| 5f | CF3 | Ph | Me-4 | 62.5 | 6e | C2F5 | Ph | Cl2-2,6 | 62.5 |
| 5g | CF3 | Ph | OMe-4 | 0.9 | 6f | C2F5 | Ph | Cl2-2,4 | >250 |
| 5h | CF3 | Ph | F-4 | 125 | 6g | C2F5 | Ph | CO2Et-4 | 15.6 |
| 5i | CF3 | Ph | Cl-2 | >250 | 6h | C2F5 | Ph | SO2Me-4 | 250 |
| 5j | CF3 | Ph | Cl2-2,6 | 31.2 | 7a | C3F7 | H | Me-4 | >250 |
| 5k | CF3 | Ph | Br-4 | 31.2 | 7b | C3F7 | Ph | Me-4 | 31.2 |
| 5l | CF3 | Ph | CO2Et-4 | 31.2 | 8a | C4F9 | H | Me-4 | 250 |
| 5m | CF3 | Ph | SO2Me-4 | 125 | 8b | C4F9 | Ph | Me-4 | >250 |
| 5o | CF3 | Ph | SO3H-4 | 62.5 | 9a | Me | H | Me-4 | >250 |
| 5p | CF3 | C6H4SO2NH2-4 | Me-4 | 62.5 | 9b | Me | Ph | Me-4 | >250 |
| 5q | CF3 | C6H4SO2Me-4 | Me-4 | >250 | Spectinomycin | 15.6 | |||
| 5r | CF3 | C6H4Me-4 | Me-4 | >250 | |||||
| No | Structure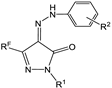 | IC50, μM | Selectivity Index (SI) * | |||
|---|---|---|---|---|---|---|
| RF | R1 | R2 | HeLa | Human Dermal Fibroblasts | ||
| 5b | CF3 | H | Me-4 | 2.26 ± 0.14 | 8.18 ± 0.73 | 3.62 |
| 5f | CF3 | Ph | Me-4 | 4.49 ± 0.18 | 20.50 ± 2.52 | 4.56 |
| 5m | CF3 | Ph | SO2Me-4 | n.a. | n.a. | - |
| 5p | CF3 | SO2NH2-4 | Me-4 | n.a. | n.a. | - |
| 5q | CF3 | SO2Me-4 | Me-4 | n.a. | n.a. | - |
| 6a | C2F5 | H | Me-4 | 1.47 ± 0.13 | 12.98 ± 2.56 | 8.83 |
| 6b | C2F5 | Ph | Me-4 | 65.88 ± 5.22 | 98.56 ± 6.32 | 1.49 |
| 7b | C3F7 | Ph | Me-4 | 59.67 ± 6.34 | 73.79 ± 5.32 | 1.24 |
| 8b | C4F9 | Ph | Me-4 | n.a. | n.a. | - |
| 9a | Me | H | Me-4 | 3.16 ± 0.25 | 24.30 ± 1.82 | 7.68 |
| 9b | Me | Ph | Me-4 | 46.45 ± 1.98 | 24.12 ± 3.21 | 0.52 |
| Camptothecin | 1.08 ± 0.98 | 5.38 ± 0.48 | 4.98 | |||
| Doxorubicin | 4.25 ± 0.55 | 2.72 ± 0.26 | 0.64 | |||
| No | Structure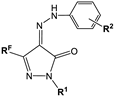 | Latent Period Prolongation, % (p-Value 1) | Acute Toxicity | ||||
|---|---|---|---|---|---|---|---|
| RF | R1 | R2 | 1 h | 2 h | Dose, mg/kg | Viability, % | |
| 5a | CF3 | H | H | 40 * | n.a. | 300 | 100 |
| 5b | CF3 | H | Me-4 | 69.5 ** | 59.7 * | 300 | 100 |
| 5c | CF3 | H | Br-4 | n.a. | 51 ** | 300 | 100 |
| 5d | CF3 | Me | Me-4 | n.a. | n.a. | 300 | 100 |
| 5e | CF3 | Ph | H | 100 *** | 129 ** | 300 | 100 |
| 5f | CF3 | Ph | Me-4 | 63 **** | 68 ** | 300 | 100 |
| 5g | CF3 | Ph | OMe-4 | n.a. | 76.9 * | 300 | 100 |
| 5h | CF3 | Ph | F-4 | 66 ** | 67 § | 300 | 100 |
| 5i | CF3 | Ph | Cl-2 | 42 * | 61 * | 300 | 100 |
| 5j | CF3 | Ph | Cl2-2,6 | n.a. | n.a. | 300 | 100 |
| 5k | CF3 | Ph | Br-4 | 89 ** | 76 *** | 300 | 100 |
| 5l | CF3 | Ph | CO2Et-4 | 68.1 ** | 107.2 ** | 300 | 100 |
| 5m | CF3 | Ph | SO2Me-4 | 116 ** | 144 *** | 300 | 100 |
| 5n | CF3 | Ph | SO2NH2-4 | n.a. | 84 **** | 300 | 100 |
| 5o | CF3 | Ph | SO3H-4 | 109 ** | 54 * | 300 | 100 |
| 5p | CF3 | C6H4SO2NH2-4 | Me-4 | 62.1 * | 70.9 * | 300 | 100 |
| 5q | CF3 | C6H4SO2Me-4 | Me-4 | 103.8 *** | 110.3 ** | 300 | 100 |
| 5r | CF3 | C6H4Me-4 | Me-4 | 63.2 * | 74.2 * | 300 | 100 |
| 6a | C2F5 | H | Me-4 | 83.9 *** | 63.3 * | 300 | 100 |
| 6b | C2F5 | Ph | Me-4 | 158.5 *** | 74.3 *** | 300 | 100 |
| 6c | C2F5 | Ph | Cl-2 | 75 ** | 51 ** | 300 | 100 |
| 6d | C2F5 | Ph | Cl-3 | 93 *** | 63 ** | 300 | 100 |
| 6e | C2F5 | Ph | Cl2-2,6 | n.a. | n.a. | 300 | 100 |
| 6f | C2F5 | Ph | Cl2-2,4 | n.a. | n.a. | 300 | 100 |
| 6g | C2F5 | Ph | CO2Et-4 | 67 ** | 73 * | 300 | 100 |
| 6h | C2F5 | Ph | SO2Me-4 | 80 ** | 98 ** | 300 | 100 |
| 7a | C3F7 | H | Me-4 | 42 ** | 52 ** | 300 | 100 |
| 7b | C3F7 | Ph | Me-4 | 144 **** | 82.3 ** | 300 | 100 |
| 8a | C4F9 | H | Me-4 | 62.9 * | 40.4 * | 300 | 100 |
| 8b | C4F9 | Ph | Me-4 | 95.2 ** | 124.8 *** | 300 | 100 |
| 9a | Me | H | Me-4 | 80.8 ** | n.a. | 300 | 100 |
| 9b | Me | Ph | Me-4 | n.a. | 140.2 ** | 300 | 100 |
| Diclofenac, 10 mg/kg | 63.5 ± 18.2 2 | 70.0 ± 20.8 2 | 100 | 66 | |||
| Metamizole, 15 mg/kg | 34.2 ± 11.3 3 | 64.6 ± 22.5 3 | 300 | 100 | |||
| No | Structure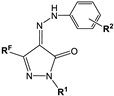 | Dose, mg/kg | Inhibition of Paw Edema Development Comparing to Control, % | ||||
|---|---|---|---|---|---|---|---|
| after 1 h | after 3 h | after 5 h | |||||
| RF | R1 | R2 | |||||
| 5f | CF3 | Ph | Me-4 | 15 | n.a. | n.a. | 30.9 * |
| 5m | CF3 | Ph | SO2Me-4 | 25 | 49.0 ** | 44.3 * | 35.3 * |
| 6a | C2F5 | H | Me-4 | 15 | n.a. | n.a. | n.a. |
| 8b | C4F9 | Ph | Me-4 | 15 | n.a. | n.a. | n.a. |
| 9b | Me | Ph | Me-4 | 15 | n.a. | n.a. | n.a. |
| Diclofenac | 10 | 35.6 ± 5.8 1 | 54.3 ± 12.1 1 | 46.2 ± 13.2 1 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burgart, Y.V.; Elkina, N.A.; Shchegolkov, E.V.; Krasnykh, O.P.; Makhaeva, G.F.; Triandafilova, G.A.; Solodnikov, S.Y.; Boltneva, N.P.; Rudakova, E.V.; Kovaleva, N.V.; et al. Powerful Potential of Polyfluoroalkyl-Containing 4-Arylhydrazinylidenepyrazol-3-ones for Pharmaceuticals. Molecules 2023, 28, 59. https://doi.org/10.3390/molecules28010059
Burgart YV, Elkina NA, Shchegolkov EV, Krasnykh OP, Makhaeva GF, Triandafilova GA, Solodnikov SY, Boltneva NP, Rudakova EV, Kovaleva NV, et al. Powerful Potential of Polyfluoroalkyl-Containing 4-Arylhydrazinylidenepyrazol-3-ones for Pharmaceuticals. Molecules. 2023; 28(1):59. https://doi.org/10.3390/molecules28010059
Chicago/Turabian StyleBurgart, Yanina V., Natalia A. Elkina, Evgeny V. Shchegolkov, Olga P. Krasnykh, Galina F. Makhaeva, Galina A. Triandafilova, Sergey Yu. Solodnikov, Natalia P. Boltneva, Elena V. Rudakova, Nadezhda V. Kovaleva, and et al. 2023. "Powerful Potential of Polyfluoroalkyl-Containing 4-Arylhydrazinylidenepyrazol-3-ones for Pharmaceuticals" Molecules 28, no. 1: 59. https://doi.org/10.3390/molecules28010059