
Recent Advances in Donepezil Delivery Systems via the Nose-to-Brain Pathway


 , , , , ,
, , , , ,  ,
, 

Abstract
1. Introduction
1.1. Donepezil Development History
1.2. Physicochemical Characteristics and Clinical Considerations of Oral DPZ
1.3. Various Strategies for Overcoming the Limitations of DPZ Oral Delivery
1.3.1. Rapid Disintegration Tablets and Oral Film
1.3.2. Patches
1.3.3. Microneedle
1.3.4. Long-Acting Injection (LAI)
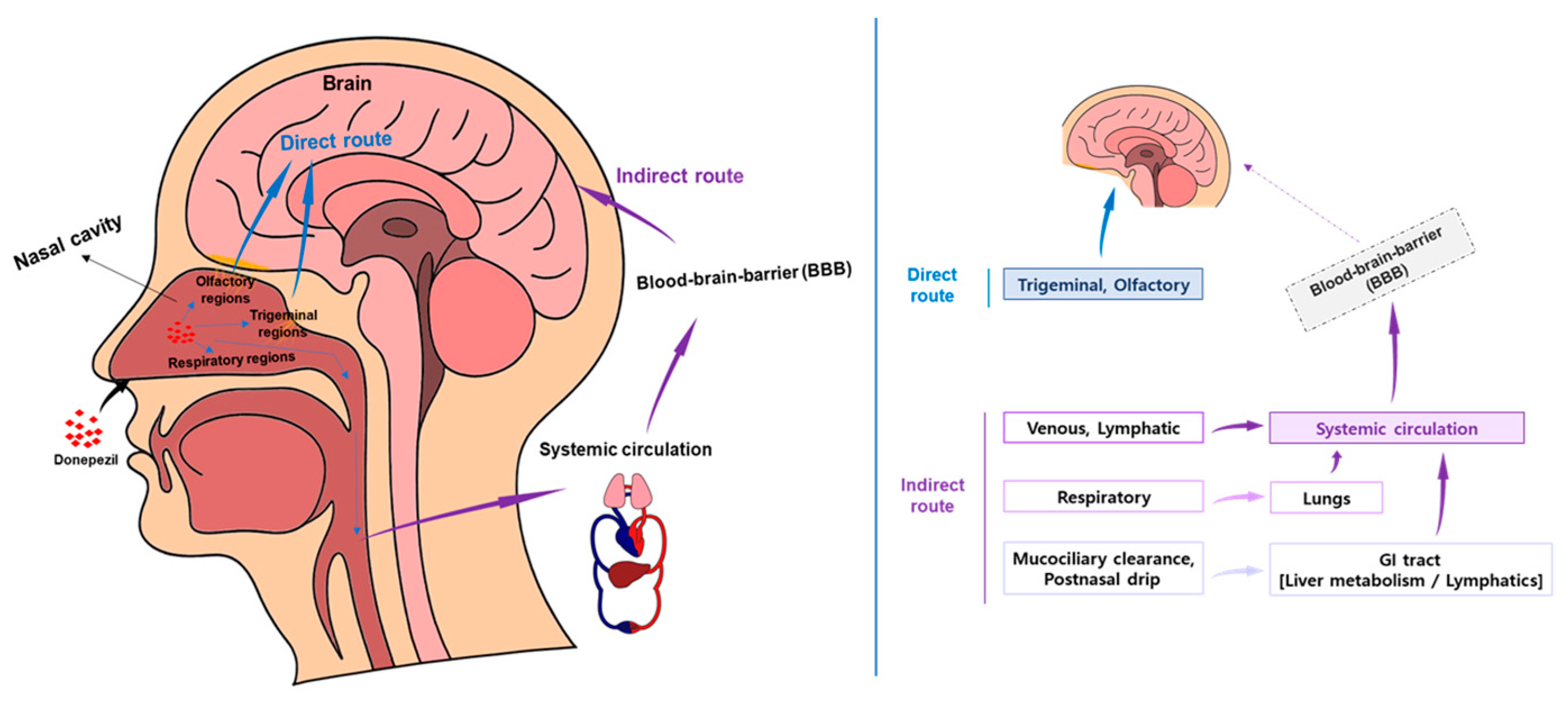
1.4. Nose-to-Brain (N2B) Drug Delivery of DPZ
1.4.1. N2B Drug Delivery Mechanism and Advantages
1.4.2. Key Issues in the Development of N2B Drug Delivery Formulation Technology
2. Case Studies of DDS for DPZ Delivery to N2B
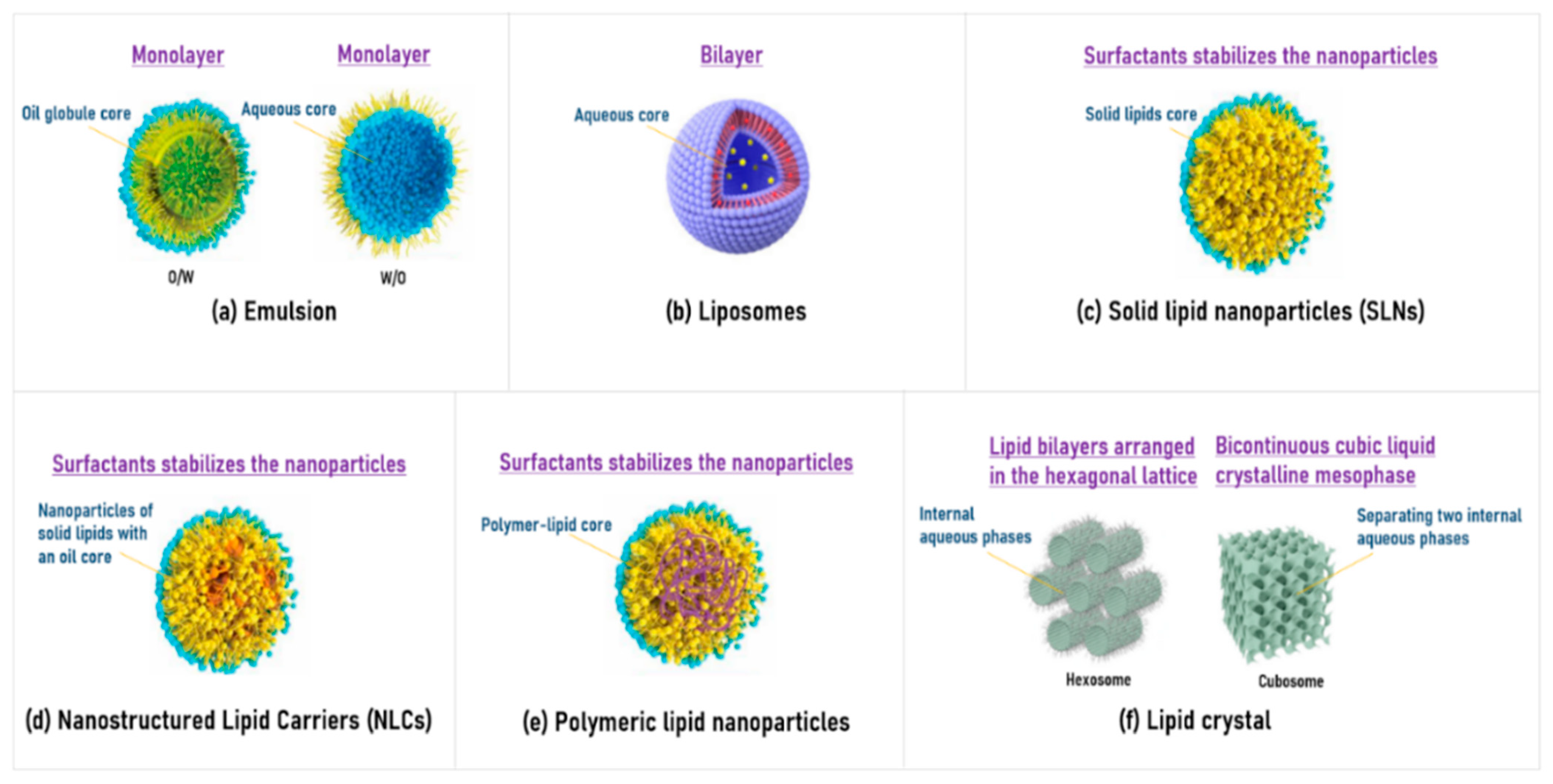
2.1. Lipid-Based Formulations
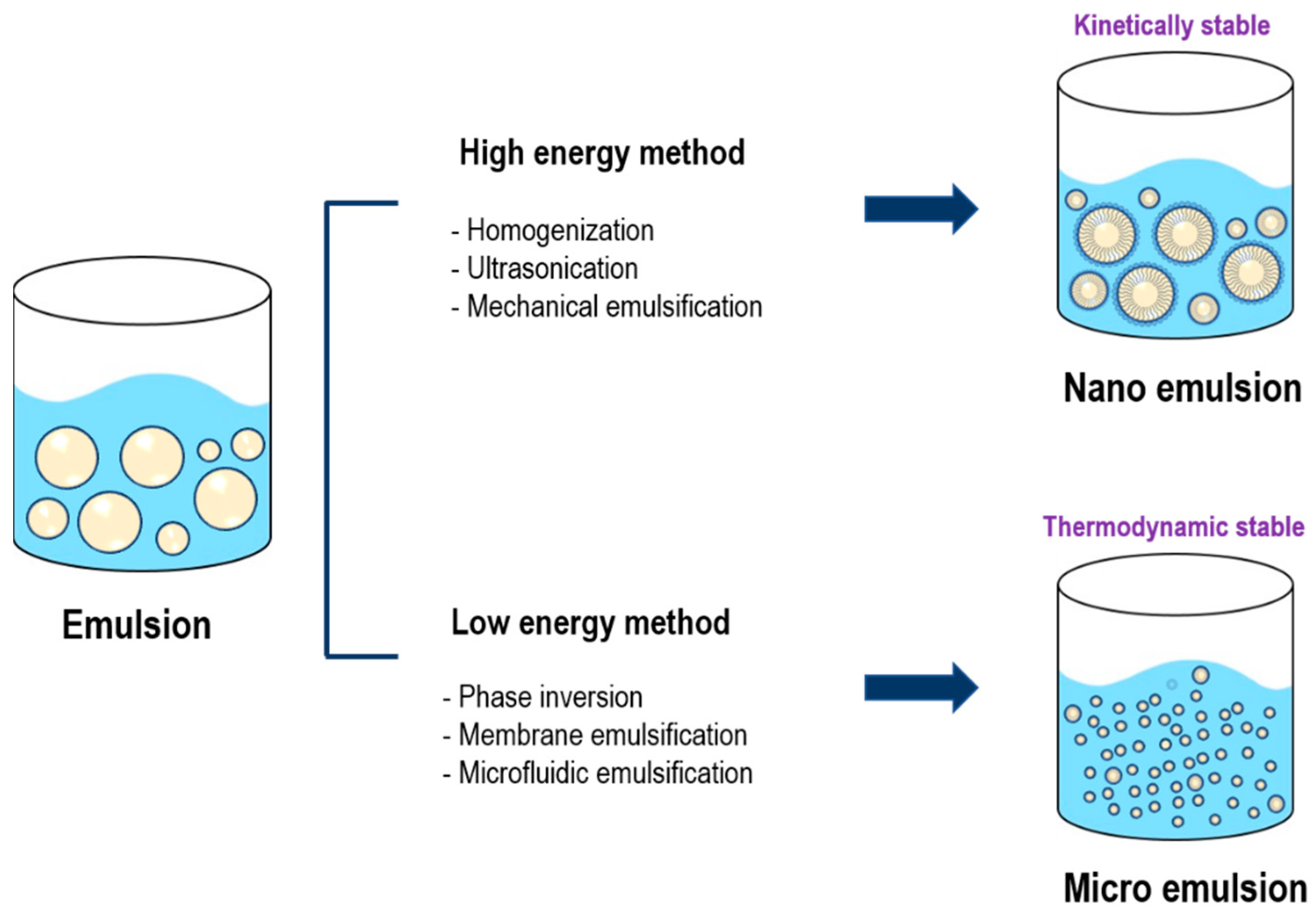
2.1.1. Emulsion
2.1.2. Liposome
2.1.3. Solid Lipid Nanoparticles (SLNs)
2.1.4. Nanostructured Lipid Carriers (NLCs)
2.1.5. Polymeric Lipid Nanoparticles
2.1.6. Liquid Crystal (Cubosome and Hexosome)
2.2. Solid Particle-Based Formulations
2.2.1. Inhalable Dry Powder
2.2.2. Suspension
- (1)
- Nanosuspension
- (2)
- Microsuspension
2.3. Solution-Based Formulation
2.4. Gel
2.5. Film
3. Expert Opinion on the Commercialization of DZP Formulation for N2B
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Agrawal, M.; Saraf, S.; Saraf, S.; Antimisiaris, S.G.; Chougule, M.B.; Shoyele, S.A.; Alexander, A. Nose-to-brain drug delivery: An update on clinical challenges and progress towards approval of anti-Alzheimer drugs. J. Control. Release 2018, 281, 139–177. [Google Scholar] [CrossRef] [PubMed]
- Kása, P.; Rakonczay, Z.; Gulya, K. The cholinergic system in Alzheimer’s disease. Prog. Neurobiol. 1997, 52, 511–535. [Google Scholar] [CrossRef] [PubMed]
- Thornton, S.; Crane, T. Laboratory confirmed massive donepezil ingestion. In Toxicology Cases for the Clinical and Forensic Laboratory; Elsevier: Amsterdam, The Netherlands, 2020; pp. 485–487. [Google Scholar]
- Bartus, R.T.; Dean, R.L., III; Beer, B.; Lippa, A.S. The cholinergic hypothesis of geriatric memory dysfunction. Science 1982, 217, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, P.J.; Price, D.L.; Struble, R.G.; Clark, A.W.; Coyle, J.T.; DeLong, M.R. Alzheimer’s disease and senile dementia: Loss of neurons in the basal forebrain. Science 1982, 215, 1237–1239. [Google Scholar] [CrossRef] [PubMed]
- Sutthapitaksakul, L.; Dass, C.R.; Sriamornsak, P. Donepezil—An updated review of challenges in dosage form design. J. Drug Deliv. Sci. Technol. 2021, 63, 102549. [Google Scholar] [CrossRef]
- Cecilia Rodrigues Simoes, M.; Pereira Dias Viegas, F.; Soares Moreira, M.; de Freitas Silva, M.; Maximo Riquiel, M.; Mattos da Rosa, P.; Rosa Castelli, M.; Henrique dos Santos, M.; Gomes Soares, M.; Viegas, C. Donepezil: An important prototype to the design of new drug candidates for Alzheimer’s disease. Mini Rev. Med. Chem. 2014, 14, 2–19. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, H.; Ogura, H.; Arai, Y.; Iimura, Y.; Yamanishi, Y. Research and development of donepezil hydrochloride, a new type of acetylcholinesterase inhibitor. Jpn. J. Pharmacol. 2002, 89, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Livingstone, N.; Hanratty, J.; McShane, R.; Macdonald, G. Pharmacological interventions for cognitive decline in people with Down syndrome. Cochrane Database Syst. Rev. 2015, 2015, CD011546. [Google Scholar] [CrossRef] [PubMed]
- Mohan, M.; Carpenter, P.K.; Bennett, C. Donepezil for dementia in people with Down syndrome. Cochrane Database Syst. Rev. 2009, 2009, CD007178. [Google Scholar] [CrossRef] [PubMed]
- Ridha, B.H.; Crutch, S.; Cutler, D.; Frost, C.; Knight, W.; Barker, S.; Epie, N.; Warrington, E.K.; Kukkastenvehmas, R.; Douglas, J. A double-blind placebo-controlled cross-over clinical trial of DONepezil in Posterior cortical atrophy due to underlying Alzheimer’s Disease: DONIPAD study. Alzheimer’s Res. Ther. 2018, 10, 44. [Google Scholar] [CrossRef] [PubMed]
- Devanand, D.P.; Pelton, G.H.; D’Antonio, K.; Ciarleglio, A.; Scodes, J.; Andrews, H.; Lunsford, J.; Beyer, J.L.; Petrella, J.R.; Sneed, J. Donepezil treatment in patients with depression and cognitive impairment on stable antidepressant treatment: A randomized controlled trial. Am. J. Geriatr. Psychiatry 2018, 26, 1050–1060. [Google Scholar] [CrossRef] [PubMed]
- Román, G.C.; Rogers, S.J. Donepezil: A clinical review of current and emerging indications. Expert Opin. Pharmacother. 2004, 5, 161–180. [Google Scholar] [CrossRef] [PubMed]
- Saluja, S.; Kasha, P.C.; Paturi, J.; Anderson, C.; Morris, R.; Banga, A.K. A novel electronic skin patch for delivery and pharmacokinetic evaluation of donepezil following transdermal iontophoresis. Int. J. Pharm. 2013, 453, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Moffat, A.C.; Osselton, M.D.; Widdop, B.; Watts, J. Clarke’s Analysis of Drugs and Poisons; Pharmaceutical Press: London, UK, 2011; Volume 3. [Google Scholar]
- Asiri, Y.; Mostafa, G. Donepezil. In Profiles of Drug Substances, Excipients and Related Methodology; Academic Press: New York, NY, USA, 2021; Volume 35, pp. 117–150. [Google Scholar]
- Ruela, A.L.M.; Carvalho, F.C.; Pereira, G.R. Exploring the phase behavior of monoolein/oleic acid/water systems for enhanced donezepil administration for Alzheimer disease treatment. J. Pharm. Sci. 2016, 105, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Geldmacher, D.S.; Farlow, M.R.; Sun, Y.; Moline, M.; Mackell, J. Efficacy and safety of donepezil 23 mg versus donepezil 10 mg for moderate-to-severe Alzheimer’s disease: A subgroup analysis in patients already taking or not taking concomitant memantine. Dement. Geriatr. Cogn. Disord. 2012, 33, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Graber, M.A.; Dachs, R.; Darby-Stewart, A. Donepezil to manage Alzheimer disease: New vs. standard Dosing. Am. Fam. Physician 2011, 83, 742–744. [Google Scholar] [PubMed]
- Begley, D.J. Delivery of therapeutic agents to the central nervous system: The problems and the possibilities. Pharmacol. Ther. 2004, 104, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Brasnjevic, I.; Steinbusch, H.W.; Schmitz, C.; Martinez-Martinez, P.; Initiative, E.N.R. Delivery of peptide and protein drugs over the blood–brain barrier. Prog. Neurobiol. 2009, 87, 212–251. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Sinko, P.J. Drug delivery across the blood–brain barrier: Why is it difficult? How to measure and improve it? Expert Opin. Drug Deliv. 2006, 3, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, M.; Tripathi, D.K.; Saraf, S.; Saraf, S.; Antimisiaris, S.G.; Mourtas, S.; Hammarlund-Udenaes, M.; Alexander, A. Recent advancements in liposomes targeting strategies to cross blood-brain barrier (BBB) for the treatment of Alzheimer’s disease. J. Control. Release 2017, 260, 61–77. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A. Drug delivery to the brain in Alzheimer’s disease: Consideration of the blood–brain barrier. Adv. Drug Deliv. Rev. 2012, 64, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Drath, I.; Richter, F.; Feja, M. Nose-to-brain drug delivery: From bench to bedside. Transl. Neurodegener. 2025, 14, 23. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; Maeng, H.J.; Yu, K.H.; Lee, K.R.; Tsuruo, T.; Kim, D.D.; Shim, C.K.; Chung, S.J. Evidence of carrier-mediated transport in the penetration of donepezil into the rat brain. J. Pharm. Sci. 2010, 99, 1548–1566. [Google Scholar] [CrossRef] [PubMed]
- Drapaca, C.S. Mathematical Modeling of Alzheimer’s Drug Donepezil Hydrochloride Transport to the Brain after Oral Administration. Fractal Fract. 2024, 8, 496. [Google Scholar] [CrossRef]
- Park, H.; Kim, J.-S.; Hong, S.; Ha, E.-S.; Nie, H.; Zhou, Q.T.; Kim, M.-S. Tableting process-induced solid-state polymorphic transition. J. Pharm. Investig. 2022, 52, 175–194. [Google Scholar] [CrossRef]
- Nam, J.-h.; Kim, B.-h.; Shafioul, A.S.M.; Jin, M.; Cho, C.-W. A comprehensive review of oral disintegrating film products, and their quality assessment and development. J. Pharm. Investig. 2025, 55, 351–373. [Google Scholar] [CrossRef]
- Nagy, Z.K.; Nyúl, K.; Wagner, I.; Molnár, K.; Marosi, G. Electrospun water soluble polymer mat for ultrafast release of Donepezil HCl. Express Polym. Lett. 2010, 4, 763–772. [Google Scholar] [CrossRef]
- Sevilla, C.; Jiménez-Caballero, P.; Alfonso, V. Orally disintegrating donepezil: Are the main caregivers of patients with Alzheimer’s disease more satisfied with this formulation of donepezil than with the traditional one? Rev. De Neurol. 2009, 49, 451–457. [Google Scholar]
- Zhao, Z.Q.; Chen, B.Z.; Zhang, X.P.; Zheng, H.; Guo, X.D. An Update on the Routes for the Delivery of Donepezil. Mol. Pharm. 2021, 18, 2482–2494. [Google Scholar] [CrossRef] [PubMed]
- Ministry of Food and Drug Safety. Available online: https://nedrug.mfds.go.kr/eng/index (accessed on 25 May 2025).
- Mahesh Kumar, T.; Vetrivel Rajan, M.; Gowsalya, K. A Comprehensive Review on Complexities and Directions of Drug Delivery Using Transdermal Patches. Int. J. Pharm. Sci. 2024, 2, 533–552. [Google Scholar] [CrossRef]
- Long, L.-y.; Zhang, J.; Yang, Z.; Guo, Y.; Hu, X.; Wang, Y. Transdermal delivery of peptide and protein drugs: Strategies, advantages and disadvantages. J. Drug Deliv. Sci. Technol. 2020, 60, 102007. [Google Scholar] [CrossRef]
- Gandhi, K.; Dahiya, A.; Kalra, T.; Singh, K. Transdermal drug delivery—A review. Int. J. Res. Pharm. Sci. 2012, 3, 379–388. [Google Scholar]
- Sozio, P.; Cerasa, L.S.; Marinelli, L.; Di Stefano, A. Transdermal donepezil on the treatment of Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2012, 8, 361–368. [Google Scholar] [PubMed]
- Valia, K.H.; Ramaraju, V.S. Transdermal Methods and Systems for Treating Alzheimer’s Disease. U.S. Patent No. 9,248,104, 2 February 2016. [Google Scholar]
- Kim, K.H.; Gwak, H.S. Effects of vehicles on the percutaneous absorption of donepezil hydrochloride across the excised hairless mouse skin. Drug Dev. Ind. Pharm. 2011, 37, 1125–1130. [Google Scholar] [CrossRef] [PubMed]
- Galipoğlu, M.; Erdal, M.S.; Güngör, S. Biopolymer-based transdermal films of donepezil as an alternative delivery approach in Alzheimer’s disease treatment. AAPS PharmSciTech 2015, 16, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Van Giau, V.; Vo, T.K. Current advances in transdermal delivery of drugs for Alzheimer’s disease. Indian J. Pharmacol. 2017, 49, 145–154. [Google Scholar] [PubMed]
- Zaid Alkilani, A.; McCrudden, M.T.; Donnelly, R.F. Transdermal drug delivery: Innovative pharmaceutical developments based on disruption of the barrier properties of the stratum corneum. Pharmaceutics 2015, 7, 438–470. [Google Scholar] [CrossRef] [PubMed]
- Kyriazanou, A.; Dallas, P.; Rekkas, D.; Choulis, N. Effect of several factors on the mechanical properties of a pressure sensitive adhesive containing penetration enhancers. STP Pharma Sci. 2002, 12, 283–286. [Google Scholar]
- Sheth, N.S.; Mistry, R.B. Formulation and evaluation of transdermal patches and to study permeation enhancement effect of eugenol. J. Appl. Pharm. Sci. 2011, 96–101. [Google Scholar]
- Das, A.; Ahmed, A.B. Natural permeation enhancer for transdermal drug delivery system and permeation evaluation: A review. Asian J. Pharm. Clin. Res. 2017, 10, 5–9. [Google Scholar] [CrossRef]
- Roy, N.; Agrawal, M.; Chaudhary, S.; Tirkey, V.; Dhwaj, A.; Mishra, N. Review article on permeation enhancers: A major breakthrough in drug delivery technology. Int. J. Pharm. Sci. Res. 2017, 8, 1001–1011. [Google Scholar]
- Kim, J.-Y.; Han, M.-R.; Kim, Y.-H.; Shin, S.-W.; Nam, S.-Y.; Park, J.-H. Tip-loaded dissolving microneedles for transdermal delivery of donepezil hydrochloride for treatment of Alzheimer’s disease. Eur. J. Pharm. Biopharm. 2016, 105, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zeng, M.; Shan, H.; Tong, C. Microneedle patches as drug and vaccine delivery platform. Curr. Med. Chem. 2017, 24, 2413–2422. [Google Scholar] [CrossRef] [PubMed]
- Waghule, T.; Singhvi, G.; Dubey, S.K.; Pandey, M.M.; Gupta, G.; Singh, M.; Dua, K. Microneedles: A smart approach and increasing potential for transdermal drug delivery system. Biomed. Pharmacother. 2019, 109, 1249–1258. [Google Scholar] [CrossRef] [PubMed]
- Ita, K. Transdermal delivery of drugs with microneedles—Potential and challenges. Pharmaceutics 2015, 7, 90–105. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, L.H.; Hesby, A. Intramuscular injection: An integrative research review and guideline for evidence-based practice. Appl. Nurs. Res. 2002, 15, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Cocoman, A.; Murray, J. Intramuscular injections: A review of best practice for mental health nurses. J. Psychiatr. Ment. Health Nurs. 2008, 15, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Lefevre, G.; Small, G.; Appel-Dingemanse, S. Pharmacokinetic rationale for the rivastigmine patch. Neurology 2007, 69, S10–S13. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Quan, P.; Fang, L.; Cun, D.; Yang, M. Sustained release donepezil loaded PLGA microspheres for injection: Preparation, In Vitro and In Vivo study. Asian J. Pharm. Sci. 2015, 10, 405–414. [Google Scholar] [CrossRef]
- An, T.; Choi, J.; Kim, A.; Lee, J.H.; Nam, Y.; Park, J.; kyung Sun, B.; Suh, H.; Kim, C.-J.; Hwang, S.-J. Sustained release of risperidone from biodegradable microspheres prepared by in-situ suspension-evaporation process. Int. J. Pharm. 2016, 503, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Fournier, E.; Passirani, C.; Montero-Menei, C.; Benoit, J. Biocompatibility of implantable synthetic polymeric drug carriers: Focus on brain biocompatibility. Biomaterials 2003, 24, 3311–3331. [Google Scholar] [CrossRef] [PubMed]
- Bode, C.; Kranz, H.; Siepmann, F.; Siepmann, J. In-situ forming PLGA implants for intraocular dexamethasone delivery. Int. J. Pharm. 2018, 548, 337–348. [Google Scholar] [CrossRef] [PubMed]
- McDonald, T.O.; Martin, P.; Patterson, J.P.; Smith, D.; Giardiello, M.; Marcello, M.; See, V.; O’Reilly, R.K.; Owen, A.; Rannard, S. Multicomponent organic nanoparticles for fluorescence studies in biological systems. Adv. Funct. Mater. 2012, 22, 2469–2478. [Google Scholar] [CrossRef]
- Giardiello, M.; McDonald, T.O.; Martin, P.; Owen, A.; Rannard, S.P. Facile synthesis of complex multi-component organic and organic–magnetic inorganic nanocomposite particles. J. Mater. Chem. 2012, 22, 24744–24752. [Google Scholar] [CrossRef]
- Kempe, S.; Mäder, K. In situ forming implants—An attractive formulation principle for parenteral depot formulations. J. Control. Release 2012, 161, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Corona, A., III; Schubert, B.; Reeder, R.; Henson, M.A. The effect of oil type on the aggregation stability of nanostructured lipid carriers. J. Colloid Interface Sci. 2014, 418, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Gapp, G.; Holzknecht, P. Risk analysis of sterile production plants: A new and simple, workable approach. PDA J. Pharm. Sci. Technol. 2011, 65, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Badkar, A.; Wolf, A.; Bohack, L.; Kolhe, P. Development of biotechnology products in pre-filled syringes: Technical considerations and approaches. AAPS PharmSciTech 2011, 12, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Owen, A.; Rannard, S. Strengths, weaknesses, opportunities and challenges for long acting injectable therapies: Insights for applications in HIV therapy. Adv. Drug Deliv. Rev. 2016, 103, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Devkar, T.B.; Tekade, A.R.; Khandelwal, K.R. Surface engineered nanostructured lipid carriers for efficient nose to brain delivery of ondansetron HCl using Delonix regia gum as a natural mucoadhesive polymer. Colloids Surf. B Biointerfaces 2014, 122, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Patil, R.P.; Pawara, D.D.; Gudewar, C.S.; Tekade, A.R. Nanostructured cubosomes in an in situ nasal gel system: An alternative approach for the controlled delivery of donepezil HCl to brain. J. Liposome Res. 2019, 29, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Chen, Y.; Zhang, W.; Xia, X.; Li, H.; Qin, M.; Gao, H. Nanotechnology for enhanced nose-to-brain drug delivery in treating neurological diseases. J. Control. Release 2024, 366, 519–534. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Shi, Y.; Zhang, Y.; Yang, Y.; Du, Y.; Yang, Z.; Wang, X.; Lei, T.; Xu, Y.; Chen, Y. Intranasal Carrier-Free Nanomodulator Addresses Both Symptomatology and Etiology of Alzheimer’s Disease by Restoring Neuron Plasticity and Reprogramming Lesion Microenvironment. ACS Nano 2024, 18, 29779–29793. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, L.C.; Silva-Abreu, M.; Clares, B.; Rodríguez-Lagunas, M.J.; Halbaut, L.; Cañas, M.-A.; Calpena, A.C. Formulation strategies to improve nose-to-brain delivery of donepezil. Pharmaceutics 2019, 11, 64. [Google Scholar] [CrossRef] [PubMed]
- Frey William, I.H. Neurologic Agents for Nasal Administration to the Brain. Patent WO1991007947A1, 13 June 1991. [Google Scholar]
- Frey, L.W.H.; Danielyan, L.; Gleiter, C.H. Methods, Pharmaceutical Compositions and Articles of Manufacture for Administering Therapeutic Cells to the Animal Central Nervous System. U.S. Patent No. 9,445,991, 20 September 2016. [Google Scholar]
- Gao, M.; Shen, X.; Mao, S. Factors influencing drug deposition in the nasal cavity upon delivery via nasal sprays. J. Pharm. Investig. 2020, 50, 251–259. [Google Scholar] [CrossRef]
- Crowe, T.P.; Greenlee, M.H.W.; Kanthasamy, A.G.; Hsu, W.H. Mechanism of intranasal drug delivery directly to the brain. Life Sci. 2018, 195, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Vyas, T.K.; Babbar, A.; Sharma, R.; Singh, S.; Misra, A. Intranasal mucoadhesive microemulsions of clonazepam: Preliminary studies on brain targeting. J. Pharm. Sci. 2006, 95, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Duong, V.-A.; Nguyen, T.-T.-L.; Maeng, H.-J. Recent advances in intranasal liposomes for drug, gene, and vaccine delivery. Pharmaceutics 2023, 15, 207. [Google Scholar] [CrossRef] [PubMed]
- Frey, W.H., II. Intranasal drug delivery bypasses the blood–brain barrier. Neurol. Rev. 2016, 24, 40–41. [Google Scholar]
- Bhattacharya, S.; Maelicke, A.; Montag, D. Nasal application of the galantamine pro-drug memogain slows down plaque deposition and ameliorates behavior in 5X familial Alzheimer’s disease mice. J. Alzheimer’s Dis. 2015, 46, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.C.; da Fonseca, C.O.; Schönthal, A.H. Bringing intranasal drug delivery for malignancies in the brain to market. Expert Opin. Drug Deliv. 2025, 22, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Nguyen, T.T.D.; Tran, N.-M.-A.; Van Vo, G. Lipid-based nanocarriers via nose-to-brain pathway for central nervous system disorders. Neurochem. Res. 2022, 47, 552–573. [Google Scholar] [CrossRef] [PubMed]
- Brako, F.; Boateng, J. Transmucosal drug delivery: Prospects, challenges, advances, and future directions. Expert Opin. Drug Deliv. 2025, 22, 525–553. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-Z.; Zhang, Y.-Q.; Wang, Z.-Z.; Wu, K.; Lou, J.-N.; Qi, X.-R. Enhanced brain distribution and pharmacodynamics of rivastigmine by liposomes following intranasal administration. Int. J. Pharm. 2013, 452, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Einer-Jensen, N.; Hunter, R. Counter-current transfer in reproductive biology. Reproduction 2005, 129, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Illum, L. Transport of drugs from the nasal cavity to the central nervous system. Eur. J. Pharm. Sci. 2000, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.-H.; Jang, J.-H.; Lee, Y.-B. Drug delivery to the brain via the nasal route of administration: Exploration of key targets and major consideration factors. J. Pharm. Investig. 2023, 53, 119–152. [Google Scholar] [CrossRef] [PubMed]
- Md, S.; Ali, M.; Ali, R.; Bhatnagar, A.; Baboota, S.; Ali, J. Donepezil nanosuspension intended for nose to brain targeting: In Vitro and In Vivo safety evaluation. Int. J. Biol. Macromol. 2014, 67, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Dhuria, S.V.; Hanson, L.R.; Frey, W.H., II. Intranasal delivery to the central nervous system: Mechanisms and experimental considerations. J. Pharm. Sci. 2010, 99, 1654–1673. [Google Scholar] [CrossRef] [PubMed]
- Alexander, A.; Dwivedi, S.; Giri, T.K.; Saraf, S.; Saraf, S.; Tripathi, D.K. Approaches for breaking the barriers of drug permeation through transdermal drug delivery. J. Control. Release 2012, 164, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Florence, A.T. The oral absorption of micro-and nanoparticulates: Neither exceptional nor unusual. Pharm. Res. 1997, 14, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Lockman, P.; Mumper, R.; Khan, M.; Allen, D. Nanoparticle technology for drug delivery across the blood-brain barrier. Drug Dev. Ind. Pharm. 2002, 28, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ugwoke, M.I.; Verbeke, N.; Kinget, R. The biopharmaceutical aspects of nasal mucoadhesive drug delivery. J. Pharm. Pharmacol. 2001, 53, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Van Den Berg, M.P.; Merkus, P.; Romeijn, S.G.; Verhoef, J.C.; Merkus, F.W. Hydroxocobalamin uptake into the cerebrospinal fluid after nasal and intravenous delivery in rats and humans. J. Drug Target. 2003, 11, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Al-Ghananeem, A.M.; Traboulsi, A.A.; Dittert, L.W.; Hussain, A.A. Targeted brain delivery of 17β-estradiol via nasally administered water soluble prodrugs. AAPS PharmSciTech 2002, 3, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Drummond, C.J.; Zhai, J.; Tran, N. Lipid Nanoparticles: Versatile Drug Delivery Vehicles for Traversing the Blood Brain Barrier to Treat Brain Cancer. Adv. Funct. Mater. 2024, 34, 2404234. [Google Scholar] [CrossRef]
- Costa, C.P.; Moreira, J.N.; Lobo, J.M.S.; Silva, A.C. Intranasal delivery of nanostructured lipid carriers, solid lipid nanoparticles and nanoemulsions: A current overview of In Vivo studies. Acta Pharm. Sin. B 2021, 11, 925–940. [Google Scholar] [CrossRef] [PubMed]
- John, R.; Monpara, J.; Swaminathan, S.; Kalhapure, R. Chemistry and Art of Developing Lipid Nanoparticles for Biologics Delivery: Focus on Development and Scale-Up. Pharmaceutics 2024, 16, 131. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.; Lim, H.; Park, H.; Yu, J.; An, J.; Yoo, H.Y.; Lee, T. Recent progress of lipid nanoparticles-based lipophilic drug delivery: Focus on surface modifications. Pharmaceutics 2023, 15, 772. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Nigam, K.; Bhatnagar, I.; Sukhpal, H.; Awasthy, S.; Shankar, S.; Tyagi, A.; Dang, S. Treatment of Alzheimer’s diseases using donepezil nanoemulsion: An intranasal approach. Drug Deliv. Transl. Res. 2020, 10, 1862–1875. [Google Scholar] [CrossRef] [PubMed]
- Handa, M.; Sanap, S.N.; Bhatta, R.S.; Patil, G.P.; Palkhade, R.; Singh, D.P.; Shukla, R. Simultaneous intranasal codelivery of donepezil and memantine in a nanocolloidal carrier: Optimization, pharmacokinetics, and pharmacodynamics studies. Mol. Pharm. 2023, 20, 4714–4728. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, L.C.; Vacacela, M.; Clares, B.; Garcia, M.L.; Fabrega, M.-J.; Calpena, A.C. Development of a Nasal Donepezil-loaded Microemulsion for the Treatment of Alzheimer’s Disease: In Vitro and Ex Vivo Characterization. CNS Neurol. Disord. Drug Targets 2018, 17, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Khunt, D.; Shrivas, M.; Polaka, S.; Gondaliya, P.; Misra, M. Role of omega-3 fatty acids and butter oil in targeting delivery of donepezil hydrochloride microemulsion to brain via the intranasal route: A comparative study. AAPS PharmSciTech 2020, 21, 45. [Google Scholar] [CrossRef] [PubMed]
- Al Asmari, A.K.; Ullah, Z.; Tariq, M.; Fatani, A. Preparation, characterization, and In Vivo evaluation of intranasally administered liposomal formulation of donepezil. Drug Des. Dev. Ther. 2016, 10, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Rajput, A.; Butani, S. Donepezil HCl liposomes: Development, characterization, cytotoxicity, and pharmacokinetic study. AAPS PharmSciTech 2022, 23, 74. [Google Scholar] [CrossRef] [PubMed]
- Salem, H.F.; Aboud, H.M.; Abdellatif, M.M.; Abou-Taleb, H.A. Nose-to-Brain Targeted Delivery of Donepezil Hydrochloride via Novel Hyaluronic Acid-Doped Nanotransfersomes for Alzheimer’s Disease Mitigation. J. Pharm. Sci. 2024, 113, 1934–1945. [Google Scholar] [CrossRef] [PubMed]
- Yasir, M.; Sara, U.V.S.; Chauhan, I.; Gaur, P.K.; Singh, A.P.; Puri, D.; Ameeduzzafar. Solid lipid nanoparticles for nose to brain delivery of donepezil: Formulation, optimization by Box–Behnken design, In Vitro and In Vivo evaluation. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1838–1851. [Google Scholar] [CrossRef]
- Topal, G.R.; Küçüktürkmen, B.; Öz, U.C.; Özkan, E.; Bakar-Ates, F.; Bozkır, A. Investigation on formulation parameters of donepezil HCl loaded solid lipid nanoparticles. Braz. J. Pharm. Sci. 2023, 59, e22330. [Google Scholar] [CrossRef]
- Yasir, M.; Chauhan, I.; Haji, M.J.; Tura, A.J.; Saxena, P.K. Formulation and evaluation of glyceryl behenate based solid lipid nanoparticles for the delivery of donepezil to brain through nasal route. Res. J. Pharm. Technol. 2018, 11, 2836–2844. [Google Scholar] [CrossRef]
- Yasir, M.; Zafar, A.; Noorulla, K.M.; Tura, A.J.; Sara, U.V.S.; Panjwani, D.; Khalid, M.; Haji, M.J.; Gobena, W.G.; Gebissa, T. Nose to brain delivery of donepezil through surface modified NLCs: Formulation development, optimization, and brain targeting study. J. Drug Deliv. Sci. Technol. 2022, 75, 103631. [Google Scholar] [CrossRef]
- Ali, M.H.; Alam, O.; Ali, A.; Ali, M.U.; Parvez, S.; Aldosari, E.; Baboota, S.; Ali, J. Donepezil and Embelin loaded nanostructured lipid carriers for direct brain delivery as an intervention for Alzheimer’s disease: Formulation design, optimization and evaluation. J. Clust. Sci. 2024, 35, 1021–1044. [Google Scholar] [CrossRef]
- Tekade, A.R.; Suryavanshi, M.R.; Shewale, A.B.; Patil, V.S. Design and development of donepezil hydrochloride loaded nanostructured lipid carriers for efficient management of Alzheimer’s disease. Drug Dev. Ind. Pharm. 2023, 49, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Shehata, M.K.; Ismail, A.A.; Kamel, M.A. Combined donepezil with astaxanthin via nanostructured lipid carriers effective delivery to brain for Alzheimer’s disease in rat model. Int. J. Nanomed. 2023, 18, 4193–4227. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, M.; Rasool, N.; Singh, Y. Polymeric lipid nanoparticles for donepezil delivery. In Polymeric Biomaterials and Bioengineering: Select Proceedings of APA Bioforum 2021; Springer: New York, NY, USA, 2022; pp. 51–63. [Google Scholar]
- de Souza, I.F.F.; Dos Santos, T.Q.; Placido, R.V.; Mangerona, B.A.; Carvalho, F.C.; Boralli, V.B.; Ruela, A.L.M.; Pereira, G.R. The liquid crystalline phase behaviour of a nasal formulation modifies the brain disposition of donepezil in rats in the treatment of Alzheimer’s disease. Colloids Surf. B Biointerfaces 2021, 203, 111721. [Google Scholar] [CrossRef] [PubMed]
- Dhiman, A.; Tandon, R.; Ahirwar, K.; Handa, M.; Srivastava, N.; Shukla, R. Optimization, characterization and In-Vitro cellular uptake of donepezil-loaded nanocrystvesicles. J. Clust. Sci. 2024, 35, 1493–1505. [Google Scholar] [CrossRef]
- Gangane, P.; Kawtikwar, P. Development of donepezil hydrochloride loaded gellan gum based nasal mucoadhesive microspheres by spray drying method. Indian J. Pharm. Educ. Res 2020, 54, 935–945. [Google Scholar] [CrossRef]
- Perkušić, M.; Nodilo, L.N.; Ugrina, I.; Špoljarić, D.; Brala, C.J.; Pepić, I.; Lovrić, J.; Matijašić, G.; Gretić, M.; Zadravec, D. Tailoring functional spray-dried powder platform for efficient donepezil nose-to-brain delivery. Int. J. Pharm. 2022, 624, 122038. [Google Scholar] [CrossRef] [PubMed]
- Gangane, P.S.; Ghormare, N.V.; Mahapatra, D.K.; Mahajan, N.M. Gellan gum assisted fabrication and characterization of donepezil hydrochloride mucoadhesive intranasal microspheres. Int. J. Curr. Res. Rev 2020, 12, 105–115. [Google Scholar] [CrossRef]
- Ambadkar, B.B.; Gaidhane, A.J.; Chandewar, A.B. Formulation and evaluation of nasal mucoadhesive microsphers by solvent evaporation technique. World J. Pharm. Res. 2020, 9, 948–963. [Google Scholar]
- Jadhav, S.M.; Mishra, S.K. Spray Dried Mucoadhesive Microparticles of Donepezil with Chitosan and Carbopol in Alzheimer’s Disease. Int. J. Pharm. Investig. 2022, 12, 205–210. [Google Scholar] [CrossRef]
- Handa, M.; Sanap, S.; Bhatta, R.; Patil, G.; Ghose, S.; Singh, D.; Shukla, R. Combining donepezil and memantine via mannosylated PLGA nanoparticles for intranasal delivery: Characterization and preclinical studies. Biomater. Adv. 2023, 154, 213663. [Google Scholar] [CrossRef] [PubMed]
- Bhavna; Md, S.; Ali, M.; Baboota, S.; Sahni, J.K.; Bhatnagar, A.; Ali, J. Preparation, characterization, In Vivo biodistribution and pharmacokinetic studies of donepezil-loaded PLGA nanoparticles for brain targeting. Drug Dev. Ind. Pharm. 2014, 40, 278–287. [Google Scholar]
- Garg, Y.; Sharma, G.; Katare, O.P.; Chopra, S.; Bhatia, A. Design and Optimization of Donepezil Hydrochloride Loaded Nanoparticles Using Doe Approach: A Novel Approach for Treating Alzheimer’s Diseases Through Nose-To-Brain Delivery. Available at SSRN 4255393. 2022. Available online: https://ssrn.com/abstract=4255393 (accessed on 20 July 2025).
- Espinoza, L.C.; Guaya, D.; Calpena, A.C.; Perotti, R.M.; Halbaut, L.; Sosa, L.; Brito-Llera, A.; Mallandrich, M. Comparative study of donepezil-loaded formulations for the treatment of Alzheimer’s disease by nasal administration. Gels 2022, 8, 715. [Google Scholar] [CrossRef] [PubMed]
- Garg, Y.; Kumar, M.; Sharma, G.; Katare, O.P.; Chopra, S.; Bhatia, A. Systematic designing and optimization of polymeric nanoparticles using central composite design: A novel approach for nose-to-brain delivery of donepezil hydrochloride. J. Clust. Sci. 2024, 35, 1007–1019. [Google Scholar] [CrossRef]
- Garg, Y.; Bhatia, A. Treating Alzheimer’s Disease via Donepezil Hydrochloride Loaded Nanoparticles: A Novel Approach for Brain Drug Delivery. Alzheimer’s Dement. 2022, 18, e069264. [Google Scholar] [CrossRef]
- Lee, K.-R.; Maeng, H.-J.; Chae, J.-B.; Chong, S.; Kim, D.-D.; Shim, C.-K.; Chung, S.-J. Lack of a primary physicochemical determinant in the direct transport of drugs to the brain after nasal administration in rats: Potential involvement of transporters in the pathway. Drug Metab. Pharmacokinet. 2010, 25, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Butani, S. Fabrication of an ion-sensitive in situ gel loaded with nanostructured lipid carrier for nose to brain delivery of donepezil. Asian J. Pharm. (AJP) 2018, 12, 293–302. [Google Scholar]
- Al Harthi, S.; Alavi, S.E.; Radwan, M.A.; El Khatib, M.M.; AlSarra, I.A. Nasal delivery of donepezil HCl-loaded hydrogels for the treatment of Alzheimer’s disease. Sci. Rep. 2019, 9, 9563. [Google Scholar] [CrossRef] [PubMed]
- Gangopadhyay, A.; Dandagi, P.M.; Sutar, K.P. Development and evaluation of thermoreversible ethosomal gel of donepezil hydrochloride for intranasal delivery. J. Pharm. Innov. 2023, 18, 238–246. [Google Scholar] [CrossRef]
- Gu, F.; Fan, H.; Cong, Z.; Li, S.; Wang, Y.; Wu, C. Preparation, characterization, and In Vivo pharmacokinetics of thermosensitive s nasal gel of donepezil hydrochloride. Acta Pharm. 2020, 70, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Perkušić, M.; Nižić Nodilo, L.; Ugrina, I.; Špoljarić, D.; Jakobušić Brala, C.; Pepić, I.; Lovrić, J.; Safundžić Kučuk, M.; Trenkel, M.; Scherließ, R. Chitosan-based thermogelling system for nose-to-brain donepezil delivery: Optimising formulation properties and nasal deposition profile. Pharmaceutics 2023, 15, 1660. [Google Scholar] [CrossRef] [PubMed]
- Adnet, T.; Groo, A.-C.; Picard, C.; Davis, A.; Corvaisier, S.; Since, M.; Bounoure, F.; Rochais, C.; Le Pluart, L.; Dallemagne, P. Pharmacotechnical development of a nasal drug delivery composite nanosystem intended for Alzheimer’s disease treatment. Pharmaceutics 2020, 12, 251. [Google Scholar] [CrossRef] [PubMed]
- Papakyriakopoulou, P.; Rekkas, D.M.; Colombo, G.; Valsami, G. Development and In Vitro-Ex Vivo evaluation of novel polymeric nasal donepezil films for potential use in Alzheimer’s disease using experimental design. Pharmaceutics 2022, 14, 1742. [Google Scholar] [CrossRef] [PubMed]
- Kaikousidis, C.; Papakyriakopoulou, P.; Dokoumetzidis, A.; Valsami, G. Donepezil brain and blood pharmacokinetic modeling after nasal film and oral solution administration in mice. Pharmaceutics 2023, 15, 1409. [Google Scholar] [CrossRef] [PubMed]
- Papakyriakopoulou, P.; Balafas, E.; Colombo, G.; Rekkas, D.M.; Kostomitsopoulos, N.; Valsami, G. Nose-to-Brain delivery of donepezil hydrochloride following administration of an HPMC-Me-β-CD-PEG400 nasal film in mice. J. Drug Deliv. Sci. Technol. 2023, 84, 104463. [Google Scholar] [CrossRef]
- Shah, B. Microemulsion as a promising carrier for nose to brain delivery: Journey since last decade. J. Pharm. Investig. 2021, 51, 611–634. [Google Scholar] [CrossRef]
- Sheth, T.; Seshadri, S.; Prileszky, T.; Helgeson, M.E. Multiple nanoemulsions. Nat. Rev. Mater. 2020, 5, 214–228. [Google Scholar] [CrossRef]
- Al-Sakkaf, M.K.; Onaizi, S.A. Effects of emulsification factors on the characteristics of crude oil emulsions stabilized by chemical and Biosurfactants: A review. Fuel 2024, 361, 130604. [Google Scholar] [CrossRef]
- Sessa, M.; Balestrieri, M.L.; Ferrari, G.; Servillo, L.; Castaldo, D.; D’Onofrio, N.; Donsì, F.; Tsao, R. Bioavailability of encapsulated resveratrol into nanoemulsion-based delivery systems. Food Chem. 2014, 147, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.; Choudhury, H.; Yeun, O.C.; Yin, H.M.; Lynn, T.W.; Tine, C.L.; Wi, N.S.; Yen, K.C.; Phing, C.S.; Kesharwani, P. Perspectives of nanoemulsion strategies in the improvement of oral, parenteral and transdermal chemotherapy. Curr. Pharm. Biotechnol. 2018, 19, 276–292. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, H.; Gorain, B.; Karmakar, S.; Biswas, E.; Dey, G.; Barik, R.; Mandal, M.; Pal, T.K. Improvement of cellular uptake, In Vitro antitumor activity and sustained release profile with increased bioavailability from a nanoemulsion platform. Int. J. Pharm. 2014, 460, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Bahadur, S.; Pardhi, D.M.; Rautio, J.; Rosenholm, J.M.; Pathak, K. Intranasal nanoemulsions for direct nose-to-brain delivery of actives for CNS disorders. Pharmaceutics 2020, 12, 1230. [Google Scholar] [CrossRef] [PubMed]
- Samaridou, E.; Alonso, M.J. Nose-to-brain peptide delivery—The potential of nanotechnology. Bioorganic Med. Chem. 2018, 26, 2888–2905. [Google Scholar] [CrossRef] [PubMed]
- Win, T.; Rajagopal, J.; Mandal, U.K.; Sengupta, P.; Chatterjee, B. Incorporation of carbopol to palm olein based analgesic cream: Effect on formulation characteristics. Lat. Am. J. Pharm. 2017, 36, 2144–2152. [Google Scholar]
- Duong, V.-A.; Nguyen, T.-T.-L.; Maeng, H.-J. Preparation of solid lipid nanoparticles and nanostructured lipid carriers for drug delivery and the effects of preparation parameters of solvent injection method. Molecules 2020, 25, 4781. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, Y.; Yu, T.; Song, G.; Xu, T.; Xin, T.; Lin, Y.; Han, B. Nano-based drug delivery systems for periodontal tissue regeneration. Pharmaceutics 2022, 14, 2250. [Google Scholar] [CrossRef] [PubMed]
- Saad, M.A.; Hasan, T. Spotlight on photoactivatable liposomes beyond drug delivery: An enabler of multitargeting of molecular pathways. Bioconjugate Chem. 2022, 33, 2041–2064. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Bisht, B.; Dutta, S.; Paul, M.K. Current advances in the use of exosomes, liposomes, and bioengineered hybrid nanovesicles in cancer detection and therapy. Acta Pharmacol. Sin. 2022, 43, 2759–2776. [Google Scholar] [CrossRef] [PubMed]
- Luiz, M.T.; Dutra, J.A.P.; Tofani, L.B.; de Araújo, J.T.C.; Di Filippo, L.D.; Marchetti, J.M.; Chorilli, M. Targeted liposomes: A nonviral gene delivery system for cancer therapy. Pharmaceutics 2022, 14, 821. [Google Scholar] [CrossRef] [PubMed]
- Müller, R.H.; Radtke, M.; Wissing, S.A. Solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) in cosmetic and dermatological preparations. Adv. Drug Deliv. Rev. 2002, 54, S131–S155. [Google Scholar] [CrossRef] [PubMed]
- Sivadasan, D.; Sultan, M.H.; Madkhali, O.; Almoshari, Y.; Thangavel, N. Polymeric lipid hybrid nanoparticles (plns) as emerging drug delivery platform—A comprehensive review of their properties, preparation methods, and therapeutic applications. Pharmaceutics 2021, 13, 1291. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Seo, M.-J.; Lim, H.-s.; Hong, J.-T. Lipid-based Liquid Crystalline Phases for Biocompatible and Versatile Drug Delivery. Yakhak Hoeji 2023, 67, 137–144. [Google Scholar] [CrossRef]
- Chavda, V.P.; Dyawanapelly, S.; Dawre, S.; Ferreira-Faria, I.; Bezbaruah, R.; Gogoi, N.R.; Kolimi, P.; Dave, D.J.; Paiva-Santos, A.C.; Vora, L.K. Lyotropic liquid crystalline phases: Drug delivery and biomedical applications. Int. J. Pharm. 2023, 647, 123546. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Ha, E.-S.; Kim, M.-S. Surface modification strategies for high-dose dry powder inhalers. J. Pharm. Investig. 2021, 51, 635–668. [Google Scholar] [CrossRef]
- Kim, J.-S.; Park, H.; Kang, K.-T.; Ha, E.-S.; Kim, M.-S.; Hwang, S.-J. Micronization of a poorly water-soluble drug, fenofibrate, via supercritical-fluid-assisted spray-drying. J. Pharm. Investig. 2022, 52, 353–366. [Google Scholar] [CrossRef]
- Djupesland, P.G. Nasal drug delivery devices: Characteristics and performance in a clinical perspective—A review. Drug Deliv. Transl. Res. 2013, 3, 42–62. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.R.; Agrawal, Y. Nanosuspension: An approach to enhance solubility of drugs. J. Adv. Pharm. Technol. Res. 2011, 2, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, A.; Jadhav, K.; Kadam, V. An overview: Microspheres as a nasal drug delivery system. Int. J. Pharm. Sci. Rev. Res. 2010, 5, 8–17. [Google Scholar]
- Adepu, S.; Ramakrishna, S. Controlled drug delivery systems: Current status and future directions. Molecules 2021, 26, 5905. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, V.S.; Shaw, C. Essential Chemistry for Formulators of Semisolid and Liquid Dosages; Academic Press: Cambridge, MA, USA, 2015. [Google Scholar]
- Jeong, J.-S.; Ha, E.-S.; Park, H.; Lee, S.-K.; Kim, J.-S.; Kim, M.-S. Measurement and correlation of solubility of rivaroxaban in dichloromethane and primary alcohol binary solvent mixtures at different temperatures. J. Mol. Liq. 2022, 357, 119064. [Google Scholar] [CrossRef]
- Aderibigbe, B.A. In situ-based gels for nose to brain delivery for the treatment of neurological diseases. Pharmaceutics 2018, 10, 40. [Google Scholar] [CrossRef] [PubMed]
- Corazza, E.; Di Cagno, M.P.; Bauer-Brandl, A.; Abruzzo, A.; Cerchiara, T.; Bigucci, F.; Luppi, B. Drug delivery to the brain: In situ gelling formulation enhances carbamazepine diffusion through nasal mucosa models with mucin. Eur. J. Pharm. Sci. 2022, 179, 106294. [Google Scholar] [CrossRef] [PubMed]
- Dalvi, A.V.; Ravi, P.R.; Uppuluri, C.T.; Mahajan, R.R.; Katke, S.V.; Deshpande, V.S. Thermosensitive nasal in situ gelling systems of rufinamide formulated using modified tamarind seed xyloglucan for direct nose-to-brain delivery: Design, physical characterization, and In Vivo evaluation. J. Pharm. Investig. 2021, 51, 199–211. [Google Scholar] [CrossRef]
- Chhajed, S.; Sangale, S.; Barhate, S. Advantageous nasal drug delivery system: A review. Int. J. Pharm. Sci. Res. 2011, 2, 1322–1336. [Google Scholar]
- Sabaghi, M.; Hoseyni, S.Z.; Tavasoli, S.; Mozafari, M.; Katouzian, I. Strategies of confining green tea catechin compounds in nano-biopolymeric matrices: A review. Colloids Surf. B Biointerfaces 2021, 204, 111781. [Google Scholar] [CrossRef] [PubMed]
- Aderibigbe, B.A.; Naki, T. Design and efficacy of nanogels formulations for intranasal administration. Molecules 2018, 23, 1241. [Google Scholar] [CrossRef] [PubMed]
- Picone, P.; Sabatino, M.A.; Ditta, L.A.; Amato, A.; San Biagio, P.L.; Mulè, F.; Giacomazza, D.; Dispenza, C.; Di Carlo, M. Nose-to-brain delivery of insulin enhanced by a nanogel carrier. J. Control. Release 2018, 270, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zou, Z.; Liu, S.; Miao, S.; Liu, H. Nanogels as novel nanocarrier systems for efficient delivery of CNS therapeutics. Front. Bioeng. Biotechnol. 2022, 10, 954470. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Indication | Clinical Research Findings | Mechanism/Characteristics | Significant Efficacy | Ref |
|---|---|---|---|---|
| Alzheimer’s Disease (AD) | Well-established as a major treatment | Acetylcholinesterase inhibition | Yes | [6,13] |
| Vascular Dementia | Improves cognitive and functional outcomes | Enhances cholinergic neurotransmission similar to AD | Yes | [6,13] |
| Autism Spectrum Disorder | Under research | Potential cholinergic enhancement | Under Study | [6,13] |
| Down Syndrome | No significant efficacy observed in clinical trials | Potential cholinergic enhancement | No | [6,9,10,13] |
| Parkinson’s Disease | Under research | Potential cholinergic enhancement | Under Study | [6,13] |
| Traumatic Brain Injury | Under research | Potential cholinergic enhancement | Under Study | [6,13] |
| Post-Stroke Cognitive Impairment | Under research | Potential cholinergic enhancement | Under Study | [6,13] |
| Multiple Sclerosis-Related Cognitive Impairment | Significant improvements in behavior, cognitive function, and daily activities compared to placebo | Cholinergic enhancement | Yes | [6,13] |
| Lewy Body Dementia | Significant improvements in behavior, cognitive function, and daily activities compared to placebo | Cholinergic enhancement | Yes | [6,13] |
| Cortical Atrophy | No significant efficacy observed in clinical trials | Potential cholinergic enhancement | No | [6,11,13] |
| Depression and Cognitive Impairment | No significant efficacy observed in clinical trials | Potential cholinergic enhancement | No | [6,12,13] |
| Disease Progression Inhibition | Potential to modify disease (neuroprotective effects) | Inhibition of oxygen-glucose deprivation, Aβ-induced cell death, and glutamate-induced cell death | Under Study | [6,13] |
| Drug Delivery System | Features | Formulation | Preparation Method | Result/Outcome | Ref | |
|---|---|---|---|---|---|---|
| In Vitro | In Vivo | |||||
| Lipid-based formulation | ||||||
| Emulsion | O/W Nanoemulsion (NE) | - Capryol 90 - Labrasol - Transcutol-P - Water - Pluronic F-127 | - Stirring | - Size: DPZ-NE (128.50 nm) - PDI: DPZ-NE (0.12) - pH: DPZ-NE (5.82), DPZ-PNE (6.14) - Transparent, DPZ-NE (monophasic), DPZ-PNE (homogeneous, thermostable) - No signs of precipitated drug, - Entrapment efficiency: DPZ-NE (94.32%), DPZ-PNE (93.85%) - Viscosity: DPZ-NE (10.69 mPa·s) - DPZ-NE (Newtonian behavior), DPZ-PNE (shear thinning behavior) - Permeation profile (porcine nasal mucosa): DPZ-PNE (532.30 µg), DPZ-NE (199.56 µg) - Mucoadhesion study: DPZ-PNE (82.43%), DPZ-NE (71.31%) | - Pigs, IN route, dose: 300 µL - No infiltration of inflammatory cells | [69] |
| - Labrasol - Cetyl pyridinium chloride - Glycerol | - Homogenization - Ultrasonication | - Size: 65.36 nm - PDI: 0.084 - Zeta potential: −10.7 mV - Transmittance (100%): 650 nm - Fast release in PBS (pH 7.4), ACSF, simulated nasal fluid - Decreased cell viability: placebo > aqueous DPZ (85%) > NE (76.3%) | - Sprague Dawley rats, IN route, dose: 0.09 mg - Drug distribution: brain > blood - Brain Distribution (1.5 h): NE IN (3.42%/g) > aqueous IN (2.34%/g) > i.v (1.88%/g) > oral (0.58%/g) - Blood Distribution (30 min): i.v (2.97%/g) > IN (2.04%/g) > oral (1.55%/g) - Brain Cmax: NE IN (3.42%/g) > i.v (1.88%/g) > Oral (0.58%/g) - Tmax: NE IN (1.5 h) > i.v (3 h) > Oral (3 h) - Brain retention: NE IN (up to 24 h) > oral (no uptake) - AUC (48.55% h/g), AUMC (619.17% h2/g), MRT (12.75 h), kel’ (0.078 h−1), CL (0.082%/g h−1), DTE (360.59%), DTP (72.23%) | [97] | ||
| - Pine Oil - Polysorbate 80 - Diethylene Glycol Monoethyl Ether | - Low-energy emulsification | - Size: 16 nm - Spherical - Zeta potential: −7.22 mV | - Swiss albino mice, dose: 1 mg/kg - IN-NE concentration (15 min): Brain (678 ng/mL) > Plasma (3 ng/mL) | [98] | ||
| - Castor oil - Labrasol - Transcutol-P - Propylene glycol | - O/W emulsification | - Size: 58.9 nm - Spherical, smooth, regular surface - PDI: 0.19 - pH: 6.38 - Viscosity: 44.69 mPa·s - Newtonian flow - High stability at temperature variations - Release (24 h): 74.66%, hyperbola kinetic model - Permeation profile (Porcine nasal mucosa): highest permeation during the first 4 h, 80% (2000 μg) - Nasal mucosa retention (6 h): 35.52% (812.9 μg/g) | [99] | |||
| O/W Microemulsion (ME) | - PEG 600, Capmul® MCM EP, Captex® 90, Capryol 90 - Labrasol, Maisine® - PaceolTM - Amyloid β (1–42) - Tween 80, 20, 60 - Transcutol-P® - Butter oil - Omega-3 fish oil | - Water titration | - Size: 52.43–163.10 nm - PDI: 0.26–0.487 - Nasal Diffusion: DPZ HCl-BO-ME (71.22%) > DPZ HCl-O3FO-ME (62.16%) > DPZ HCl-ME (59.69%) > DPZ HCl solution (55.01%) | - Sprague Dawley rats, dose: 0.5 mg/kg - Bioavailability: DPZ HCl-O3FO-ME-IN (361.73%) > DPZ HCl-BO-ME-IN (313.59%) > DPZ HCl-ME-i.v (168.62%) > DPZ HCl solution-IN (8.96%) | [100] | |
| Liposomes | Liposomes | - Carboxymethyl cellulose - 1,2-distearyl-sn-glycero-3-phosphocholine - Cholesterol, PEG - Chloroform - Sodium dihydrogen phosphate | - Thin layer hydration | - Size: 102 nm - spherical shape, single unilamellar vesicle - PDI: 0.28 - Zeta potential: −28.31 mV - Entrapment efficiency: 84.91% - Stability (3 months): stable at 4°, 25 °C | - Male Wistar rats, IN route, dose: 1 mg/kg body weight - Higher AUC0–t, AUC0–∞, Cmax - Sustained release - Higher bioavailability in plasma and brain - Free from toxicity - Half-life: loposome (6.90 h) > free drug (5.55 h) | [101] |
| Liposome–based in situ gel | - Hydrogenated soy phosphatidyl cholin - Cholesterol - Ethanol - Ammonium sulfate | - Ethanol injection | - Size: 103 nm - Spherical - PDI: 0.108 - Entrapment efficiency: 93% - Nasal mucosa permeation: DPZ HCl-loaded liposomes (80.11%) > DPZ HCl solution-based in situ gel (13.12%) | - Sprague Dawley rats, IN route, dose: 1 mg/kg - Biodistribution: 1239.61 pg/g, High brain distribution - Brain AUC, Tmax: 1239.61 pg/g, 0.5 h - DTE: 314.29% | [102] | |
| Transfersomes | - Hyaluronic acid | - Thin film hydration | - Size: 227.5 nm - Entrapment efficiency: 75.83% - Release (8 h): 37.94% - Adequate stability - Nontoxic and tolerable for IN delivery - Permeation profile (nasal mucosa, 24 h): 547.49 µg/cm2 | - Drug targeting index: 5.08 - DTE: 508.25% - Direct N2B DTE: 80.32% | [103] | |
| Solid lipid nanoparticles (SLNs) | - Glyceryl monostearate - Tween 80, poloxamer 188 (1:1) | - Solvent emulsification diffusion | - Size: 121.0 nm - Zeta potential: −24.1 mV - Release: 89.35% - Entrapment efficiency: 67.95% - Drug loading: 12.15% - DSC melting peak: 59.58 C, 54.53 C - Amorphous form - Release: Initial burst, followed by slow release | - Male albino Wistar, IN route, dose: 0.09 mg - AUC0–∞: nanoparticle > control group (2.61-fold higher) - Significant (p < 0.05) enhancement in bioavailability of DPZ in the brain - Localization (kidney, spleen, liver): nanoparticles > control | [104] | |
| - Tween 80 - Lecithin | - Homogenization - Sonication | - Size: 87.2 nm - Spherical, smooth - PDI: 0.22 - Zeta potential: −17.0 mV - Encapsulation efficiency: 93.84% - Release (24 h): 70% - Toxicity: no toxic effect on cells - Particle size and PDI increased with increasing lipid concentration but decreased with increasing amounts of surfactant and co-surfactant | [105] | |||
| - Glyceryl behenate - Tween 80, poloxamer 188 (1:1) | - Solvent emulsification diffusion | - Release (24 h): 96.72% - Stability (6 months): no significant change at 4, 25 °C/60% RH (p > 0.05), significant particle size increase at 40 °C/75% RH (p < 0.001) | - Albino wistar rats, dose: 0.09 mg - AUC0-∞: DPZ-SLN >DPZ-Solution (1.92-fold higher) - DTE: 288.75% - DTP: 65.37% | [106] | ||
| Nanostructured lipid carriers (NLCs), | - Mixture of Compritol and Capryol 90 - Poloxamer 188 - Chitosan | - Homogenization - Sonication | - Size: 192.5 nm - Spherical shape (TEM. DSC) - PDI: 0.298 - Zeta potential: 38.9 mV - Entrapment efficiency: 89.85% | - Albino Wistar rats, dose: 0.18 mg - Bioavailability (IN): DPZ-chitosan-NLCs > DPZ-Solution - Bioavailability (IN, IV): DPZ-Chitosan-NLCs (IN) > DPZ-Chitosan-NLCs (IV) - Brain Targeting (DTE/DTP): DPZ-chitosan-NLCs (321.21%/74.55%) > DPZ-Solution (158.52%/36.92%) | [107] | |
| - DPZ HCl and Embelin-loaded | - Hot emulsification probe sonication | - Size: 180.2 nm - Zeta potential: −12 mV - PDI: 0.37 - Release: 90.72% - Embelin: 81.30 - Permeation (goat nasal mucosa): NLCs > suspension - HET CAM score: 0.68 - Cellular uptake study: high cellular uptake of NLC via N2A cells | [108] | |||
| - Glyceryl monostearate - Tween 80 - Poloxamer 407 - Nigella sativa oil | - High-pressure homogenization - Ultrasonication | - Size: 107.4 nm - PDI: 0.25 - Zeta potential: −41.7 mV - Entrapment efficiency: 70.20% - Drug content: 89.05% | - Cmax: brain 4.597 µg/mL > blood 2.258 µg/mL - Tmax: 1 h | [109] | ||
| - Glyceryl palmitostearate - Oleic acid - Astaxanthin - Poloxamer 188 - Polysorbate 80 | - High-shear homogenization | - Size: 149.9 nm - Spherical - PDI: 0.224 - Zeta potential: −33.7 mV - Entrapment efficiency: 93.85% - Release (24 h): sustained - Stability (6 months): 4–8 °C | [110] | |||
| Polymeric Lipid Nanoparticles | - Soy lecithin - Methanol - Chloroform - Glutaraldehyde - Chitosan - Glacial acetic acid - Ethanol - Gelatin - Acetone | - Homogenization - Desolvation | - Size: CLN (237.43 nm, GLN (278.86 nm) - Drug loading: CLN (10.24%), GLN (8.77%) - Release: CLN (burst release of up to 99.99% drug for 5 days), GLN (sustained release of 33.31% drug for 30 days under acidic conditions) - Cell viability studies; safe toward mouse fibroblast cells (L929) - Mucoadhesive | [111] | ||
| Liquid crystal | Cubosomes | - GMO - Poloxamer 407 - Glucomannan - Gellan gum | - O/W emulsification | - Size: 137.8–231.4 nm - PDI: 0.38–0.48 - Zeta potential: −40 mV - pH 6.4 - Entrapment efficiency: 30.85–48.48% - Viscosity: 180 cps - Release (pH 6.6): Initial burst, followed by slow release (24.52% at 2 h, 53.73% at 6 h) - Irregular polyangular (Nearly spherical) - Degree of gelation: Immediate gelation remains for a few hours (less stiff gel) - Mucoadhesive strength: 138.6 g - Gel strength: 34 s - Drug content: 86.07–92.40% | - Male Sprague Dawley rats, IN route, dose: 1 mg/kg - Cmax: OCG 24.01 µg/mL > OCD 14.34 µg/mL > solution 3.96 µg/mL - Bioavailability: OCG, OCD > solution (p value < 0.001) - Extended residence time in the nasal cavity - Increased permeation | [66] |
| Lyotropic liquid crystal mesophases | - CETETH-10 - Oleic acid - Water | - Mixing - Stirring | - Mucoadhesive Strength (Work of adhesion/Peak of adhesion): M1 (5.67 Ns, 0.93 N), M2 (6.41 Ns, 0.79 N) - Hexagonal phases (12% and 20% nasal fluid): no significant difference in adhesion (p > 0.05) - Drug release (6 h): 25% | - Wistar rats, dose: 25 mg.kg−1 - Phase transition (12–20% ANF): isotropic to anisotropic - Brain concentration (4 h): sustained above 1000 ng/g | [112] | |
| Lyotropic liquid crystalline mesophases | - Monoolein/oleic acid/water - Oleic acid - GMO | - Direct mixing - Dissolving | - Swelling study: W/O ME phase to reverse hexagonal mesophase transition - Viscosity increase in situ - Mucoadhesive - Release (24 h): controlled - Dissolution efficiency (90 min): pH 1.2 (61.67%), pH 4.5 (4.33%), pH 6.8 (1.16%) | [17] | ||
| Nano-liquidcrystal | - Pine oil | - Film hydration - Sonication | - Size: 129 nm - PDI: 0.19 - Zeta potential: −27.5 mV - Stability: stable at 4°, 25 °C - Crystalline cubic shape - AChE inhibition activity: higher in combination than others | [113] | ||
| Solid particle-based formulation | ||||||
| Microspheres | - Gellan gum | - Conventional spray drying | - Mucoadhesive strength increased as the gellan gum amount increased. - Mucoadhesion range: 80.30–94.43% - Release: zero-order release, likely due to low gellan gum viscosity - Permeation profile (Sheep nasal mucosa): enhanced in smaller microspheres | [114] | ||
| - Chitosan - Mannitol | - Spray drying | - Size: Dv10 (6.7–11.6 μm), Dv50 (17.1–35.7 μm), Dv90 (34.1–72.7 μm) - Hausner ratio: 1.15–1.30 - Spray cone angle range: 22.5–28.3° - Olfactory deposition: 13.9–65.5% - Turbinate deposition: 19.4–47.6% - Drug release (5 h): 100% | [115] | |||
| - Gellan gum | - W/O emulsification cross-linking - Gentle heating - Constant agitation | - Size: 14.3–18.3 µm - Production yield: 40.26–55.93% - Drug loading: 49.21–74.60% - Entrapment efficiency: 34.5–53.6% - Swelling: 82–91% - Mucoadhesion: 45.6–79.6% - Release: 76.92–98.26% - Permeation profile (goat nasal mucosa): 60.76–95.73% | [116] | |||
| - Gellan Gum - N-Octanol - Span 80 | - W/O emulsification cross-linking | - Size: 157.65–373.87 μm - Entrapment efficiency: 34.5–53% - Production yield: 44.26–55.93% - Drug loading: 3.45–5.23% - As the stirring rate increased, the particle size decreased - Swelling property: 0.82–0.91% - Release (7 h): 98% | [117] | |||
| Microparticles | - Chitosan - Carbopol 934 | - Spray drying | - Size: chitosan (18.3–21.4 µm), carbopol (14.7–18.3 µm) - Spherical or ellipsoid - Yield: chitosan (43.96–68.12%), carbopol 56.46–63.23%) - Drug content: chitosan 89.87–96.05%), carbopol (91.5–95.07%) - Drug release: chitosan (66.57–85.74%), carbopol (69.54–91.53%) | - In-bred Albino rats, IN route, dose: 20 µL PBS solution (concentration 50 mg/mL) - Brain distribution: carbopol 934 (129.51%) > Chitosan (110.87%) > Pure DPZ HCl | [118] | |
| Nanoparticles | - India:PLGA (50:50) - Memantine - Polyvinyl alcohol or Tween 80 - span 80 - D-mannose | - Simple emulsification | - Size: 179.4 nm - PDI: 0.22 - Zeta potential: −33.1 mV - SEM: 200 nm - Release: burst release initially, followed by extended release | - Brain distribution (15 min): coated IN (573.61 ng/mL) > uncoated IN (247.77 ng/mL) > coated peroral (168.08 ng/mL) > uncoated peroral (138.27 ng/mL) - Brain AUC, Tmax: 3470.34 ng.h/mL, 0.25 h - Plasma AUC, Tmax: 2526.01 ng.h/mL, 2 h | [119] | |
| Nanosuspension | - PLGA 50:50 block copolymer (35–40 kDa) -Dichloromethane | - Solvent emulsification, diffusion, evaporation | - Size: 89.67 nm - Spherical, smooth (TEM, SEM) - Release: Initial burst, followed by slow release | - Brain distribution: nanoparticles > drug solution | [120] | |
| - Chitosan - Sodium tripolyphosphate - Glacial acetic acid | - Ionotropic gelation | - Size: 177.8 nm - Drug payload: DPZ 22.2 mg/chitosan 100 mg - Spherical - PDI: 0.593 - Zeta potential: +16.6 mV - Process yield: 91.96% - Release (24 h): sustained, 90.82% - Adhesive force: 9.26 g - Stability (3 months): no significant changes in particle size, drug content, or zeta potential | - Wistar rats, IN route, dose: 40 µL - Brain distribution: nanoparticles > control - High blood clearance | [121] | ||
| - Chitosan - Polysorbate-80 - Acetic acid - Tripolyphosphate | - Ionic cross-linking | - Size: 150–200 nm - PDI: 0.341 - Particle stability (PBS): stable short-term, potential aggregation/size increase long-term - Drug loading capacity: 40–48% - Release (300 min): 56.17–96.74% | - Male Sprague Dawley rats, IN route, dose: 0.5 mg/mL, 1 mg/mL, 1.5 mg/mL (groups I–III) - Plasma, brain AUC: nanosuspension (684.83 ng h/mL, 352.75 ng h/mL) > suspension (440.20 ng h/mL, 95.216 ng h/mL) - Systemic AUC, Cmax, Tmax: Nanosuspension > control | [85] | ||
| Suspension | Gel-based suspension | - Chitosan - Transcutol® P - N-Methylpyrrolidone - Water - Pluronic F-127 | - O/W emulsification | - pH: DPZ-CGEL (5.9), DPZ-PGel1 (6.2), DPZ-PGel2 (6.3) - Viscosity: at 25 °C, DPZ-CGel > DPZ-PGel1 > DPZ-PGel2; at 35 °C, DPZ-PGel > DPZ-CGel - Swelling (first-order model): DPZ-PGel1 (k = 0.15 min−1), DPZ-PGel2 (0.12 min−1) - Stability (30 days): no significant changes in its appearance (25, 40 °C) - Release: DPZ-PGel1 (98%) > DPZ-CGel (81.8%) - Maximum release amount: DPZ-PGel1 (2249 µg) > DPZ-PGel2 (1913 µg) > DPZ-Cgel (1615 µg) - DPZ-Pgel1 showed higher values of flux, Kp, partition coefficient vehicle/tissue, diffusion coefficient, and Css - Gelation temperature: 32–33 °C - Nasal mucosa retention, Permeation profile (porcine nasal mucosa): DPZ-PGel1 > DPZ-CGel, DPZ-PGel2 | [122] | |
| Polymeric Nanoparticles | - Chitosan | - Size: 180.2 nm - Spherical (confocal laser) - PDI: 0.282 - Zeta potential: +16.6 mV - Drug release: 90% - Permeation profile (24 h): 70% | - Wistar rats, IN route - Brain distribution (6 h): IN > oral, nanoparticles > solution | [123] | ||
| Nanoparticle | - Chitosan | - Ionic gelation | - Size: 177.8 nm - Zeta potential: +16.6 mV - Drug payload: 22.2 mg/100 mg of chitosan - Process yield: 91.96% - Mucoadhesive strength: 9.26 g - Release (24 h): 90% - Ex vivo release: 70% | - Rats, IN route - 3-fold higher drug delivery to the brain | [124] | |
| Solution-based formulation | ||||||
| Solution | - PEG 400 - Ethanol - Water - Dimethyl sulfoxide | - Dissolving | - Male Sprague Dawley rats, IN route, dose: 4–16 mg/kg - Transporter expression: rOAT3, rOCT2 detected in olfactory epithelium - Brain delivery: direct pathway | [125] | ||
| Gel-based formulation | ||||||
| Gels | Ion-sensitive in situ nano gel | - Glyceryl distearate - Capmul MCM - AcrysolK150 - Poloxamer 188 - Tween 80 - Gellan gum - Xanthan gum | - Melt emulsification-probe sonication | - Size: <200 nm - PDI: <0.300 - Zeta potential: −35 mV - Effect of liquid lipid: higher concentration reduced particle size - Expansion coefficient: <3% - Viscosity: 2–11 cp | - Male Sprague Dawley rats, IN route, dose: 1 mg/kg - Tmax: 1 h - Brain Cmax: gel > marketed, - Plasma Cmax: marketed, > gel - Plasma, brain residence time: gel > marketed - Brain drug concentration: IN > oral (2-fold higher) | [126] |
| Hydrogel | - Chitosan solution - 2-iminothiolane HCl - 2-mercaptoethanol - HCl - NaCl | - Radiation-Induced crosslinking | - Size: 438.7 nm - Entrapment efficiency: 62.5% - Half-life: 3.5 h - Stability: 4 > 20 °C - Inverse correlation between cross-linking density and swelling degree - Swelling degree: chitosan (21%) hydrogels > thiolated chitosan (19.8%) - Viscosity: chitosan hydrogels > thiolated chitosan hydrogels - Disulfide bonds enhance mucoadhesiveness of thiolated chitosan - TCH no significant histological changes | - New Zealand white rabbits, IN route, dose: 5 mg - Cmax: nasal (12 ng/mL) > oral (8.2 ng/mL) - Brain drug content: IN gel > oral tablets (2-fold higher) | [127] | |
| Ethosomal nano gel | - Phospholipon - Ethanol - Poloxamer 407, Poloxamer 188 - Carbopol 934 | - Ethanol injection | - Size: 110.06 - Entrapment efficiency: 70.02% - Gelation temperature: 31.7 °C - Mucoadhesive strength: 3332 - In vitro drug release - Permeation profile (sheep nasal mucosa): 100% | [128] | ||
| Thermosensitive in situ gel | - Poloxamer 407 - Poloxamer 188 - hydroxypropyl-β-cyclodextrin - Ethylparaben | - Agitation, stirring | - Gelation temperature: 32.5 °C - Gelation time: 40 s - Release (45 min): 90%, sustained, zero-order model | - Sprague Dawley male rats, dose: 10 mg kg–1 - Rapid absorption - Bioavailability: 385.58% - Brain DTE: 151.2% - Plasma Cmax, Tmax: gel (4113.41 ng mL–1,10 min) > solution (269.35 ng mL–1, 55 min) - AUC0-t: gel (2718.25 ng h mL–1) >solution (735.68 ng h mL–1) | [129] | |
| Thermoresponsive in situ gel | - Chitosan - Acetic acid - β-glycerophosphate solution | - Dropwise, mixing | - Gelation temperature: 34 °C - Olfactory deposition: 71.8% - Release: prolonged drug release (t1/2 about 90 min) - Mucoadhesive behavior - Permeation profile (Calu-3 cells): reversible permeation - Mucoadhesion, Permeation: gel > solution | - Arion lusitanicus slugs - Safety: acceptable slug mucosal irritability profile | [130] | |
| Thermosensitive gel | - Poloxamer 407, 188 - Phosphatidylcholine/cholesterol (8:1) - Chloroform/methanol (4:1) | - Thin film hydration | - Solubility: higher aqueous solubility - Gelation temperature: 32–35 °C - Osmolarity: 280 mOsm - pH 6 - Viscosity: Shear-thinning behavior with increasing shear rate | [131] | ||
| Film-based formulation | ||||||
| Film | Polymeric nasal film | - HPMC E50 nasal films - PEG 400 - Methyl-β-Cyclodextrin | - Continuous magnetic stirring - Cooled for Polymer hydration | - Drug content: 90.0–99.8% - Tthickness: 19.6–170.8 µm - Stability (6 months): stable (airtight), low residual moisture (< 3%) - Enhanced mucoadhesion and nasal permeability | [132] | |
| Nasal film | - Eight-week-old C57BL/6J mice, IN route, dose: 4 mg/kg - Brain distribution: 0.021 - Blood distribution: 0.005 - Tlag: 4.06 min, Tk0: 11.3 min, Mtt: 56 min, ktr: 0.171 1/min, P1: 0.543, P2: 0.3, Ka: 0.173 1/min, K20: 0.046 1/min | [133] | ||||
| HPMC- Methyl-β-Cyclodextrin-PEG 400 nasal film | - HPMC E50 - PEG 400 - Methyl-β-Cyclodextrin | - Dispersion - Stirring - Cooled for polymer hydration | - Eight-week-old C57BL/6J mice, IN route, dose: 4 mg/kg - Cmax: IN (1.46 μg/mL) > oral (0.37 μg/mL) - AUC0~t (min μg)/mL): IN (43.3) > oral (31.8) - Tmax: IN (15 min) > oral (30 min) - Brain distribution: Film > control | [134] | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jon, J.; Jeong, J.; Jung, J.; Cho, H.; Song, K.; Kim, E.-S.; Lee, S.H.; Han, E.; Chung, W.-H.; Moon, A.; et al. Recent Advances in Donepezil Delivery Systems via the Nose-to-Brain Pathway. Pharmaceutics 2025, 17, 958. https://doi.org/10.3390/pharmaceutics17080958
Jon J, Jeong J, Jung J, Cho H, Song K, Kim E-S, Lee SH, Han E, Chung W-H, Moon A, et al. Recent Advances in Donepezil Delivery Systems via the Nose-to-Brain Pathway. Pharmaceutics. 2025; 17(8):958. https://doi.org/10.3390/pharmaceutics17080958
Chicago/Turabian StyleJon, Jiyoon, Jieun Jeong, Joohee Jung, Hyosun Cho, Kyoung Song, Eun-Sook Kim, Sang Hyup Lee, Eunyoung Han, Woo-Hyun Chung, Aree Moon, and et al. 2025. "Recent Advances in Donepezil Delivery Systems via the Nose-to-Brain Pathway" Pharmaceutics 17, no. 8: 958. https://doi.org/10.3390/pharmaceutics17080958
APA StyleJon, J., Jeong, J., Jung, J., Cho, H., Song, K., Kim, E.-S., Lee, S. H., Han, E., Chung, W.-H., Moon, A., Kang, K.-T., Kim, M.-S., & Park, H. (2025). Recent Advances in Donepezil Delivery Systems via the Nose-to-Brain Pathway. Pharmaceutics, 17(8), 958. https://doi.org/10.3390/pharmaceutics17080958