
In Vitro Oral Cavity Permeability Assessment to Enable Simulation of Drug Absorption


Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Cell Culture
2.2.2. Permeability Assay Standardization
2.2.3. Transepithelial Permeation Assessment
2.2.4. Computational Permeation Prediction
2.2.5. HPLC Analysis
2.3. Statistical Analysis
3. Results and Discussion
3.1. Human-Derived In Vitro Oral Cavity Tissue Models
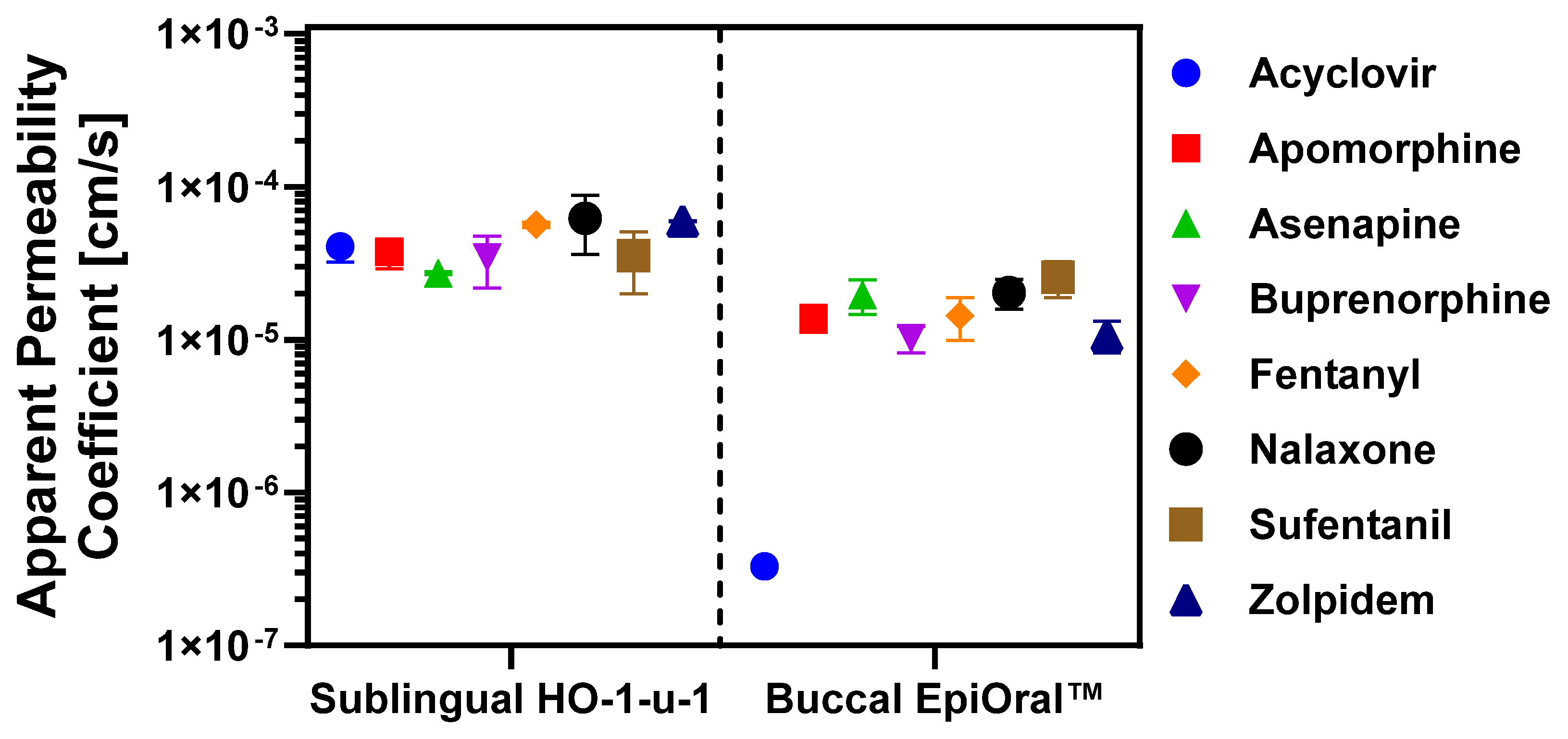
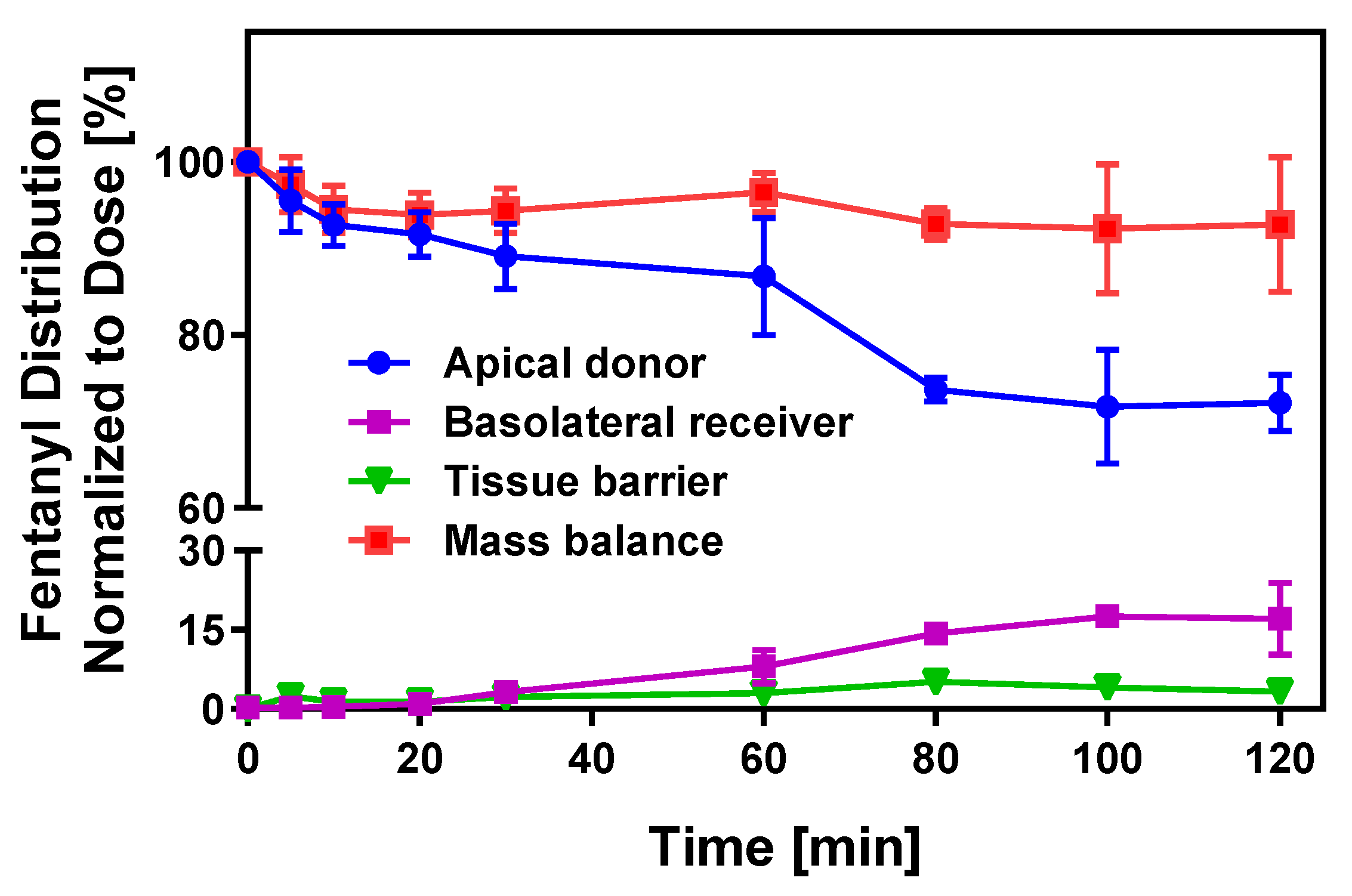
3.2. Permeation Properties of APIs Across In Vitro Oral Cavity Tissue Models
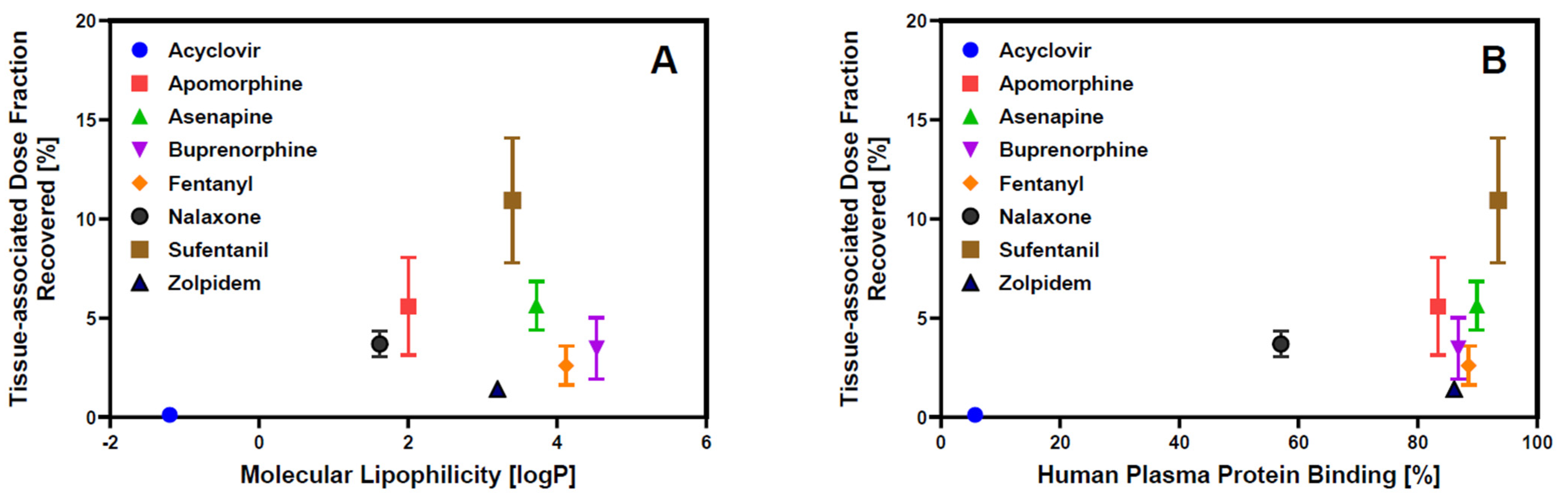
3.3. Tissue Binding
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ibarra, M.; Trocóniz, I.F.; Fagiolino, P. Enteric reabsorption processes and their impact on drug pharmacokinetics. Sci. Rep. 2021, 11, 5794. [Google Scholar] [CrossRef] [PubMed]
- Gancher, S.T.; Nutt, J.G.; Woodward, W.R. Absorption of apomorphine by various routes in parkinsonism. Mov. Disord. Off. J. Mov. Disord. Soc. 1991, 6, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Minassian, A.; Young, J.W. Evaluation of the clinical efficacy of asenapine in schizophrenia. Expert Opin. Pharmacother. 2010, 11, 2107–2115. [Google Scholar] [CrossRef] [PubMed]
- Downing, C.; Jonathan, M.; Aparna, T.; Tyring, S. Acyclovir Lauriad®: A muco-adhesive buccal tablet for the treatment of recurrent herpes labialis. Expert Rev. Anti-Infect. Ther. 2014, 12, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Hamman, J.H.; Enslin, G.M.; Kotzé, A.F.J.B. Oral delivery of peptide drugs: Barriers and developments. BioDrugs 2005, 19, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Bierbaumer, L.; Schwarze, U.Y.; Gruber, R.; Neuhaus, W. Cell culture models of oral mucosal barriers: A review with a focus on applications, culture conditions and barrier properties. Tissue Barriers 2018, 6, 1479568. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-L.H.; Chetty, D.J.; Chien, Y.W. A mechanistic analysis to characterize oramucosal permeation properties. Int. J. Pharm. 1999, 184, 63–72. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration (FDA). Approved Drug Products with Therapeutic Equivalence Evaluations. In Orange Book; FDA: Silver Spring, MD, USA, 2025. [Google Scholar]
- Attia, I.A.; El-Gizawy, S.A.; Fouda, M.A.; Donia, A.M. Influence of a niosomal formulation on the oral bioavailability of acyclovir in rabbits. AAPS PharmSciTech 2007, 8, 106. [Google Scholar] [CrossRef] [PubMed]
- Marquet, P. Pharmacology of high-dose buprenorphine. In Buprenorphine Therapy of Opiate Addiction; Humana Press: Totowa, NJ, USA, 2002; pp. 1–11. [Google Scholar]
- Streisand, J.B.; Varvel, J.R.; Stanski, D.R.; Le Maire, L.; Ashburn, M.A.; Hague, B.I.; Tarver, S.D.; Stanley, T.H. Absorption and bioavailability of oral transmucosal fentanyl citrate. Anesthesiology 1991, 75, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.; Hopp, M.; Mundin, G.; Bond, S.; Bailey, P.; Woodward, J.; Bell, D. Low absolute bioavailability of oral naloxone in healthy subjects. Int. J. Clin. Pharmacol. Ther. 2012, 50, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Willsie, S.K.; Evashenk, M.A.; Hamel, L.G.; Hwang, S.S.; Chiang, Y.-K.; Palmer, P.P. Pharmacokinetic properties of single- and repeated-dose sufentanil sublingual tablets in healthy volunteers. Clin. Ther. 2015, 37, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Langtry, H.D.; Benfield, P. Zolpidem. Drugs 1990, 40, 291–313. [Google Scholar] [CrossRef] [PubMed]
- Myoken, Y.; Moroyama, T.; Miyauchi, S.; Takada, K.; Namba, M. Monoclonal antibodies against human oral squamous cell carcinoma reacting with keratin proteins. Cancer 1987, 60, 2927–2937. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zuo, Z.; Chow, M.S.S. HO-1-u-1 model for screening sublingual drug delivery—Influence of pH, osmolarity and permeation enhancer. Int. J. Pharm. 2009, 370, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Amal, A.S.S.; Hussain, S.; Jalaluddin, M. Preparation of artificial saliva formulation. Pharm. Technol. 2015, 2, 6–12. [Google Scholar]
- Muralidharan, S.; Kalaimani, J.; Parasuraman, S.; Dhanaraj, S.A. Development and Validation of Acyclovir HPLC External Standard Method in Human Plasma: Application to Pharmacokinetic Studies. Adv. Pharm. 2014, 2014, 284652. [Google Scholar] [CrossRef]
- Ang, Z.Y.; Boddy, M.; Liu, Y.; Sunderland, B. Stability of apomorphine in solutions containing selected antioxidant agents. Drug Des. Dev. Ther. 2016, 10, 3253–3265. [Google Scholar] [CrossRef] [PubMed]
- Chhalotiya, U.K.; Bhatt, K.K.; Shah, D.A.; Patel, J.R. Stability-indicating liquid chromatographic method for the quantification of the new antipsychotic agent asenapine in bulk and in pharmaceutical formulation. Sci. Pharm. 2012, 80, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Mostafavi, A.; Abedi, G.; Jamshidi, A.; Afzali, D.; Talebi, M. Development and validation of a HPLC method for the determination of buprenorphine hydrochloride, naloxone hydrochloride and noroxymorphone in a tablet formulation. Talanta 2009, 77, 1415–1419. [Google Scholar] [CrossRef] [PubMed]
- Lambropoulos, J.; Spanos, G.A.; Lazaridis, N.V. Development and validation of an HPLC assay for fentanyl, alfentanil, and sufentanil in swab samples. J. Pharm. Biomed. Anal. 2000, 23, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Meka, V.S.; Battu, J.R.; Dharmalingam, S.R.; Gorajana, A.; Kolapalli, V.R.M. Development of a Validated HPLC Method for the Estimation of Metformin HCl and Propranolol HCl. J. Pharm. Res. Int. 2014, 4, 1909–1922. [Google Scholar] [CrossRef]
- Konoz, E.; Sarrafi, A.H.M.; Abdolahnejad, R.; Bahrami-Zonoz, M. Development and Validation of a Reversed-Phase HPLC Method for the Estimation of Zolpidem in Bulk Drug and Tablets. J. Chem. 2013, 2013, 357890. [Google Scholar] [CrossRef]
- Yamamoto, K.; Kurihara, M.; Matsusue, Y.; Imanishi, M.; Tsuyuki, M.; Kirita, T. Whole saliva flow rate and body profile in healthy young adults. Arch. Oral Biol. 2009, 54, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Eleraky, N.E.; Swarnakar, N.K.; Mohamed, D.F.; Attia, M.A.; Pauletti, G.M. Permeation-Enhancing Nanoparticle Formulation to Enable Oral Absorption of Enoxaparin. AAPS PharmSciTech 2020, 21, 88. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Whitney-Pickett, C.; Umland, J.P.; Zhang, H.; Zhang, X.; Gebhard, D.F.; Lai, Y.; Federico, J.J., III; Davidson, R.E.; Smith, R.J.; et al. Development of a new permeability assay using low-efflux MDCKII cells. J. Pharm. Sci. 2011, 100, 4974–4985. [Google Scholar] [CrossRef] [PubMed]
- Huth, F.; Domange, N.; Poller, B.; Vapurcuyan, A.; Durrwell, A.; Hanna, I.D.; Faller, B. Predicting Oral Absorption for Compounds Outside the Rule of Five Property Space. J. Pharm. Sci. 2021, 110, 2562–2569. [Google Scholar] [CrossRef] [PubMed]
- Klausner, M.; Ayehunie, S.; Breyfogle, B.A.; Wertz, P.W.; Bacca, L.; Kubilus, J. Organotypic human oral tissue models for toxicological studies. Toxicol. Vitr. 2007, 21, 938–949. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, V.M.; Shertzer, H.G.; Menon, A.G.; Pauletti, G.M. High glucose concentration in isotonic media alters caco-2 cell permeability. AAPS PharmSciTech 2003, 5, E24. [Google Scholar]
- Wertz, P.W.; Squier, C.A. Cellular and molecular basis of barrier function in oral epithelium. Crit. Rev. Ther. Drug Carr. Syst. 1991, 8, 237–269. [Google Scholar]
- Volpe, D.A. Application of method suitability for drug permeability classification. AAPS J. 2010, 12, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Kaur, N.; Sharma, P.; Li, X.; Jasti, B. Sublingual permeability of model drugs in New Zealand White Rabbits: In Vitro-In vivo correlation. Int. J. Pharm. 2025, 668, 124998. [Google Scholar] [CrossRef] [PubMed]
- Wanasathop, A.; Patel, P.B.; Choi, H.A.; Li, S.K. Permeability of Buccal Mucosa. Pharmaceutics 2021, 13, 1814. [Google Scholar] [CrossRef] [PubMed]
- Lanevskij, K.; Didziapetris, R. Physicochemical QSAR analysis of passive permeability across Caco-2 monolayers. J. Pharm. Sci. 2019, 108, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, J.; Streisand, J.B. Oral mucosal drug delivery: Clinical pharmacokinetics and therapeutic applications. Clin. Pharmacokinet. 2002, 41, 661–680. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, J.A.; van der Voort Maarschalk, K. Understanding the oral mucosal absorption and resulting clinical pharmacokinetics of asenapine. AAPS PharmSciTech 2012, 13, 1110–1115. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Active Pharmaceutical Ingredient | Molecular Weight [Da] | pKa 1) | Lipophilicity [logP] 1) | Human Plasma Protein Binding 1) | U.S.-Marketed Single Dose 2) | Oral Bioavailability | Generic Equivalent |
|---|---|---|---|---|---|---|---|
| Acyclovir | 225.3 | 2.53; 1.19 | −1.37 | 5.7% | 50 mg (buccal) | 15–30% [9] | No |
| Apomorphine hydrochloride | 303.8 | 7.64 | 2.81 | 83.4% | 30 mg (sublingual) | 1.7% [2] | No |
| Asenapine maleate | 401.8 | 8.57 | 4.20 | 89.9% | 10 mg (sublingual) | 2% [3] | Yes |
| Buprenorphine hydrochloride | 504.1 | 8.21 | 4.92 | 86.8% | 8 mg (sublingual) | >20% [10] | No |
| Fentanyl citrate | 528.6 | 8.15 | 3.99 | 88.5% | 800 µg (sublingual) | <10% [11] | No |
| Naloxone hydrochloride | 363.8 | 7.31 | 1.25 | 57.0% | 2 mg (buccal/sublingual) | <2% [12] | No |
| Sufentanil citrate | 578.7 | 7.94 | 3.86 | 93.5% | 30 µg (sublingual) | <10% [13] | No |
| Zolpidem tartrate | 764.9 | 5.53 | 2.77 | 86.1% | 10 mg (sublingual) | 45–70% [14] | Yes |
| Analyte | Stationary Phase | Mobile Phase | Retention Time | Detection Wavelength | Ref. |
|---|---|---|---|---|---|
| Acyclovir | C18 (250 mm × 4.6 mm, 5 µm particle size) | 10 mM phosphate buffer, pH 4.5 supplemented with 0.05% (w/v) of 1-decane sulfonic acid/ACN (95:5) | 4.1 min | 254 nm | [18] |
| Apomorphine HCl | C18 (250 mm × 4.6 mm, 5 µm particle size) | 50 mM o-phosphoric acid, pH 3.1/ACN (80:20) | 5.8 min | 272 nm | [19] |
| Asenapine maleate | C18 (250 mm × 4.6 mm, 5 µm particle size) | 25 mM phosphate buffer, pH 3.2/ACN (95:5) | 3.7 min | 232 nm | [20] |
| Buprenorphine HCl | C8 (150 mm × 4.6 mm, 5 µm particle size) | 10 mM phosphate buffer, pH 6.0 supplemented with 0.05% (w/v) of 1-decane sulfonic acid/ACN (35:65) | 7.5 min | 214 nm | [21] |
| Fentanyl citrate | C18 (250 mm × 4.6 mm, 5 µm particle size) | 50 mM phosphate buffer, pH 4.5/ACN (65:35) | 4.5 min | 230 nm | [22] |
| Naloxone HCl | C18 (250 mm × 4.6 mm, 5 µm particle size) | 10 mM phosphate buffer, pH 5.0/ACN (80:20) | 4.8 min | 283 nm | [21] |
| Propranolol | C18 (250 mm × 4.6 mm, 5 µm particle size) | 50 mM phosphate buffer, pH 4.5/ACN (65:35) | 6 min | 214 nm | [23] |
| Sufentanil citrate | C18 (250 mm × 4.6 mm, 5 µm particle size) | 130 mM ammonium acetate buffer, pH 7.2/ACN (40:60) | 6.8 min | 230 nm | [22] |
| Zolpidem tartrate | C18 (250 mm × 4.6 mm, 5 µm particle size) | 20 mM ammonium acetate buffer, pH 8.0/ACN (40:60) | 5.2 min | 245 nm | [24] |
| Active Pharmaceutical Ingredient | Sublingual Papp/Ranking 1) [×105 cm/s] | Buccal Papp/Ranking 2) [×105 cm/s] | Papp MDCK-LE Predicted/Ranking 3) [×105 cm/s] |
|---|---|---|---|
| Acyclovir | 4.07 ± 0.87 (#4) | 0.03 ± 0.01 (#8) | 0.15 (#8) |
| Apomorphine HCl | 3.73 ± 0.82 (#5) | 1.38 ± 0.27 (#5) | 1.90 (#4) |
| Asenapine maleate | 2.72 ± 0.06 (#8) | 1.96 ± 0.50 (#3) | 1.89 (#3) |
| Buprenorphine HCl | 3.45 ± 1.28 (#7) | 1.02 ± 0.21 (#7) | 1.25 (#7) |
| Fentanyl citrate | 5.63 ± 0.23 (#3) | 1.43 ± 0.45 (#4) | 2.32 (#1) |
| Naloxone HCl | 6.21 ± 2.60 (#1) | 2.30 ± 0.45 (#2) | 1.76 (#6) |
| Sufentanil citrate | 3.54 ± 1.54 (#6) | 2.56 ± 0.68 (#1) | 1.87 (#5) |
| Zolpidem tartrate | 5.97 ± 0.05 (#2) | 1.07 ± 0.25 (#6) | 2.25 (#2) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dwivedi, P.; Kalra, P.; Zhou, H.; Alam, K.; Tsakalozou, E.; Al-Ghabeish, M.; Kelchen, M.; Pauletti, G.M. In Vitro Oral Cavity Permeability Assessment to Enable Simulation of Drug Absorption. Pharmaceutics 2025, 17, 924. https://doi.org/10.3390/pharmaceutics17070924
Dwivedi P, Kalra P, Zhou H, Alam K, Tsakalozou E, Al-Ghabeish M, Kelchen M, Pauletti GM. In Vitro Oral Cavity Permeability Assessment to Enable Simulation of Drug Absorption. Pharmaceutics. 2025; 17(7):924. https://doi.org/10.3390/pharmaceutics17070924
Chicago/Turabian StyleDwivedi, Pankaj, Priyata Kalra, Haiying Zhou, Khondoker Alam, Eleftheria Tsakalozou, Manar Al-Ghabeish, Megan Kelchen, and Giovanni M. Pauletti. 2025. "In Vitro Oral Cavity Permeability Assessment to Enable Simulation of Drug Absorption" Pharmaceutics 17, no. 7: 924. https://doi.org/10.3390/pharmaceutics17070924
APA StyleDwivedi, P., Kalra, P., Zhou, H., Alam, K., Tsakalozou, E., Al-Ghabeish, M., Kelchen, M., & Pauletti, G. M. (2025). In Vitro Oral Cavity Permeability Assessment to Enable Simulation of Drug Absorption. Pharmaceutics, 17(7), 924. https://doi.org/10.3390/pharmaceutics17070924