

Tyrosine Kinase Inhibitors for Gastrointestinal Stromal Tumor After Imatinib Resistance


Abstract
1. Introduction
2. Relevance and Distinction from Existing Literature
3. Methods of Literature Selection
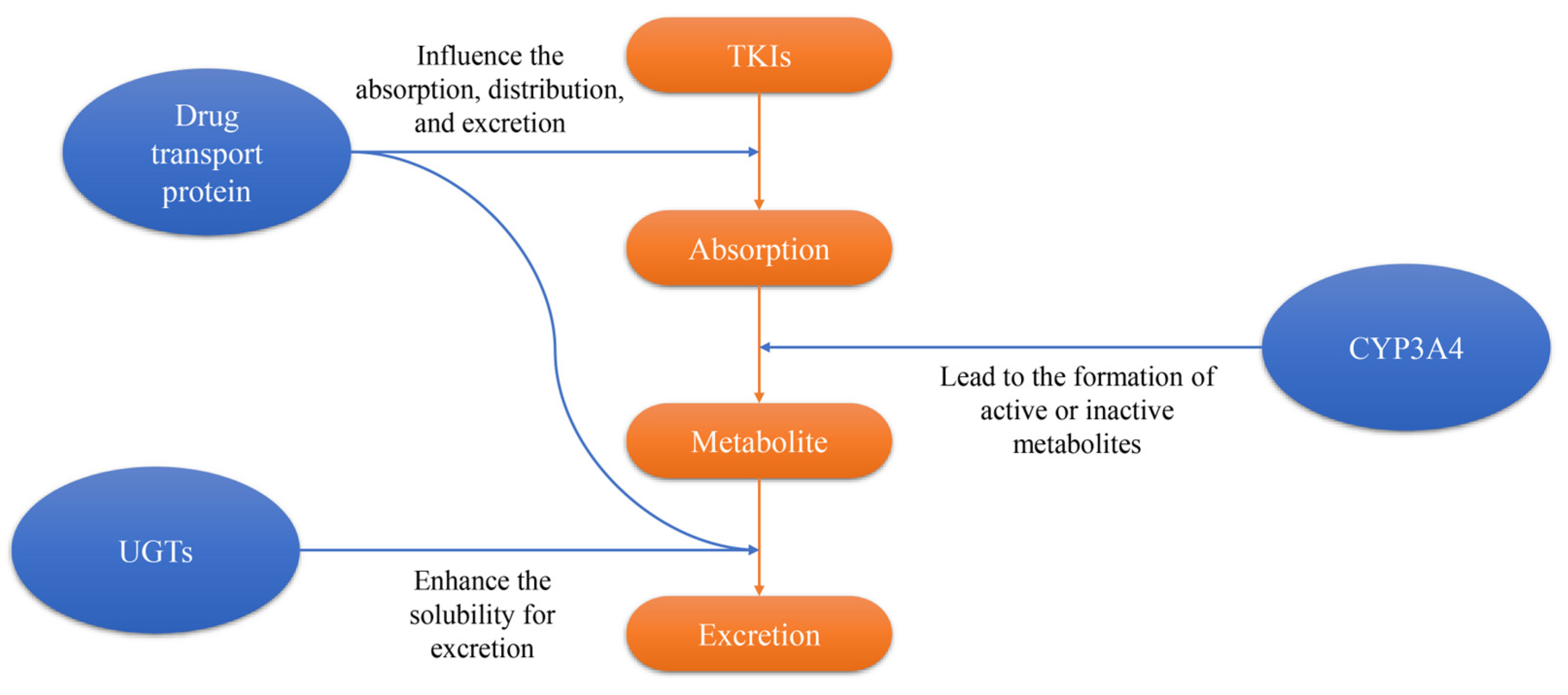
4. Imatinib Resistance
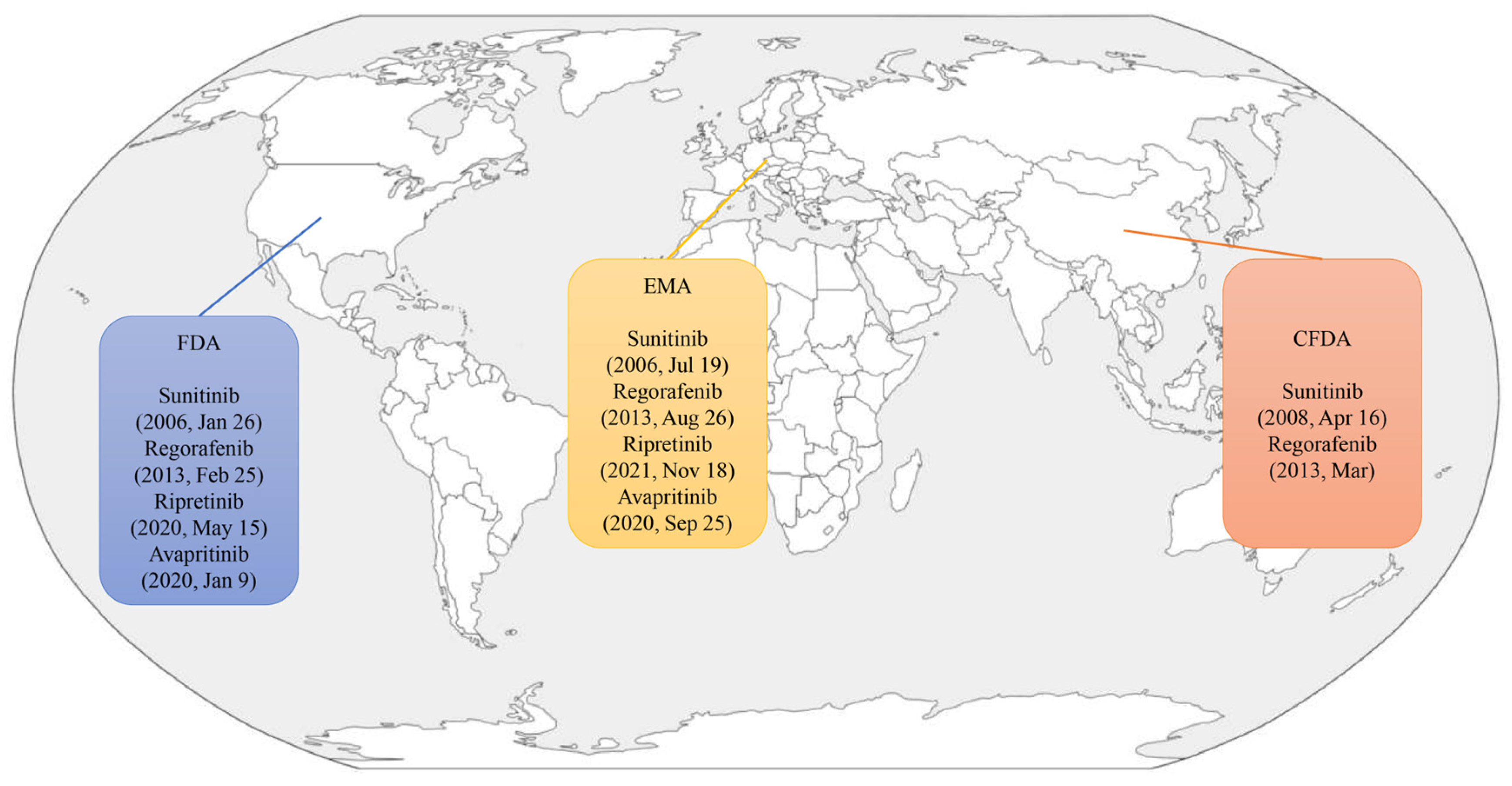
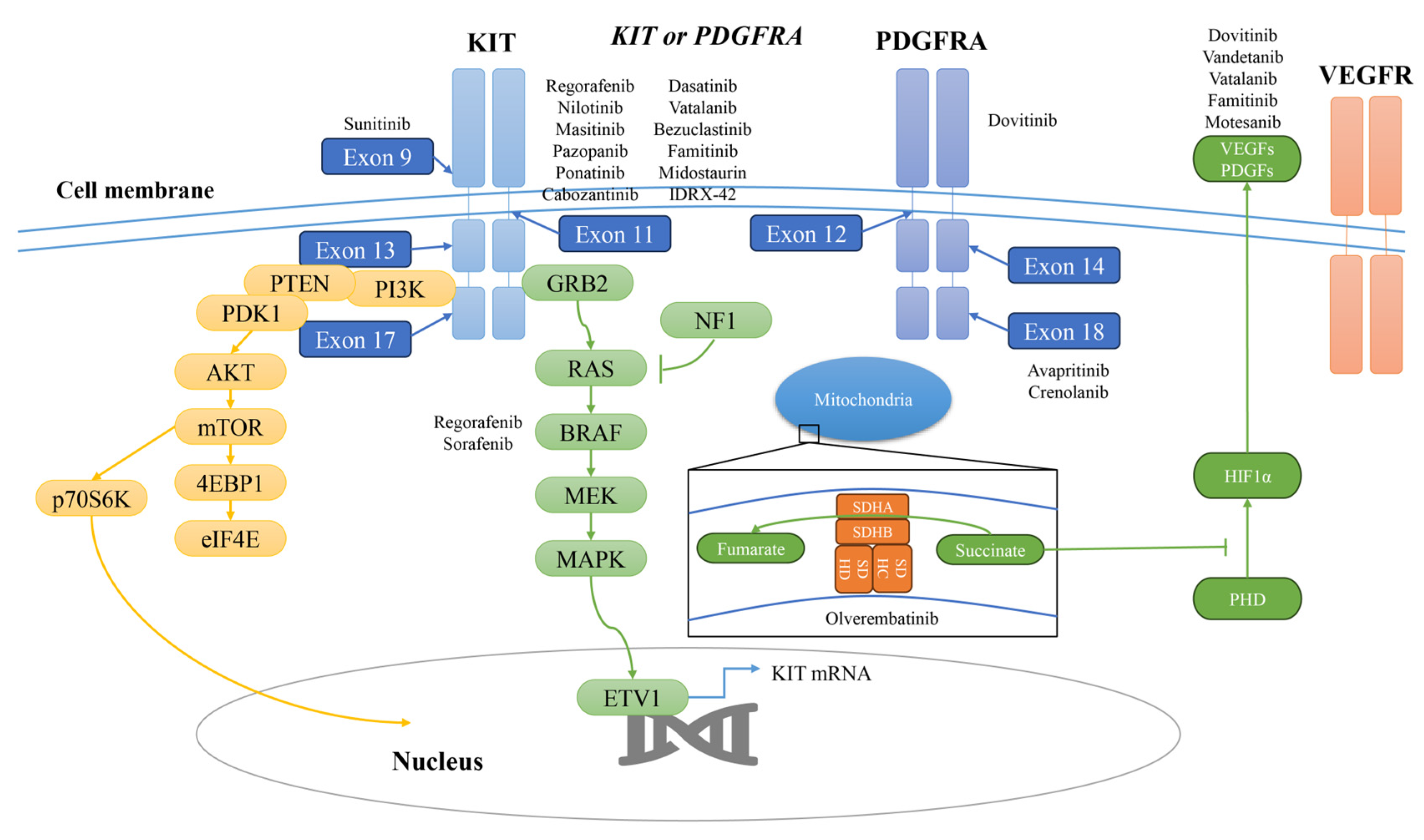
5. Approved Agents
5.1. Sunitinib
5.2. Regorafenib
5.3. Ripretinib
5.4. Avapritinib
6. Alternative Agents
6.1. Nilotinib
6.2. Masitinib
6.3. Sorafenib
6.4. Dovitinib
6.5. Pazopanib
6.6. Ponatinib
6.7. Cabozantinib
6.8. Vandetanib
6.9. Dasatinib
6.10. Vatalanib
6.11. Crenolanib
7. Investigational Agents
7.1. Bezuclastinib
7.2. Famitinib
7.3. Motesanib
7.4. Midostaurin
8. Other Potential TKIs
9. Overview
Author Contributions
Funding
Conflicts of Interest
References
- Nilsson, B.; Bümming, P.; Meis-Kindblom, J.M.; Odén, A.; Dortok, A.; Gustavsson, B.; Sablinska, K.; Kindblom, L.-G. Gastrointestinal stromal tumors: The incidence, prevalence, clinical course, and prognostication in the preimatinib mesylate era--a population-based study in western Sweden. Cancer 2005, 103, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Joensuu, H. Second line therapies for the treatment of gastrointestinal stromal tumor. Curr. Opin. Oncol. 2007, 19, 353–358. [Google Scholar] [CrossRef]
- Huizinga, J.D.; Thuneberg, L.; Klüppel, M.; Malysz, J.; Mikkelsen, H.B.; Bernstein, A. W/kit gene required for interstitial cells of Cajal and for intestinal pacemaker activity. Nature 1995, 373, 347–349. [Google Scholar] [CrossRef]
- Kindblom, L.G.; Remotti, H.E.; Aldenborg, F.; Meis-Kindblom, J.M. Gastrointestinal pacemaker cell tumor (GIPACT): Gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am. J. Pathol. 1998, 152, 1259–1269. [Google Scholar] [PubMed]
- Hirota, S.; Isozaki, K.; Moriyama, Y.; Hashimoto, K.; Nishida, T.; Ishiguro, S.; Kawano, K.; Hanada, M.; Kurata, A.; Takeda, M.; et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998, 279, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Corless, C.L.; Duensing, A.; McGreevey, L.; Chen, C.-J.; Joseph, N.; Singer, S.; Griffith, D.J.; Haley, A.; Town, A.; et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003, 299, 708–710. [Google Scholar] [CrossRef]
- Dematteo, R.P.; Heinrich, M.C.; El-Rifai, W.M.; Demetri, G. Clinical management of gastrointestinal stromal tumors: Before and after STI-571. Hum. Pathol. 2002, 33, 466–477. [Google Scholar] [CrossRef]
- Dematteo, R.P.; Ballman, K.V.; Antonescu, C.R.; Maki, R.G.; Pisters, P.W.T.; Demetri, G.D.; Blackstein, M.E.; Blanke, C.D.; von Mehren, M.; Brennan, M.F.; et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: A randomised, double-blind, placebo-controlled trial. Lancet 2009, 373, 1097–1104. [Google Scholar] [CrossRef]
- Kee, D.; Zalcberg, J.R. Current and emerging strategies for the management of imatinib-refractory advanced gastrointestinal stromal tumors. Ther. Adv. Med. Oncol. 2012, 4, 255–270. [Google Scholar] [CrossRef]
- Bauer, S.; Joensuu, H. Emerging Agents for the Treatment of Advanced, Imatinib-Resistant Gastrointestinal Stromal Tumors: Current Status and Future Directions. Drugs 2015, 75, 1323–1334. [Google Scholar] [CrossRef]
- Corless, C.L.; Schroeder, A.; Griffith, D.; Town, A.; McGreevey, L.; Harrell, P.; Shiraga, S.; Bainbridge, T.; Morich, J.; Heinrich, M.C. PDGFRA mutations in gastrointestinal stromal tumors: Frequency, spectrum and in vitro sensitivity to imatinib. J. Clin. Oncol. 2005, 23, 5357–5364. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Besmer, P.; Guo, T.; Arkun, K.; Hom, G.; Koryotowski, B.; Leversha, M.A.; Jeffrey, P.D.; Desantis, D.; Singer, S.; et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin. Cancer Res. 2005, 11, 4182–4190. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Corless, C.L.; Blanke, C.D.; Demetri, G.D.; Joensuu, H.; Roberts, P.J.; Eisenberg, B.L.; von Mehren, M.; Fletcher, C.D.; Sandau, K.; et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J. Clin. Oncol. 2006, 24, 4764–4774. [Google Scholar] [CrossRef] [PubMed]
- Serrano, C.; Martín-Broto, J.; Asencio-Pascual, J.M.; López-Guerrero, J.A.; Rubió-Casadevall, J.; Bagué, S.; García-del-Muro, X.; Fernández-Hernández, J.Á.; Herrero, L.; López-Pousa, A.; et al. 2023 GEIS Guidelines for gastrointestinal stromal tumors. Ther. Adv. Med. Oncol. 2023, 15, 17588359231192388. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Nakamura, Y.; Komatsu, Y.; Yuki, S.; Takahashi, N.; Okano, N.; Hirano, H.; Ohtsubo, K.; Ohta, T.; Oki, E.; et al. Different efficacy of tyrosine kinase inhibitors by KIT and PGFRA mutations identified in circulating tumor DNA for the treatment of refractory gastrointestinal stromal tumors. BJC Rep. 2024, 2, 54. [Google Scholar] [CrossRef]
- Yip, D.; Zalcberg, J.; Blay, J.-Y.; Eriksson, M.; Espinoza, D.; Price, T.; Marreaud, S.; Italiano, A.; Steeghs, N.; Boye, K.; et al. Imatinib alternating with regorafenib compared to imatinib alone for the first-line treatment of advanced gastrointestinal stromal tumor: The AGITG ALT-GIST intergroup randomized phase II trial. Br. J. Cancer 2025, 132, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Corless, C.L.; Demetri, G.D.; Blanke, C.D.; von Mehren, M.; Joensuu, H.; McGreevey, L.S.; Chen, C.-J.; Van den Abbeele, A.D.; Druker, B.J.; et al. Kinase Mutations and Imatinib Response in Patients With Metastatic Gastrointestinal Stromal Tumor. J. Clin. Oncol. 2023, 41, 4829–4836. [Google Scholar] [CrossRef]
- Debiec-Rychter, M.; Dumez, H.; Judson, I.; Wasag, B.; Verweij, J.; Brown, M.; Dimitrijevic, S.; Sciot, R.; Stul, M.; Vranck, H.; et al. Use of c-KIT/PDGFRA mutational analysis to predict the clinical response to imatinib in patients with advanced gastrointestinal stromal tumours entered on phase I and II studies of the EORTC Soft Tissue and Bone Sarcoma Group. Eur. J. Cancer 2004, 40, 689–695. [Google Scholar] [CrossRef]
- Wardelmann, E.; Merkelbach-Bruse, S.; Pauls, K.; Thomas, N.; Schildhaus, H.U.; Heinicke, T.; Speidel, N.; Pietsch, T.; Buettner, R.; Pink, D.; et al. Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clin. Cancer Res. 2006, 12, 1743–1749. [Google Scholar] [CrossRef]
- Desai, J.; Shankar, S.; Heinrich, M.C.; Fletcher, J.A.; Fletcher, C.D.; Manola, J.; Morgan, J.A.; Corless, C.L.; George, S.; Tuncali, K.; et al. Clonal evolution of resistance to imatinib in patients with metastatic gastrointestinal stromal tumors. Clin. Cancer Res. 2007, 13, 5398–5405. [Google Scholar] [CrossRef]
- Nishida, T.; Kanda, T.; Nishitani, A.; Takahashi, T.; Nakajima, K.; Ishikawa, T.; Hirota, S. Secondary mutations in the kinase domain of the KIT gene are predominant in imatinib-resistant gastrointestinal stromal tumor. Cancer Sci. 2008, 99, 799–804. [Google Scholar] [CrossRef]
- Corless, C.L.; Barnett, C.M.; Heinrich, M.C. Gastrointestinal stromal tumours: Origin and molecular oncology. Nat. Rev. Cancer 2011, 11, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, K.; Nagashima, T.; Fujiya, K.; Hatakeyama, K.; Watanabe, Y.; Morimoto, K.; Kamada, F.; Shimoda, Y.; Ohnami, S.; Naruoka, A.; et al. Whole-genome and Epigenomic Landscapes of Malignant Gastrointestinal Stromal Tumors Harboring KIT Exon 11 557-558 Deletion Mutations. Cancer Res. Commun. 2023, 3, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Boichuk, S.; Galembikova, A.; Dunaev, P.; Valeeva, E.; Shagimardanova, E.; Gusev, O.; Khaiboullina, S. A Novel Receptor Tyrosine Kinase Switch Promotes Gastrointestinal Stromal Tumor Drug Resistance. Molecules 2017, 22, 2152. [Google Scholar] [CrossRef] [PubMed]
- Li, D.-G.; Jiang, J.-P.; Chen, F.-Y.; Wu, W.; Fu, J.; Wang, G.-H.; Li, Y.-B. Insulin-like growth factor 2 targets IGF1R signaling transduction to facilitate metastasis and imatinib resistance in gastrointestinal stromal tumors. World J. Gastrointest. Oncol. 2024, 16, 3585–3599. [Google Scholar] [CrossRef]
- Kelly, C.M.; Shoushtari, A.N.; Qin, L.X.; D’Angelo, S.P.; Dickson, M.A.; Gounder, M.M.; Keohan, M.L.; Mcfadyen, C.; Sjoberg, A.; Singer, S.; et al. A phase Ib study of BGJ398, a pan-FGFR kinase inhibitor in combination with imatinib in patients with advanced gastrointestinal stromal tumor. Investig. New Drugs 2019, 37, 282–290. [Google Scholar] [CrossRef]
- Zhang, Z.; Jiang, N.Y.; Guan, R.Y.; Zhu, Y.K.; Jiang, F.Q.; Piao, D. Identification of critical microRNAs in gastrointestinal stromal tumor patients treated with Imatinib. Neoplasma 2018, 65, 683–692. [Google Scholar] [CrossRef]
- Cui, Z.; Sun, H.; Gao, Z.; Li, C.; Xiao, T.; Bian, Y.; Liu, Z.; Gu, T.; Zhang, J.; Li, T.; et al. TRIM21/USP15 balances ACSL4 stability and the imatinib resistance of gastrointestinal stromal tumors. Br. J. Cancer 2024, 130, 526–541. [Google Scholar] [CrossRef]
- Shima, T.; Taniguchi, K.; Tokumaru, Y.; Inomata, Y.; Arima, J.; Lee, S.W.; Takabe, K.; Yoshida, K.; Uchiyama, K. Glucose transporter-1 inhibition overcomes imatinib resistance in gastrointestinal stromal tumor cells. Oncol. Rep. 2022, 47, 8218. [Google Scholar] [CrossRef]
- Rausch, J.L.; Ali, A.A.; Lee, D.M.; Gebreyohannes, Y.K.; Mehalek, K.R.; Agha, A.; Patil, S.S.; Tolstov, Y.; Wellens, J.; Dhillon, H.S.; et al. Differential antitumor activity of compounds targeting the ubiquitin-proteasome machinery in gastrointestinal stromal tumor (GIST) cells. Sci. Rep. 2020, 10, 5178. [Google Scholar] [CrossRef]
- Demetri, G.D.; van Oosterom, A.T.; Garrett, C.R.; Blackstein, M.E.; Shah, M.H.; Verweij, J.; McArthur, G.; Judson, I.R.; Heinrich, M.C.; Morgan, J.A.; et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: A randomised controlled trial. Lancet 2006, 368, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Abrams, T.J.; Lee, L.B.; Murray, L.J.; Pryer, N.K.; Cherrington, J.M. SU11248 inhibits KIT and platelet-derived growth factor receptor beta in preclinical models of human small cell lung cancer. Mol. Cancer Ther. 2003, 2, 471–478. [Google Scholar]
- Mendel, D.B.; Laird, A.D.; Xin, X.; Louie, S.G.; Christensen, J.G.; Li, G.; Schreck, R.E.; Abrams, T.J.; Ngai, T.J.; Lee, L.B.; et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: Determination of a pharmacokinetic/pharmacodynamic relationship. Clin. Cancer Res. 2003, 9, 327–337. [Google Scholar]
- Mulet-Margalef, N.; Garcia-Del-Muro, X. Sunitinib in the treatment of gastrointestinal stromal tumor: Patient selection and perspectives. Onco Targets Ther. 2016, 9, 7573–7582. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Maki, R.G.; Corless, C.L.; Antonescu, C.R.; Harlow, A.; Griffith, D.; Town, A.; McKinley, A.; Ou, W.B.; Fletcher, J.A.; et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J. Clin. Oncol. 2008, 26, 5352–5359. [Google Scholar] [CrossRef]
- Judson, I.R. Prognosis, imatinib dose, and benefit of sunitinib in GIST: Knowing the genotype. J. Clin. Oncol. 2008, 26, 5322–5325. [Google Scholar] [CrossRef]
- Ferrari, S.M.; Centanni, M.; Virili, C.; Miccoli, M.; Ferrari, P.; Ruffilli, I.; Ragusa, F.; Antonelli, A.; Fallahi, P. Sunitinib in the Treatment of Thyroid Cancer. Curr. Med. Chem. 2019, 26, 963–972. [Google Scholar] [CrossRef]
- Shirao, K.; Nishida, T.; Doi, T.; Komatsu, Y.; Muro, K.; Li, Y.; Ueda, E.; Ohtsu, A. Phase I/II study of sunitinib malate in Japanese patients with gastrointestinal stromal tumor after failure of prior treatment with imatinib mesylate. Investig. New Drugs 2010, 28, 866–875. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, C.; Zhang, T.; Liu, H.; Zhong, J.; Wang, Z.; Wang, L.; Hong, L. Second-line sunitinib for Chinese patients with advanced gastrointestinal stromal tumor: 37.5 mg schedule outperformed 50 mg schedule in adherence and prognosis. Transl. Cancer Res. 2021, 10, 3206–3217. [Google Scholar] [CrossRef]
- Sasaki, K.; Kanda, T.; Matsumoto, Y.; Ishikawa, T.; Hirota, S.; Saijo, Y. Sunitinib therapy for imatinib-resistant and/or intolerant gastrointestinal stromal tumors: Comparison of safety and efficacy between standard and reduced dosage regimens. Jpn. J. Clin. Oncol. 2023, 53, 297–303. [Google Scholar] [CrossRef]
- George, S.; Blay, J.Y.; Casali, P.G.; Le Cesne, A.; Stephenson, P.; Deprimo, S.E.; Harmon, C.S.; Law, C.N.; Morgan, J.A.; Ray-Coquard, I.; et al. Clinical evaluation of continuous daily dosing of sunitinib malate in patients with advanced gastrointestinal stromal tumour after imatinib failure. Eur. J. Cancer 2009, 45, 1959–1968. [Google Scholar] [CrossRef]
- Sutton, T.L.; Walker, B.S.; Billingsley, K.G.; Sheppard, B.C.; Corless, C.L.; Heinrich, M.C.; Mayo, S.C. Imatinib-resistant gastrointestinal stromal tumors in the era of second- and third-line tyrosine kinase inhibitors: Does surgical resection have a role? Surgery 2021, 170, 1481–1486. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, P.; Bylina, E.; Klimczak, A.; Świtaj, T.; Falkowski, S.; Kroc, J.; Ługowska, I.; Brzeskwiniewicz, M.; Melerowicz, W.; Osuch, C.; et al. The outcome and predictive factors of sunitinib therapy in advanced gastrointestinal stromal tumors (GIST) after imatinib failure–one institution study. BMC Cancer 2012, 12, 1–9. [Google Scholar] [CrossRef]
- Guo, T.; Hajdu, M.; Agaram, N.P.; Shinoda, H.; Veach, D.; Clarkson, B.D.; Maki, R.G.; Singer, S.; Dematteo, R.P.; Besmer, P.; et al. Mechanisms of sunitinib resistance in gastrointestinal stromal tumors harboring KITAY502-3ins mutation: An in vitro mutagenesis screen for drug resistance. Clin. Cancer Res. 2009, 15, 6862–6870. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Jones, R.L.; George, S.; Gelderblom, H.; Schöffski, P.; von Mehren, M.; Zalcberg, J.R.; Kang, Y.K.; Razak, A.A.; Trent, J.; et al. Ripretinib versus sunitinib in gastrointestinal stromal tumor: ctDNA biomarker analysis of the phase 3 INTRIGUE trial. Nat. Med. 2024, 30, 498–506. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Blay, J.Y.; Chi, P.; Jones, R.L.; Serrano, C.; Somaiah, N.; Gelderblom, H.; Zalcberg, J.R.; Reichmann, W.; Sprott, K.; et al. The INSIGHT study: A randomized, Phase III study of ripretinib versus sunitinib for advanced gastrointestinal stromal tumor with KIT exon 11 + 17/18 mutations. Future Oncol. 2024, 20, 1973–1982. [Google Scholar] [CrossRef]
- Westerdijk, K.; Desar, I.M.E.; Steeghs, N.; van der Graaf, W.T.A.; van Erp, N.P. Dutch Pharmacology and Oncology Group (DPOG). Imatinib, sunitinib and pazopanib: From flat-fixed dosing towards a pharmacokinetically guided personalized dose. Br. J. Clin. Pharmacol. 2020, 86, 258–273. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Dumas, J.; Adnane, L.; Lynch, M.; Carter, C.A.; Schütz, G.; Thierauch, K.; Zopf, D. Regorafenib (BAY 73-4506): A new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int. J. Cancer 2011, 129, 245–255. [Google Scholar] [CrossRef]
- Mross, K.; Frost, A.; Steinbild, S.; Hedbom, S.; Büchert, M.; Fasol, U.; Unger, C.; Krätzschmar, J.; Heinig, R.; Boix, O.; et al. A phase I dose-escalation study of regorafenib (BAY 73-4506), an inhibitor of oncogenic, angiogenic, and stromal kinases, in patients with advanced solid tumors. Clin. Cancer Res. 2012, 18, 2658–2667. [Google Scholar] [CrossRef]
- Ferraro, D.; Zalcberg, J. Regorafenib in gastrointestinal stromal tumors: Clinical evidence and place in therapy. Ther. Adv. Med. Oncol. 2014, 6, 222–228. [Google Scholar] [CrossRef]
- Demetri, G.D.; Reichardt, P.; Kang, Y.K.; Blay, J.Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.; von Mehren, M.; Joensuu, H.; et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; George, S.; von Mehren, M.; Heinrich, M.C. Early and Next-Generation KIT/PDGFRA Kinase Inhibitors and the Future of Treatment for Advanced Gastrointestinal Stromal Tumor. Front. Oncol. 2021, 11, 672500. [Google Scholar] [CrossRef]
- Takahashi, T.; Nishida, T.; Kudo, T.; Boku, N.; Honma, Y.; Komatsu, Y.; Nakatsumi, H.; Matsumoto, K.; Onoe, T.; Oki, E.; et al. Regorafenib as second-line therapy for imatinib-resistant gastrointestinal stromal tumor (GIST). J. Clin. Oncol. 2020, 38 (Suppl. S4), 823. [Google Scholar] [CrossRef]
- Martin-Broto, J.; Valverde, C.; Hindi, N.; Vincenzi, B.; Martinez-Trufero, J.; Grignani, G.; Italiano, A.; Lavernia, J.; Vallejo, A.; Tos, P.D.; et al. REGISTRI: Regorafenib in first-line of KIT/PDGFRA wild type metastatic GIST: A collaborative Spanish (GEIS), Italian (ISG) and French Sarcoma Group (FSG) phase II trial. Mol. Cancer 2023, 22, 127. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, Y.; Doi, T.; Sawaki, A.; Kanda, T.; Yamada, Y.; Kuss, I.; Demetri, G.D.; Nishida, T. Regorafenib for advanced gastrointestinal stromal tumors following imatinib and sunitinib treatment: A subgroup analysis evaluating Japanese patients in the phase III GRID trial. Int. J. Clin. Oncol. 2015, 20, 905–912. [Google Scholar] [CrossRef]
- de Man, F.; Hussaarts, K.; de With, M.; Hoop, E.O.-D.; van Halteren, H.; Graauw, N.v.d.B.-D.; Eskens, F.; van Gelder, T.; van Leeuwen, R.; Mathijssen, R. Influence of the proton pump inhibitor esomeprazole on the bioavailability of regorafenib. Ann. Oncol. 2018, 29, viii158. [Google Scholar] [CrossRef]
- Smith, B.D.; Hood, M.M.; Wise, S.C.; Kaufman, M.D.; Lu, W.-P.; Rutkoski, T.; Flynn, D.L.; Heinrich, M.C. Abstract 2690: DCC-2618 is a potent inhibitor of wild-type and mutant KIT, including refractory Exon 17 D816 KIT mutations, and exhibits efficacy in refractory GIST and AML xenograft models. Cancer Res. 2015, 75, 2690. [Google Scholar] [CrossRef]
- Smith, B.D.; Kaufman, M.D.; Lu, W.-P.; Gupta, A.; Leary, C.B.; Wise, S.C.; Rutkoski, T.J.; Ahn, Y.M.; Al-Ani, G.; Bulfer, S.L.; et al. Ripretinib (DCC-2618) Is a Switch Control Kinase Inhibitor of a Broad Spectrum of Oncogenic and Drug-Resistant KIT and PDGFRA Variants. Cancer Cell 2019, 35, 738–751.e9. [Google Scholar] [CrossRef]
- Blay, J.Y.; Serrano, C.; Heinrich, M.C.; Zalcberg, J.; Bauer, S.; Gelderblom, H.; Schöffski, P.; Jones, R.L.; Attia, S.; D’Amato, G.; et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 923–934. [Google Scholar] [CrossRef]
- Bauer, S.; Jones, R.L.; Blay, J.Y.; Gelderblom, H.; George, S.; Schöffski, P.; von Mehren, M.; Zalcberg, J.R.; Kang, Y.K.; Razak, A.A.; et al. Ripretinib Versus Sunitinib in Patients with Advanced Gastrointestinal Stromal Tumor After Treatment with Imatinib (INTRIGUE): A Randomized, Open-Label, Phase III Trial. J. Clin. Oncol. 2022, 40, 3918–3928. [Google Scholar] [CrossRef]
- Gelderblom, H.; Jones, R.L.; Blay, J.-Y.; George, S.; von Mehren, M.; Zalcberg, J.R.; Kang, Y.-K.; Razak, A.A.; Trent, J.; Attia, S.; et al. Patient-reported outcomes and tolerability in patients receiving ripretinib versus sunitinib after treatment with imatinib in INTRIGUE, a phase 3, open-label study. Eur. J. Cancer 2023, 192, 113245. [Google Scholar] [CrossRef]
- Evans, E.K.; Hodous, B.L.; Gardino, A.K.; Davis, A.; Zhu, J.; Shutes, A.; Kim, J.L.; Wilson, K.J.; Wilson, D.; Zhang, Y.; et al. Abstract 791: BLU-285, the first selective inhibitor of PDGFRα D842V and KIT Exon 17 mutants. Cancer Res. 2015, 75 (Suppl. S15), 791. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Jones, R.L.; von Mehren, M.; Schöffski, P.; Serrano, C.; Kang, Y.K.; Cassier, P.A.; Mir, O.; Eskens, F.; Tap, W.D.; et al. Avapritinib in advanced PDGFRA D842V-mutant gastrointestinal stromal tumour (NAVIGATOR): A multicentre, open-label, phase 1 trial. Lancet Oncol. 2020, 21, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Henriques-Abreu, M.; Serrano, C. Avapritinib in unresectable or metastatic gastrointestinal stromal tumor with PDGFRA exon 18 mutation: Safety and efficacy. Expert. Rev. Anticancer. Ther. 2021, 21, 1081–1088. [Google Scholar] [CrossRef]
- Kang, Y.-K.; George, S.; Jones, R.L.; Rutkowski, P.; Shen, L.; Mir, O.; Patel, S.; Zhou, Y.; von Mehren, M.; Hohenberger, P.; et al. Avapritinib Versus Regorafenib in Locally Advanced Unresectable or Metastatic GI Stromal Tumor: A Randomized, Open-Label Phase III Study. J. Clin. Oncol. 2021, 39, 3128–3139. [Google Scholar] [CrossRef]
- Sako, H.; Fukuda, K.; Saikawa, Y.; Nakamura, R.; Takahashi, T.; Wada, N.; Kawakubo, H.; Takeuchi, H.; Ohmori, T.; Kitagawa, Y. Antitumor effect of the tyrosine kinase inhibitor nilotinib on gastrointestinal stromal tumor (GIST) and imatinib-resistant GIST cells. PLoS ONE 2014, 9, e107613. [Google Scholar] [CrossRef] [PubMed]
- Abbaspour Babaei, M.; Kamalidehghan, B.; Saleem, M.; Huri, H.Z.; Ahmadipour, F. Receptor tyrosine kinase (c-Kit) inhibitors: A potential therapeutic target in cancer cells. Drug Des. Devel Ther. 2016, 10, 2443–2459. [Google Scholar] [CrossRef]
- Blay, J.-Y.; Shen, L.; Kang, Y.-K.; Rutkowski, P.; Qin, S.; Nosov, D.; Wan, D.; Trent, J.; Srimuninnimit, V.; Pápai, Z.; et al. Nilotinib versus imatinib as first-line therapy for patients with unresectable or metastatic gastrointestinal stromal tumours (ENESTg1): A randomised phase 3 trial. Lancet Oncol. 2015, 16, 550–560. [Google Scholar] [CrossRef]
- Cauchi, C.; Somaiah, N.; Engstrom, P.F.; Litwin, S.; Lopez, M.; Lee, J.; Davey, M.; Bove, B.; von Mehren, M. Evaluation of nilotinib in advanced GIST previously treated with imatinib and sunitinib. Cancer Chemother. Pharmacol. 2012, 69, 977–982. [Google Scholar] [CrossRef]
- Reichardt, P.; Blay, J.Y.; Gelderblom, H.; Schlemmer, M.; Demetri, G.D.; Bui-Nguyen, B.; McArthur, G.A.; Yazji, S.; Hsu, Y.; Galetic, I.; et al. Phase III study of nilotinib versus best supportive care with or without a TKI in patients with gastrointestinal stromal tumors resistant to or intolerant of imatinib and sunitinib. Ann. Oncol. 2012, 23, 1680–1687. [Google Scholar] [CrossRef]
- Dubreuil, P.; Letard, S.; Ciufolini, M.; Gros, L.; Humbert, M.; Castéran, N.; Borge, L.; Hajem, B.; Lermet, A.; Sippl, W.; et al. Masitinib (AB1010), a potent and selective tyrosine kinase inhibitor targeting KIT. PLoS ONE 2009, 4, e7258. [Google Scholar] [CrossRef] [PubMed]
- Anastassiadis, T.; Deacon, S.W.; Devarajan, K.; Ma, H.; Peterson, J.R. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1039–1045. [Google Scholar] [CrossRef]
- Le Cesne, A.; Blay, J.-Y.; Bui, B.N.; Bouché, O.; Adenis, A.; Domont, J.; Cioffi, A.; Ray-Coquard, I.; Lassau, N.; Bonvalot, S.; et al. Phase II study of oral masitinib mesilate in imatinib-naïve patients with locally advanced or metastatic gastro-intestinal stromal tumour (GIST). Eur. J. Cancer 2010, 46, 1344–1351. [Google Scholar] [CrossRef] [PubMed]
- Adenis, A.; Blay, J.Y.; Bui-Nguyen, B.; Bouché, O.; Bertucci, F.; Isambert, N.; Bompas, E.; Chaigneau, L.; Domont, J.; Ray-Coquard, I.; et al. Masitinib in advanced gastrointestinal stromal tumor (GIST) after failure of imatinib: A randomized controlled open-label trial. Ann. Oncol. 2014, 25, 1762–1769. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef]
- Kefeli, U.; Benekli, M.; Sevinc, A.; Yildiz, R.; Kaplan, M.A.; Ciltas, A.; Balakan, O.; Isikdogan, A.; Coskun, U.; Dane, F.; et al. Efficacy of sorafenib in patients with gastrointestinal stromal tumors in the third- or fourth-line treatment: A retrospective multicenter experience. Oncol. Lett. 2013, 6, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Ryu, M.H.; Ryoo, B.Y.; Im, S.A.; Kwon, H.C.; Lee, S.S.; Park, S.R.; Kang, B.Y.; Kang, Y.K. Sorafenib in patients with metastatic gastrointestinal stromal tumors who failed two or more prior tyrosine kinase inhibitors: A phase II study of Korean gastrointestinal stromal tumors study group. Investig. New Drugs 2012, 30, 2377–2383. [Google Scholar] [CrossRef]
- Rutkowski, P.; Jagielska, B.; Andrzejuk, J.; Bylina, E.; Lugowska, I.; Switaj, T.; Kosela-Paterczyk, H.; Kozak, K.; Falkowski, S.; Klimczak, A. The analysis of the long-term outcomes of sorafenib therapy in routine practice in imatinib and sunitinib resistant gastrointestinal stromal tumors (GIST). Contemp Oncol. 2017, 21, 285–289. [Google Scholar] [CrossRef]
- Joensuu, H.; Blay, J.Y.; Comandone, A.; Martin-Broto, J.; Fumagalli, E.; Grignani, G.; Del, M.X.; Adenis, A.; Valverde, C.; Pousa, A.L.; et al. Dovitinib in patients with gastrointestinal stromal tumour refractory and/or intolerant to imatinib. Br. J. Cancer 2017, 117, 1278–1285. [Google Scholar] [CrossRef]
- Ganjoo, K.N.; Villalobos, V.M.; Kamaya, A.; Fisher, G.A.; Butrynski, J.E.; Morgan, J.A.; Wagner, A.J.; D’Adamo, D.; McMillan, A.; Demetri, G.D.; et al. A multicenter phase II study of pazopanib in patients with advanced gastrointestinal stromal tumors (GIST) following failure of at least imatinib and sunitinib. Ann. Oncol. 2014, 25, 236–240. [Google Scholar] [CrossRef]
- Mir, O.; Cropet, C.; Toulmonde, M.; Cesne, A.L.; Molimard, M.; Bompas, E.; Cassier, P.; Ray-Coquard, I.; Rios, M.; Adenis, A.; et al. Pazopanib plus best supportive care versus best supportive care alone in advanced gastrointestinal stromal tumours resistant to imatinib and sunitinib (PAZOGIST): A randomised, multicentre, open-label phase 2 trial. Lancet Oncol. 2016, 17, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, M.; Moura, S.; Parra, L.; Vasconcellos, V.; Costa, G.; Leite, D.; Dias, M.; Fernandes, T.V.A.; Hoelz, L.; Pimentel, L.; et al. Ponatinib: A Review of the History of Medicinal Chemistry behind Its Development. Pharmaceuticals 2024, 17, 1361. [Google Scholar] [CrossRef]
- George, S.; von Mehren, M.; Fletcher, J.A.; Sun, J.; Zhang, S.; Pritchard, J.R.; Hodgson, J.G.; Kerstein, D.; Rivera, V.M.; Haluska, F.G.; et al. Phase II Study of Ponatinib in Advanced Gastrointestinal Stromal Tumors: Efficacy, Safety, and Impact of Liquid Biopsy and Other Biomarkers. Clin. Cancer Res. 2022, 28, 1268–1276. [Google Scholar] [CrossRef]
- Markowitz, J.N.; Fancher, K.M. Cabozantinib: A Multitargeted Oral Tyrosine Kinase Inhibitor. Pharmacotherapy 2018, 38, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Schöffski, P.; Mir, O.; Kasper, B.; Papai, Z.; Blay, J.Y.; Italiano, A.; Benson, C.; Kopeckova, K.; Ali, N.; Dileo, P.; et al. Activity and safety of the multi-target tyrosine kinase inhibitor cabozantinib in patients with metastatic gastrointestinal stromal tumour after treatment with imatinib and sunitinib: European Organisation for Research and Treatment of Cancer phase II trial 1317 “CaboGIST”. Eur. J. Cancer 2020, 134, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Glod, J.; Arnaldez, F.I.; Wiener, L.; Spencer, M.; Killian, J.K.; Meltzer, P.; Dombi, E.; Derse-Anthony, C.; Derdak, J.; Srinivasan, R.; et al. A Phase II Trial of Vandetanib (ZD6474, Caprelsa®) in Children and Adults with Succinate Dehydrogenase Deficient Gastrointestinal Stromal Tumor. Clin. Cancer Res. 2019, 25, 6302–6308. [Google Scholar] [CrossRef]
- Lindauer, M.; Hochhaus, A. Dasatinib. Recent. Results Cancer Res. 2010, 184, 83–102. [Google Scholar] [CrossRef]
- Zeng, C.; Zhu, L.; Jia, X.; Pang, Y.; Li, Z.; Lu, X.; Xie, F.; Duan, L.; Wang, Y. Spectrum of activity of dasatinib against mutant KIT kinases associated with drug-sensitive and drug-resistant gastrointestinal stromal tumors. Gastric Cancer 2020, 23, 837–847. [Google Scholar] [CrossRef]
- Montemurro, M.; Cioffi, A.; Dômont, J.; Rutkowski, P.; Roth, A.D.; von Moos, R.; Inauen, R.; Toulmonde, M.; Burkhard, R.O.; Knuesli, C.; et al. Long-term outcome of dasatinib first-line treatment in gastrointestinal stromal tumor: A multicenter, 2-stage phase 2 trial (Swiss Group for Clinical Cancer Research 56/07). Cancer 2018, 124, 1449–1454. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, X.; Wu, X.; Zhou, Y.; Zhang, B.; Liu, X.; Wu, X.; Li, Y.; Shen, L.; Li, J. A prospective multicenter phase II study on the efficacy and safety of dasatinib in the treatment of metastatic gastrointestinal stromal tumors failed by imatinib and sunitinib and analysis of NGS in peripheral blood. Cancer Med. 2020, 9, 6225–6233. [Google Scholar] [CrossRef]
- Wood, J.M.; Bold, G.; Buchdunger, E.; Cozens, R.; Ferrari, S.; Frei, J.; Hofmann, F.; Mestan, J.; Mett, H.; O’Reilly, T.; et al. PTK787/ZK 222584, a novel and potent inhibitor of vascular endothelial growth factor receptor tyrosine kinases, impairs vascular endothelial growth factor-induced responses and tumor growth after oral administration. Cancer Res. 2000, 60, 2178–2189. [Google Scholar]
- Joensuu, H.; De Braud, F.; Grignagni, G.; De Pas, T.; Spitalieri, G.; Coco, P.; Spreafico, C.; Boselli, S.; Toffalorio, F.; Bono, P.; et al. Vatalanib for metastatic gastrointestinal stromal tumour (GIST) resistant to imatinib: Final results of a phase II study. Br. J. Cancer 2011, 104, 1686–1690. [Google Scholar] [CrossRef] [PubMed]
- Mathias, T.J.; Natarajan, K.; Shukla, S.; Doshi, K.A.; Singh, Z.N.; Ambudkar, S.V.; Baer, M.R. The FLT3 and PDGFR inhibitor crenolanib is a substrate of the multidrug resistance protein ABCB1 but does not inhibit transport function at pharmacologically relevant concentrations. Investig. New Drugs 2015, 33, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Griffith, D.; McKinley, A.; Patterson, J.; Presnell, A.; Ramachandran, A.; Debiec-Rychter, M. Crenolanib inhibits the drug-resistant PDGFRA D842V mutation associated with imatinib-resistant gastrointestinal stromal tumors. Clin. Cancer Res. 2012, 18, 4375–4384. [Google Scholar] [CrossRef]
- Cicala, C.M.; Olivares-Rivas, I.; Aguirre-Carrillo, J.A.; Serrano, C. KIT/PDGFRA inhibitors for the treatment of gastrointestinal stromal tumors: Getting to the gist of the problem. Expert. Opin. Investig. Drugs 2024, 33, 159–170. [Google Scholar] [CrossRef]
- Huang, C.; Ma, X.; Wang, M.; Cao, H. Drugs in the GIST Field (Therapeutic Targets and Clinical Trial Staging). Curr. Drug Deliv. 2023, 21, 80–90. [Google Scholar] [CrossRef]
- Naito, Y.; Nishida, T.; Doi, T. Current status of and future prospects for the treatment of unresectable or metastatic gastrointestinal stromal tumours. Gastric Cancer 2023, 26, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.; Zhang, W.; Chang, C.; Chen, X.; Zhong, D.; Qin, Q.; Lou, D.; Jiang, H.; Wang, J. Phase I study of the safety, pharmacokinetics and antitumor activity of famitinib. Cancer Chemother. Pharmacol. 2013, 72, 1043–1053. [Google Scholar] [CrossRef]
- Caenepeel, S.; Renshaw-Gegg, L.; Baher, A.; Bush, T.L.; Baron, W.; Juan, T.; Manoukian, R.; Tasker, A.S.; Polverino, A.; Hughes, P.E. Motesanib inhibits Kit mutations associated with gastrointestinal stromal tumors. J. Exp. Clin. Cancer Res. 2010, 29, 96. [Google Scholar] [CrossRef]
- Benjamin, R.S.; Schöffski, P.; Hartmann, J.T.; Van Oosterom, A.; Bui, B.N.; Duyster, J.; Schuetze, S.; Blay, J.Y.; Reichardt, P.; Rosen, L.S.; et al. Efficacy and safety of motesanib, an oral inhibitor of VEGF, PDGF, and Kit receptors, in patients with imatinib-resistant gastrointestinal stromal tumors. Cancer Chemother. Pharmacol. 2011, 68, 69–77. [Google Scholar] [CrossRef]
- Sawaki, A.; Yamada, Y.; Komatsu, Y.; Kanda, T.; Doi, T.; Koseki, M.; Baba, H.; Sun, Y.N.; Murakami, K.; Nishida, T. Phase II study of motesanib in Japanese patients with advanced gastrointestinal stromal tumors with prior exposure to imatinib mesylate. Cancer Chemother. Pharmacol. 2010, 65, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Growney, J.D.; Clark, J.J.; Adelsperger, J.; Stone, R.; Fabbro, D.; Griffin, J.D.; Gilliland, D.G. Activation mutations of human c-KIT resistant to imatinib mesylate are sensitive to the tyrosine kinase inhibitor PKC412. Blood 2005, 106, 721–724. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, E.; Wright, R.D.; Jiang, J.; Ray, A.; Moreno, D.; Manley, P.W.; Fabbro, D.; Hall-Meyers, E.; Catley, L.; Podar, K.; et al. Effects of PKC412, nilotinib, and imatinib against GIST-associated PDGFRA mutants with differential imatinib sensitivity. Gastroenterology 2006, 131, 1734–1742. [Google Scholar] [CrossRef]
- Debiec-Rychter, M.; Cools, J.; Dumez, H.; Sciot, R.; Stul, M.; Mentens, N.; Vranckx, H.; Wasag, B.; Prenen, H.; Roesel, J.; et al. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology 2005, 128, 270–279. [Google Scholar] [CrossRef]
- Reichardt, P.; Pink, D.; Lindner, T.; Heinrich, M.C.; Cohen, P.S.; Wang, Y.; Yu, R.; Tsyrlova, A.; Dimitrijevic, S.; Blanke, C. A phase I/II trial of the oral PKC-inhibitor PKC412 (PKC) in combination with imatinib mesylate (IM) in patients (pts) with gastrointestinal stromal tumor (GIST) refractory to IM. J. Clin. Oncol. 2005, 23, 3016. [Google Scholar] [CrossRef]
- Esdar, C.; LInde, N.; Blum, A.; Schieferstein, H.; Drechsler, C.; Sherbetjian, E.; Petersson, C.; Ross, E.; Leuthner, B.; Grädler, U.; et al. M4205 (IDRX-42) is a highly selective and potent inhibitor of relevant oncogenic driver and resistance variants of KIT in cancer. Mol. Cancer Ther. 2025, OF1–OF14. [Google Scholar] [CrossRef]
- De Sutter, L.; Wozniak, A.; Verreet, J.; Vanleeuw, U.; De Cock, L.; Linde, N.; Drechsler, C.; Esdar, C.; Sciot, R.; Schöffski, P. Antitumor Efficacy of the Novel KIT Inhibitor IDRX-42 (Formerly M4205) in Patient- and Cell Line-Derived Xenograft Models of Gastrointestinal Stromal Tumor (GIST). Clin. Cancer Res. 2023, 29, 2859–2868. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhou, Y.; Chen, J.; Wan, X.; Li, N.; Tao, K.; Li, Y.; Wu, X.; Chen, Z.; Liu, L.; et al. Updated efficacy results of olverembatinib (HQP1351) in patients with tyrosine kinase inhibitor (TKI)-resistant succinate dehydrogenase (SDH)-deficient gastrointestinal stromal tumors (GIST) and paraganglioma. J. Clin. Oncol. 2024, 42, 11502. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agent | Key Molecular Targets | Manufacturer | Setting Tested | Current Phase | Recommended Dosage | Frequent Adverse Effects | Efficacy | Safety |
|---|---|---|---|---|---|---|---|---|
| Approved TKIs | ||||||||
| Sunitinib | KIT, PDGFRs, VEGFRs, RTK, FLT3 | Pfizer, New York, NY, USA | Second line | Phase III | 50 mg QD with schedule 4/2 | Hypertension, diarrhea, stomatitis, palmar-plantar erythrodysesthesia syndrome | (PFS) 27.3 weeks | 168 (83%) of any severity grade; 40 (20%) severe AEs |
| Regorafenib | KIT, VEGFR1-3, FGFR, PDGFRA, TIE2, BRAF, RET, RAF, p38 MAPK signaling pathways | Bayer HealthCare Pharmaceuticals, Berlin, Germany | Third line | Phase III | 160 mg QD with schedule 3/1 | Hypertension, abdominal pain, diarrhea, palmar-plantar erythrodysesthesia syndrome | (PFS) 4.8 months | 61 (92.4%) of any grade; 41 (61.4%) severe AEs |
| Ripretinib | KIT, PDGFRA | Deciphera Pharmaceuticals, Waltham, MA, USA | Fourth line | Phase III | 150 mg QD | Fatigue, alopecia, myalgia, constipation | (PFS) 6.3 months | |
| Avapritinib | KIT, PDGFRA D842V | Blueprint Medicines, Cambridge, MA, USA | First line for D842V mutation | Phase III | 300 mg QD | Nausea, fatigue, edema, diarrhea | (PFS) 4.2 months | 221 (92.5%) of any grade; 132 (55.2%) severe AEs |
| Alternative TKIs | ||||||||
| Nilotinib | KIT, PDGFRs | Novartis, Basel, Switzerland | Third line | Phase III | 400 mg BID | Abdominal pain, fatigue, vomiting, anorexia, asthenia | (PFS) 119 days | 242 (97.6%) of any grade; 39 (15.7%) severe AEs |
| Masitinib | KIT, PDFGRs, FAK | AB Science, Paris, France | Second line | Phase III | 12 mg/kg QD | Asthenia, diarrhea, nausea, muscle spasms, cutaneous rash | (RFS) 3.71 months | 22 (96%) of any grade; 12 (52%) severe AEs |
| Sorafenib | KIT, VEGFR, PDGFRB, BRAF | Bayer-Schering Pharma, Berlin, Germany | Third or fourth line | Phase II | 400 mg BID | Hand-foot skin reaction, fatigue, hypertension, abdominal pain | (RFS) 7.2 months | 72% of any grade |
| Dovitinib | KIT, FGFR, VEGFR, PDGFRB | Allarity Therapeutics, Boston, MA, USA | Second line | Phase II | 500 mg QD 5 day-on and 2-day-off | Asthenia, neutropenia, thrombocytopenia, hypertriglyceridemia | (PFS) 4.6 months | 37 (94.8%) of any grade; 25 (64.1%) severe AEs |
| Pazopanib | KIT, VEGFR1-3, PDGFRs | Teva Pharmaceuticals, Tel Aviv, Isreal | Third line | Phase II | 800 mg QD | Hypertension, fatigue, diarrhea, aspartate aminotransferase increase, nausea | (PFS) 3.4 months | 55 (72%) severe AEs |
| Ponatinib | KIT, PDGFRA | Takeda Pharmaceuticals, Tokyo, Japan | At least second line | Phase II | 45 mg QD | Fatigue, myalgia, headache, abdominal pain, hypertension | (PFS) 4 months | |
| Cabozantinib | KIT, VEGFR2, MET, AXL | Exelixis Inc., Alameda, CA, USA | Third line | Phase II | 60 mg QD | Diarrhea, palpar–plantar erythrodysesthesia syndrome, fatigue, hypertension, oral mucositis | (PFS) 5.5 months | 22 (44%) severe AEs |
| Vandetanib | VEGFR2, EGFR | Sanofi Genzyme, Cambridge, MA, USA | First line for SDH-deficient | Phase II | 300 mg QD | Diarrhea, hypertension, seizure, pneumonitis | (PFS) 5.1 months | |
| Dasatinib | KIT, PDGFR, SRC, EPH | Bristol-Myer Squibb, Princeton, NJ, USA | Third line | Phase II | 70 mg QD | Anemia, proteinuria, fatigue, neutropenia, diarrhea | (PFS) 13.6 months | 43% severe AEs |
| Vatalanib | KIT, PDFGRs, VEGFRs | Schering AG, Berlin, Germany | Second or third line | Phase II | 1250 mg QD | Hypertension, nausea, dizziness, proteinuria, abdominal pain, diarrhea | (PFS) 4.5 months | |
| Crenolanib | PDGFR D842V | Arog Pharmaceuticals, Dallas, TX, USA | Second line mainly for D842V mutation | Early-phase | NA | Insufficient information | ||
| Investigational TKIs | ||||||||
| Famitinib | KIT, VEGFR2, FLT3, FLT4 | Jiangsu Hengrui Medicine, Lianyungang, China | NA | Phase II | 25 mg QD | Hypertension, hand-foot skin reaction, oral mucositis, bone marrow suppression, diarrhea, fatigue | (PFS) 31.5 months | |
| Bezuclastinib | KIT | Cogent Biosciences, Waltham, MA, USA | NA | Phase III | NA | Insufficient information | ||
| Motesanib | KIT, VEGFRs, PDGFRs | Amgen, Thousand Oaks, CA, USA | NA | Phase II | NA | Hypertension, diarrhea, fatigue, nausea | (PFS) 16 weeks | 92% of any grade; 52% severe AEs |
| Midostaurin | KIT, VEGFRs, PDGFRs, FLT3 | Novartis, Basel, Switzerland | NA | Early-phase | NA | Nausea, vomiting, diarrhea, fatigue | ||
| IDRX-42 | KIT | GlaxoSmithKline, London, UK | NA | Phase I/II | NA | Insufficient information | ||
| Olverembatinib | FGFR1, VEGFR, HIF-2a | Ascentage Pharma, Suzhou, China | NA | Early-phase | NA | Anemia, neutropenia |
| Agent | Pharmacokinetic Properties | Drug-Drug Interactions | Formulation |
|---|---|---|---|
| Approved TKIs | |||
| Sunitinib | Dose proportional PK, large interpatient PK variability (34–60%), modest intrapatient PK variability (29–52%). Bioavailability = 41–58% Tmax = 6–12 h Protein binding = 95% Distribution volume = 2200 Metabolism by CYP3A4 Clearance = 37.2 L/h | Co-administration with strong CYP3A4 inhibitors increases sunitinib exposure | Hard gelatin capsule or tablet |
| Regorafenib | Tmax = 3–4 h Bioavailability = ~69 for 60 mg; ~83% for 100 mg Protein binding = ~99.5% Food effect: increase 36% low-fat; increase 48% high-fat | Strong CYP3A4 inhibitors are expected to increase regorafenib levels; PPIs influence the bioavailability | Tablet |
| Ripretinib | Tmax = ~4 h Protein binding = 99% Food effect: no clinical change | Strong CYP3A4 inhibitors are expected to increase ripretinib exposure | 50 mg immediate-release oral tablet |
| Avapritinib | Tmax = 2–4.1 h Metabolism majorly by CYP3A4/5; minorly by CYP2C9 | Strong CYP3A4 inhibitors are expected to increase avapritinib exposure | Film-coated oral tablet |
| Alternative TKIs | |||
| Nilotinib | Tmax = ~3 h Bioactivity = 30% Protein binding = ~98% Metabolism majorly by CYP3A4; minorly by CYP2C8 Food effect: increase up to 82% with high-fat meals | Strong CYP3A4 inhibitors are expected to increase nilotinib exposure; Antacids and H2 antagonists affect absorption | Hard gelatin capsule |
| Masitinib | Tmax = ~1.7–4.7 h Bioactivity = ~80% Protein binding = 90–93% Metabolism by CYP3A4/5 and CYP2C8 | Data limited; Potential interactions with organic cation transporters | Immediate-release film-coated tablet |
| Sorafenib | Tmax = ~3 h Protein binding = 99.5–99.7% Metabolism by CYP3A4 and UGT1A9 Food effect: decrease up to 29% with high-fat meals | Co-administration with CYP enzymes may affect metabolism | Film-coated, round, biconvex tablet |
| Dovitinib | Tmax = ~4 h Bioactivity = ~75% Metabolism majorly by CYP-mediated oxidative | Potential interactions with CYP1A2 inhibitors | Capsule |
| Pazopanib | Large interpatient PK variability (36–67%), large intrapatient PK variability (75%). Bioavailability = 14–39% Tmax = 2–4 h Protein binding = >99% Distribution volume = 9–13 Metabolism mainly by CYP3A4, also by CYP1A2 and CYP2C8 Clearance = 0.21–0.35 L/h | Limited data | Film-coated tablet |
| Ponatinib | Tmax = ~5–6 h Protein binding = >99% Metabolism mainly by CYP3A4; minorly by CYP2C8/2D6/3A5, esterases or amidases Food effect: No significant impact | CYP3A inhibitors increase AUC; inducers decrease AUC; acid reducers may decrease BA | Film-coated tablet |
| Cabozantinib | Tmax = ~2–5 h Protein binding = >99.7% Metabolism majorly by CYP3A4; minorly by CYP2C8/2C9/2C19 Food effect: increase up to 41% with high-fat meals | CYP3A4 inhibitors increase AUC; inducers decrease AUC | Film-coated tablet or hard gelatin capsules |
| Vandetanib | Tmax = ~6 h Protein binding = 90–96% Metabolism majorly by CYP3A4 | CYP3A4 inducers decrease exposure; inhibitors minimal effect | Film-coated tablet |
| Dasatinib | Tmax = 0.5–3 h Bioactivity = estimated to be 14–51% Protein binding = ~96% Metabolism majorly by CYP3A4; minorly by FMO-3, UGT Food effect: increase up to 82% with high-fat meals | CYP34A inhibitors increase AUC; inducers decrease AUC | Film-coated tablet |
| Vatalanib | Tmax = ~1.5 h | Limited data | Tablet |
| Crenolanib | Tmax = ~2–4 h Metabolism majorly by CYP3A4 | Significant with CYP3A4 modulators and acid reducers | White to pale-yellow crystalline powder; oral administrated as tablet |
| Investigational TKIs | |||
| Famitinib | Metabolism majorly by CYP3A4 Food effect: Not significant | CYP3A4 inducers decrease exposure; inhibitors increase exposure | Capsule or tablet |
| Bezuclastinib | No Detailed information published | Combination with sunitinib appears well-tolerated | Coated tablet |
| Motesanib | Tmax = ~1–4 h | Act as a CYP3A4 inhibitor and CYP1A2 inducer; increase the metabolism of erlotinib | Film-coated tablet |
| Midostaurin | Tmax = ~1.7 h Protein binding = >99.8% Metabolism majorly by CYP3A4 | CYP3A4 inhibitors increase the AUC; inducers reduce the exposure | 25 mg oral soft gelatin capsule |
| IDRX-42 | No Detailed information published | Limited data | 400 mg capsule or 300 mg tablet |
| Olverembatinib | Tmax = ~4–8 h Bioactivity = 30% Protein binding = ~98% Metabolism majorly by CYP3A4; minorly by CYP2C9 | CYP3A4 inhibitors increase AUC; inducers decrease AUC | Tablet |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, X.-H.; Zhang, Q.-S.; Hu, J.-Y.; Zhang, Y.-X.; Song, H. Tyrosine Kinase Inhibitors for Gastrointestinal Stromal Tumor After Imatinib Resistance. Pharmaceutics 2025, 17, 923. https://doi.org/10.3390/pharmaceutics17070923
Xiao X-H, Zhang Q-S, Hu J-Y, Zhang Y-X, Song H. Tyrosine Kinase Inhibitors for Gastrointestinal Stromal Tumor After Imatinib Resistance. Pharmaceutics. 2025; 17(7):923. https://doi.org/10.3390/pharmaceutics17070923
Chicago/Turabian StyleXiao, Xian-Hao, Qian-Shi Zhang, Ji-Yuan Hu, Yin-Xu Zhang, and He Song. 2025. "Tyrosine Kinase Inhibitors for Gastrointestinal Stromal Tumor After Imatinib Resistance" Pharmaceutics 17, no. 7: 923. https://doi.org/10.3390/pharmaceutics17070923
APA StyleXiao, X.-H., Zhang, Q.-S., Hu, J.-Y., Zhang, Y.-X., & Song, H. (2025). Tyrosine Kinase Inhibitors for Gastrointestinal Stromal Tumor After Imatinib Resistance. Pharmaceutics, 17(7), 923. https://doi.org/10.3390/pharmaceutics17070923