
Advanced QbD-Based Process Optimization of Clopidogrel Tablets with Insights into Industrial Manufacturing Design

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Clopidogrel Tablets
2.3. Physical Characterization of Granules, Blends, and Tablets
2.4. Assessment of Blend Uniformity Using HPLC
2.5. Quantitative Determination of Clopidogrel Content
2.6. Evaluation of Content Uniformity in Tablet Dosage Form
2.7. Drug Release Profiling and Dissolution Testing
3. Results
3.1. Determination of CQA
3.2. Process Parameter Adjustment for Pre-Blending Process
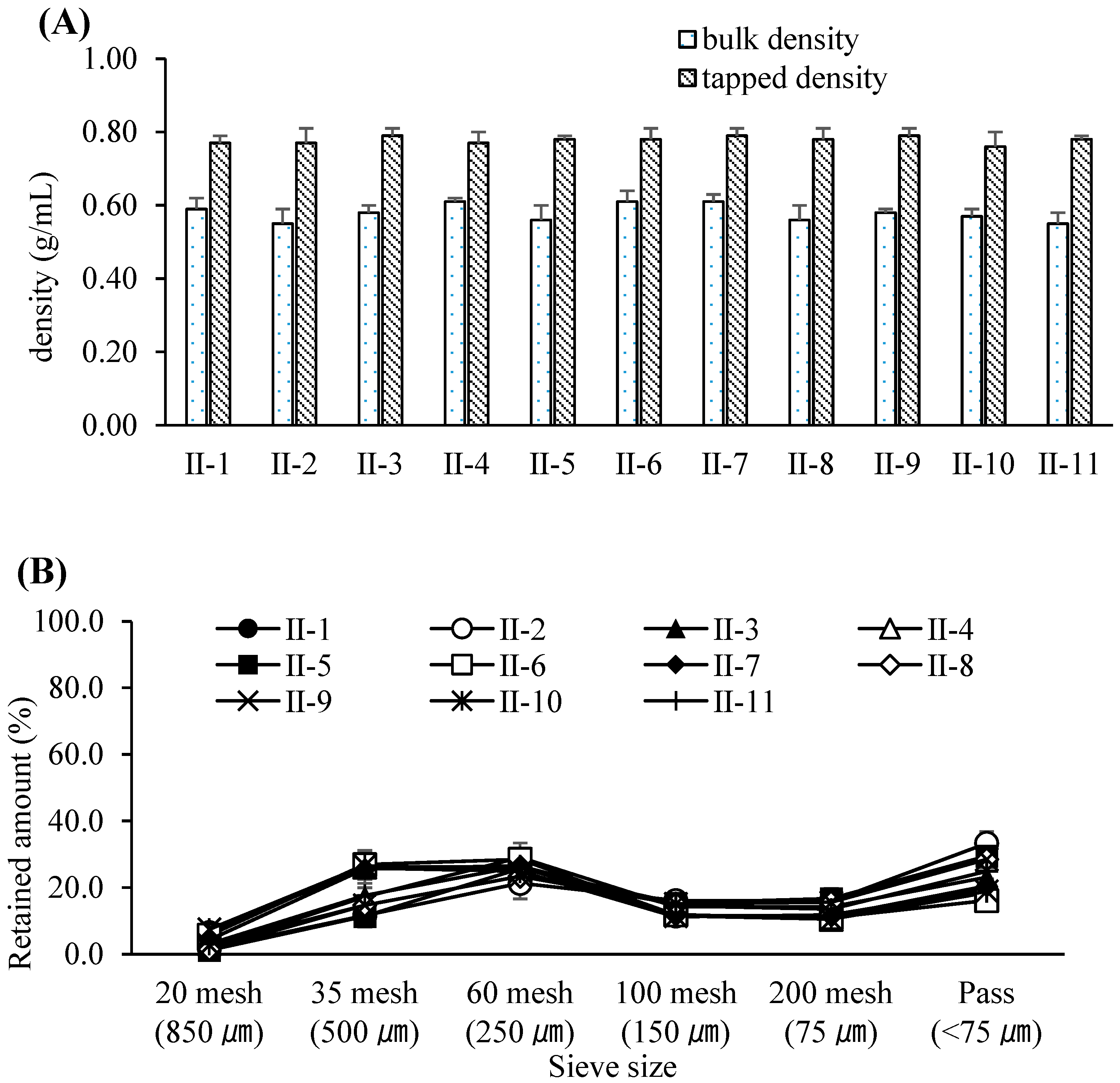
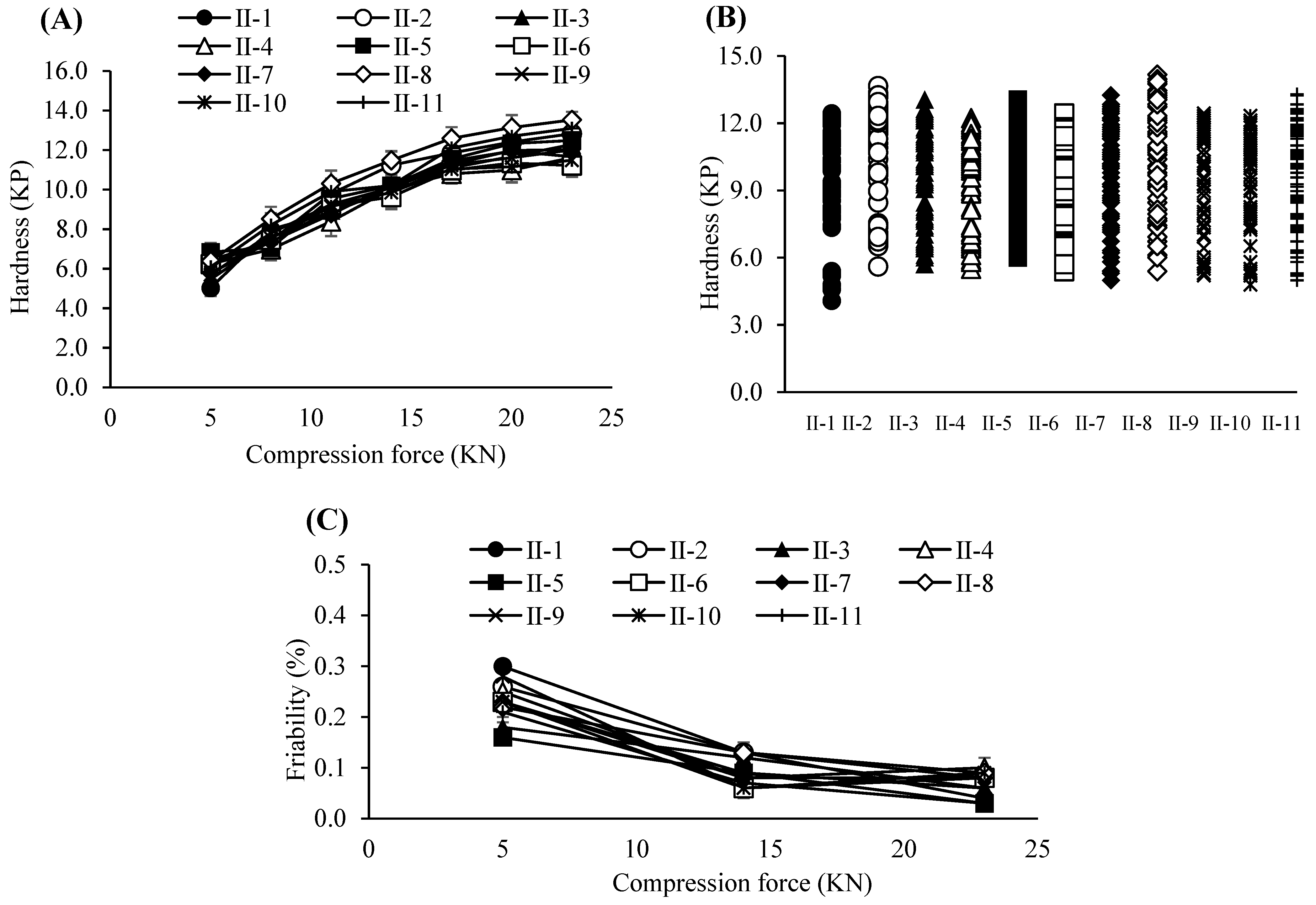
3.3. Optimization of the Dry Granulation Process
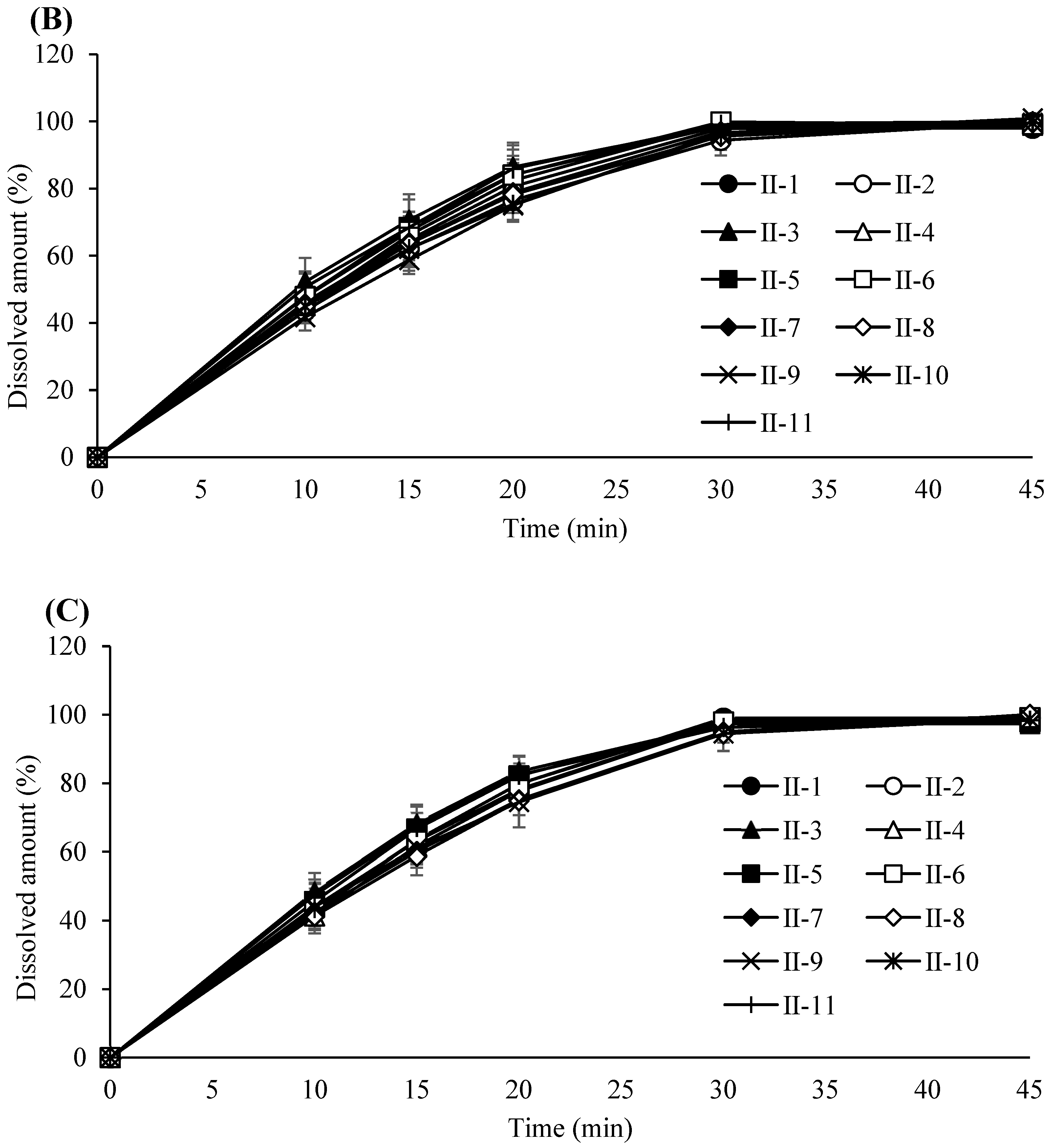
3.4. Evaluation and Refinement of Post-Blending Conditions
3.5. Impact of Lubrication Parameters on Tablet Performance
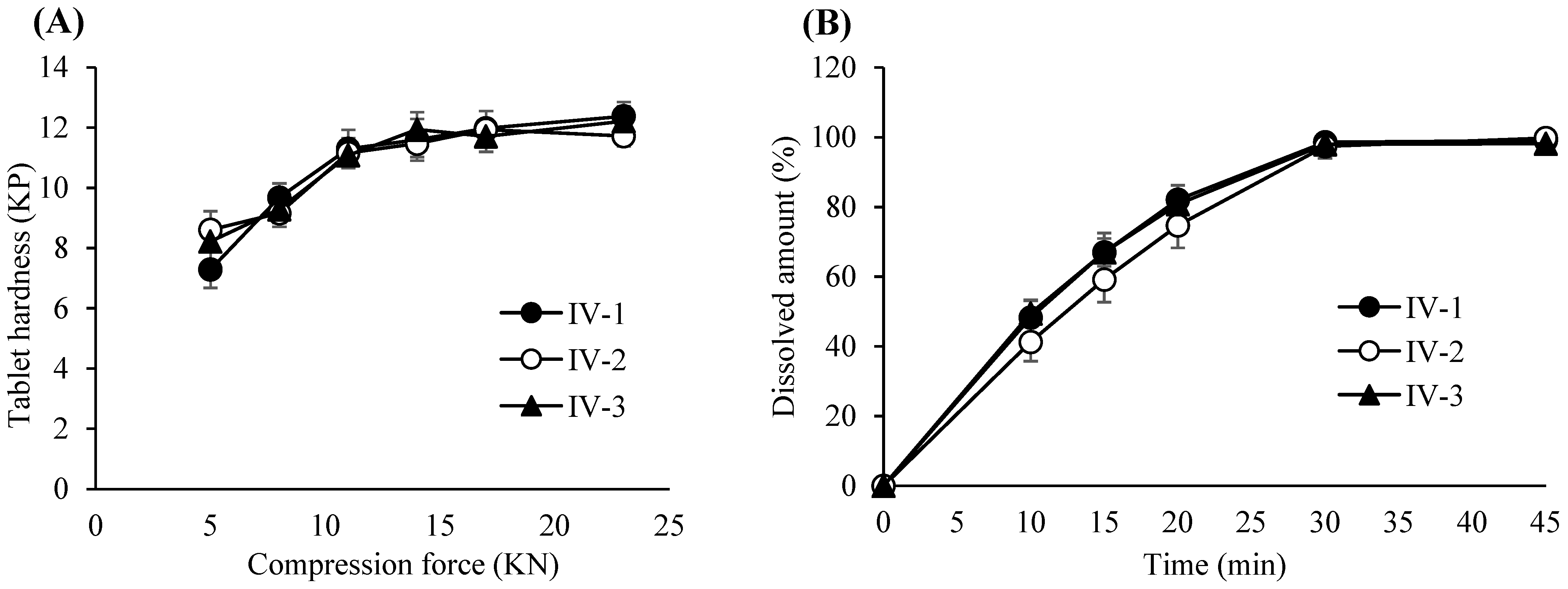
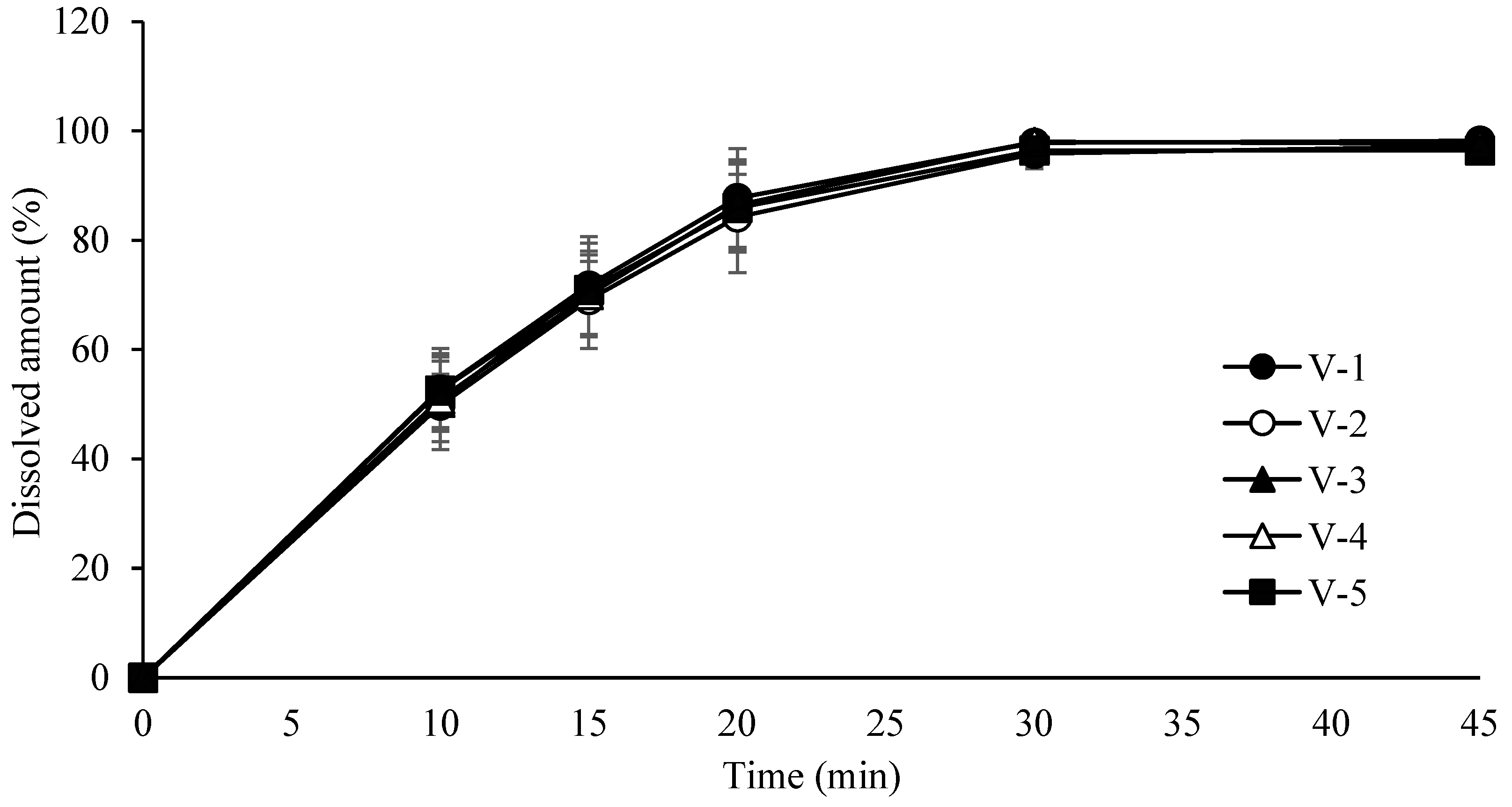
3.6. Compression Process Control for Tablet Robustness
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Grangeia, H.B.; Silva, C.; Simões, S.P.; Reis, M.S. Quality by design in pharmaceutical manufacturing: A systematic review of current status, challenges and future perspectives. Eur. J. Pharm. Biopharm. 2020, 147, 19–37. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.X.; Amidon, G.; Khan, M.A.; Hoag, S.W.; Poli, J.; Raju, G.K.; Woodcock, J. Understanding pharmaceutical Quality by Design. AAPS J. 2014, 16, 771–783. [Google Scholar] [CrossRef]
- ICH. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, Pharmaceutical Development Q8(R2); ICH: Geneva, Switzerland, 2009. [Google Scholar]
- ICH. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, Pharmaceutical Quality System Q10; ICH: Geneva, Switzerland, 2008. [Google Scholar]
- Swain, S.; Parhi, R.; Jena, B.R.; Babu, S.M. Quality by Design: Concept to applications. Curr. Drug Discov. Technol. 2019, 16, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Namjoshi, S.; Dabbaghi, M.; Roberts, M.S.; Grice, J.E.; Mohammed, Y. Quality by Design: Development of the Quality Target Product Profile (QTPP) for semisolid topical products. Pharmaceutics 2020, 12, 287. [Google Scholar] [CrossRef]
- Mohseni-Motlagh, S.F.; Dolatabadi, R.; Baniassadi, M.; Baghani, M. Application of the Quality by Design concept (QbD) in the development of hydrogel-based drug delivery systems. Polymers 2023, 15, 4407. [Google Scholar] [CrossRef]
- Teng, K.; Fu, H.; Wu, G.; Gong, P.; Xie, Y.; Zhou, P.; Gong, X.; Qu, H. QbD-guided traditional Chinese medicine manufacturing process: Development and optimization of fluid-bed granulation and drying processes for Xiaochaihu capsules. AAPS PharmSciTech 2023, 24, 210. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, B.; Liu, X.; Zhang, H.; Yao, J.; Gong, X.; Yan, J. Process optimization for the synthesis of functionalized Au@AgNPs for specific detection of Hg2+ based on Quality by Design (QbD). RSC Adv. 2022, 12, 9121–9129. [Google Scholar] [CrossRef]
- Rampado, R.; Peer, D. Design of experiments in the optimization of nanoparticle-based drug delivery systems. J. Control. Release 2023, 358, 398–419. [Google Scholar] [CrossRef]
- Shukla, A.; Dumpa, N.R.; Thakkar, R.; Shettar, A.; Ashour, E.; Bandari, S.; Repka, M.A. Influence of Poloxamer on the dissolution and stability of hot-melt extrusion-based amorphous solid dispersions using design of experiments. AAPS PharmSciTech 2023, 24, 107. [Google Scholar] [CrossRef]
- Ter Horst, J.P.; Turimella, S.L.; Metsers, F.; Zwiers, A. Implementation of Quality by Design (QbD) principles in regulatory dossiers of medicinal products in the European Union (EU) between 2014 and 2019. Ther. Innov. Regul. Sci. 2021, 55, 583–590. [Google Scholar] [CrossRef]
- Zagalo, D.M.; Sousa, J.; Simões, S. Quality by Design (QbD) approach in marketing authorization procedures of non-biological complex drugs: A critical evaluation. Eur. J. Pharm. Biopharm. 2022, 178, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Chaurasia, A.; Gupta, N.; Rajput, D.S. Effect of formulation parameters on enalapril maleate mucoadhesive buccal tablet using Quality by Design (QbD) approach. Zhongguo Ying Yong Sheng Li Xue Za Zhi 2024, 40, e20240003. [Google Scholar] [CrossRef] [PubMed]
- Gurba-Bryśkiewicz, L.; Maruszak, W.; Smuga, D.A.; Dubiel, K.; Wieczorek, M. Quality by Design (QbD) and design of experiments (DOE) as a strategy for tuning lipid nanoparticle formulations for RNA delivery. Biomedicines 2023, 11, 2752. [Google Scholar] [CrossRef]
- Osmanović Omerdić, E.; Alagić-Džambić, L.; Krstić, M.; Pašić-Kulenović, M.; Medarević, Đ.; Ivković, B.; Vasiljević, D. Long-term stability of clopidogrel solid dispersions—Importance of in vitro dissolution test. PLoS ONE 2022, 17, e0266237. [Google Scholar] [CrossRef]
- Singh, A.; Sharma, R.; Chaudhary, S.; Rana, V. Preparation and characterization of clopidogrel bisulfate-hydroxypropyl-β-cyclodextrin mixed inclusion complex for improved intestinal solubility and anti-thrombotic efficacy. J. Pharm. Sci. 2023, 112, 166–181. [Google Scholar] [CrossRef]
- Correa Soto, C.E.; Gao, Y.; Indulkar, A.S.; Ueda, K.; Zhang, G.G.Z.; Taylor, L.S. Impact of surfactants on the performance of clopidogrel-copovidone amorphous solid dispersions: Increased drug loading and stabilization of nanodroplets. Pharm. Res. 2022, 39, 167–188. [Google Scholar] [CrossRef]
- Shekhawat, P.; Pokharkar, V. Risk assessment and QbD based optimization of an Eprosartan mesylate nanosuspension: In-vitro characterization, PAMPA and in-vivo assessment. Int. J. Pharm. 2019, 567, 118415. [Google Scholar] [CrossRef] [PubMed]
- Oh, G.H.; Kim, J.E.; Park, Y.J. Development of stabilized tenofovir disoproxil tablet: Degradation profile, stabilization, and bioequivalence in beagle dogs. Drug Dev. Ind. Pharm. 2018, 44, 757–766. [Google Scholar] [CrossRef]
- Reddy, J.P.; Phanse, R.; Nesarikar, V. Parameter estimation for roller compaction process using an instrumented Vector TF mini roller compactor. Pharm. Dev. Technol. 2019, 24, 1250–1257. [Google Scholar] [CrossRef]
- Guo, Y.; Martinez, L.; Palanisamy, A.; Gururajan, B.; Sun, C.C. An evaluation of six techniques for measuring porosity of ribbons produced by roller compaction. Int. J. Pharm. 2024, 667 Pt A, 124855. [Google Scholar] [CrossRef]
- Wang, C.; Wang, Z.; Friedrich, A.; Sun, C.C. Effect of deaeration on processability of poorly flowing powders by roller compaction. Int. J. Pharm. 2022, 621, 121803. [Google Scholar] [CrossRef]
- Panda, B.K.; Chellampillai, B.; Ghodake, S.; Mali, A.J.; Kamble, R. Investigation of magnesium aluminometasilicate (Neusilin US2) based surface solid dispersion of sorafenib tosylate using QbD approach: In vitro and in vivo pharmacokinetic study. ADMET DMPK 2024, 12, 687–702. [Google Scholar] [PubMed]
- Alshaer, W.; Nsairat, H.; Lafi, Z.; Hourani, O.M.; Al-Kadash, A.; Esawi, E.; Alkilany, A.M. Quality by Design approach in liposomal formulations: Robust product development. Molecules 2022, 28, 10. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Bai, Y.; Liu, J.; Wang, Y.; He, Q.; Zhang, X.; Cheng, F.; Xu, M.; Mao, Q.; Liang, Z. Research progress on the quality control of mRNA vaccines. Expert Rev. Vaccines 2024, 23, 570–583. [Google Scholar] [CrossRef]
- Pourseif, M.M.; Masoudi-Sobhanzadeh, Y.; Azari, E.; Parvizpour, S.; Barar, J.; Ansari, R.; Omidi, Y. Self-amplifying mRNA vaccines: Mode of action, design, development and optimization. Drug Discov. Today 2022, 27, 103341. [Google Scholar] [CrossRef]
- Birla, D.; Khandale, N.; Bashir, B.; ShahbazAlam, M.; Vishwas, S.; Gupta, G.; Dureja, H.; Kumbhar, P.S.; Disouza, J.; Patravale, V.; et al. Application of quality by design in optimization of nanoformulations: Principle, perspectives and practices. Drug Deliv. Transl. Res. 2025, 15, 798–830. [Google Scholar] [CrossRef]
- Kapoor, D.; Sharma, S.; Verma, K.; Bisht, A.; Sharma, M.; Singhai, N.J.; Raval, N.; Maheshwari, R. Quality-by-design-based engineered liposomal nanomedicines to treat cancer: An in-depth analysis. Nanomedicine 2022, 17, 1173–1189. [Google Scholar] [CrossRef] [PubMed]
- Uner, B.; Ozdemir, S.; Tas, C.; Uner, M.; Ozsoy, Y. Loteprednol-loaded nanoformulations for corneal delivery by Quality-by-Design concepts: Optimization, characterization, and anti-inflammatory activity. AAPS PharmSciTech 2023, 24, 92. [Google Scholar] [CrossRef]
- Ralbovsky, N.M.; Smith, J.P. Process analytical technology and its recent applications for asymmetric synthesis. Talanta 2023, 252, 123787. [Google Scholar] [CrossRef]
- Zhong, L.; Gao, L.; Li, L.; Zang, H. Trends—Process analytical technology in solid oral dosage manufacturing. Eur. J. Pharm. Biopharm. 2020, 153, 187–199. [Google Scholar] [CrossRef]
- Su, Q.; Bommireddy, Y.; Shah, Y.; Ganesh, S.; Moreno, M.; Liu, J.; Gonzalez, M.; Yazdanpanah, N.; O’Connor, T.; Reklaitis, G.V.; et al. Data reconciliation in the Quality-by-Design (QbD) implementation of pharmaceutical continuous tablet manufacturing. Int. J. Pharm. 2019, 563, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Vo, A.Q.; Kutz, G.; He, H.; Narala, S.; Bandari, S.; Repka, M.A. Continuous manufacturing of ketoprofen delayed-release pellets using melt extrusion technology: Application of QbD design space, inline near infrared, and inline pellet size analysis. J. Pharm. Sci. 2020, 109, 3598–3607. [Google Scholar] [CrossRef]
- Fiedler, D.; Fink, E.; Aigner, I.; Leitinger, G.; Keller, W.; Roblegg, E.; Khinast, J.G. A multi-step machine learning approach for accelerating QbD-based process development of protein spray drying. Int. J. Pharm. 2023, 642, 123133. [Google Scholar] [CrossRef]
- Munir, N.; Nugent, M.; Whitaker, D.; McAfee, M. Machine learning for process monitoring and control of hot-melt extrusion: Current state of the art and future directions. Pharmaceutics 2021, 13, 1432. [Google Scholar] [CrossRef] [PubMed]
- Jakubowska, E.; Ciepluch, N. Blend segregation in tablets manufacturing and its effect on drug content uniformity—A review. Pharmaceutics 2021, 13, 1909. [Google Scholar] [CrossRef] [PubMed]
- Pathodiya, M. Cross-contamination risk assessment using FMEA tool. J. Drug Deliv. Ther. 2024, 14, 164. [Google Scholar] [CrossRef]
- Remy, B.; Glasser, B.J.; Khinast, J.G. The effect of mixer properties and fill level on granular flow in a bladed mixer. AIChE J. 2009, 55, 2804–2818. [Google Scholar] [CrossRef]
- Razavi, S.M.; Scicolone, J.; Snee, R.D.; Kumar, A.; Bertels, J.; Cappuyns, P.; Van Assche, I.; Cuitiño, A.M.; Muzzio, F. Prediction of tablet weight variability in continuous manufacturing. Int. J. Pharm. 2020, 575, 118727. [Google Scholar] [CrossRef]
- Goodwin, D.J.; van den Ban, S.; Denham, M.; Barylski, I. Real time release testing of tablet content and content uniformity. Int. J. Pharm. 2018, 537, 183–192. [Google Scholar] [CrossRef]
- Arndt, O.-R.; Baggio, R.; Adam, A.K.; Harting, J.; Franceschinis, E.; Kleinebudde, P. Impact of different dry and wet granulation techniques on granule and tablet properties: A comparative study. J. Pharm. Sci. 2018, 107, 3143–3152. [Google Scholar] [CrossRef]
- Han, J.K.; Shin, B.S.; Choi, D.H. Comprehensive study of intermediate and critical quality attributes for process control of high-shear wet granulation using multivariate analysis and the Quality by Design approach. Pharmaceutics 2019, 11, 252. [Google Scholar] [CrossRef] [PubMed]
- Borchert, D.; Zahel, T.; Thomassen, Y.E.; Herwig, C.; Suarez-Zuluaga, D.A. Quantitative CPP evaluation from risk assessment using integrated process modeling. Bioengineering 2019, 6, 114. [Google Scholar] [CrossRef]
- Atanaskova, E.; Kostovski, D.; Anevska-Stojanovska, N. Investigation of the influence of critical process parameters in roller compaction process on physical properties of granules and tablets using design of experiments. Arh. Farm. 2020, 70, 34–53. [Google Scholar] [CrossRef]
- Hudson-Curtis, B.; Novick, S. Assessing content uniformity. In Nonclinical Statistics for Pharmaceutical and Biotechnology Industries; Springer: Cham, Switzerland, 2016; pp. 631–651. [Google Scholar]
- Patel, S.; Kaushal, A.M.; Bansal, A.K. Compression physics in the formulation development of tablets. Crit. Rev. Ther. Drug Carrier Syst. 2006, 23, 1–66. [Google Scholar] [CrossRef]
- Alyami, H.; Dahmash, E.; Bowen, J.; Mohammed, A.R. An investigation into the effects of excipient particle size, blending techniques and processing parameters on the homogeneity and content uniformity of a blend containing low-dose model drug. PLoS ONE 2017, 12, e0178772. [Google Scholar] [CrossRef]
- Bekaert, B.; Grymonpré, W.; Novikova, A.; Vervaet, C.; Vanhoorne, V. Impact of blend properties and process variables on the blending performance. Int. J. Pharm. 2022, 613, 121421. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Sun, C.C. Systematic evaluation of common lubricants for optimal use in tablet formulation. Eur. J. Pharm. Sci. 2018, 117, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Jiwa, N.; Ozalp, Y.; Yegen, G.; Aksu, B. Critical tools in tableting research: Using compaction simulator and Quality by Design (QbD) to evaluate lubricants’ effect in direct compressible formulation. AAPS PharmSciTech 2021, 22, 151. [Google Scholar] [CrossRef]
- Matji, A.; Donato, N.; Gagol, A.; Morales, E.; Carvajal, L.; Serrano, D.R.; Worku, Z.A.; Healy, A.M.; Torrado, J.J. Predicting the critical quality attributes of ibuprofen tablets via modelling of process parameters for roller compaction and tabletting. Int. J. Pharm. 2019, 565, 209–218. [Google Scholar] [CrossRef]
- Van Snick, B.; Grymonpré, W.; Dhondt, J.; Pandelaere, K.; Di Pretoro, G.; Remon, J.P.; De Beer, T.; Vervaet, C.; Vanhoorne, V. Impact of blend properties on die filling during tableting. Int. J. Pharm. 2018, 549, 476–488. [Google Scholar] [CrossRef]
- Osei-Yeboah, F.; Sun, C.C. Validation and applications of an expedited tablet friability method. Int. J. Pharm. 2015, 484, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Schomberg, A.K.; Kwade, A.; Finke, J.H. The challenge of die filling in rotary presses—A systematic study of material properties and process parameters. Pharmaceutics 2020, 12, 248. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Application Area | QbD Concept Applied | Key Emphasis of QbD | Reference |
|---|---|---|---|
| Amorphous solid dispersion (ASD) formulation development | Optimization of process parameters and enhancement of dissolution performance | Improved solubility and stability of the final formulation | [11] |
| Solid dispersion of sorafenib tosylate (SFN) | Design of experiment (DoE)-based optimization for enhanced pharmacokinetics | Over 20-fold improvement in solubility and oral bioavailability | [24] |
| Liposomal nano-drug delivery systems | Systematic experimental design and risk assessment | Enhanced reproducibility and efficiency in complex nanoformulation development | [25] |
| mRNA vaccine development | Establishment of standardized quality control protocols | Improved process control and impurity profiling for high-quality mRNA vaccines | [26] |
| Self-amplifying mRNA (saRNA) vaccines | Optimization of stability, immunogenicity, and safety | Enhanced antigen expression and immune modulation | [27] |
| Nanoformulation-based drug delivery systems (NDDSs) | Optimization of critical parameters such as ZP 3, particle size, EE 4, and PDI 5 | Improved quality and reliability in nanoparticle manufacturing | [28] |
| Nanoparticle liposomes for anticancer therapy | Evaluation of formulation variables including drug–lipid ratio and processing conditions | Achievement of predefined quality attributes for enhanced therapeutic performance | [29] |
| Ophthalmic delivery using SLN 1, NLC 2, and nanoemulsion systems | D-optimal mixture design and physicochemical characterization | Anti-inflammatory efficacy and robust formulation development | [30] |
| Manufacturing Process | Ingredients | mg/T |
|---|---|---|
| Pre-blending and roller compaction | Clopidogel napadisilate—loaded solid dispersion | 171.2 1 |
| Fumaric acid | 35.0 | |
| Crospovidone | 25.0 | |
| Copovidone | 5.0 | |
| Colloidal silica | 5.0 | |
| Post-blending | D-mannitol | 31.8 |
| Crospovidone | 10.0 | |
| Colloidal silica | 12.5 | |
| Lubrication | Sucrose esters of fatty acids | 9.5 |
| Total weight | 305.0 |
| Critical Quality Attribute (CQA) | Specification |
|---|---|
| Appearance | Off-white round tablet |
| Identification | Retention time of standard solution and test solution is the same |
| Drug content | 95.0–105.0% of label claim |
| Uniformity of drug content | Acceptance value within 15.0% |
| Drug release | Not less than 80.0% in 30 min |
| Batch | I-1 | I-2 | I-3 |
|---|---|---|---|
| Blending speed (rpm) | 17 | 17 | 17 |
| Blending time (min) | 9 | 11 | 13 |
| Blending revolution number (rev.) * | 153 | 187 | 221 |
| Batch | I-1 | I-2 | I-3 | |
|---|---|---|---|---|
| Blend uniformity (10 locations, %) | Average | 99.3 | 99.4 | 98.7 |
| RSD | 1.6 | 1.3 | 1.2 | |
| Min | 98.0 | 98.1 | 97.7 | |
| Max | 101.2 | 100.5 | 99.9 | |
| Batch | II-1 | II-2 | II-3 | II-4 | II-5 | II-6 | II-7 | II-8 | II-9 | II-10 | II-11 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Roller pressure (MPa) | 9 | 9 | 8 | 9 | 7 | 9 | 8 | 7 | 7 | 7 | 8 |
| Roller speed (rpm) | 20 | 30 | 25 | 20 | 20 | 30 | 25 | 30 | 20 | 30 | 25 |
| Roller gap (mm) | 3.0 | 3.0 | 2.5 | 2.0 | 3.0 | 2.0 | 2.5 | 3.0 | 2.0 | 2.0 | 2.5 |
| Batch | II-1 | II-2 | II-3 | II-4 | II-5 | II-6 | II-7 | II-8 | II-9 | II-10 | II-11 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Blend uniformity (10 locations, %) | Average | 101.0 | 99.8 | 99.7 | 99.8 | 101.0 | 100.4 | 99.8 | 101.0 | 101.3 | 100.2 | 100.1 |
| RSD | 1.0 | 1.2 | 1.8 | 1.7 | 1.4 | 0.8 | 0.7 | 1.1 | 0.9 | 0.8 | 0.9 | |
| Min | 99.9 | 97.0 | 96.8 | 96.9 | 98.4 | 99.0 | 98.5 | 99.0 | 100.2 | 98.6 | 98.8 | |
| Max | 103.2 | 101.2 | 102.5 | 101.8 | 102.9 | 101.3 | 100.7 | 102.2 | 102.8 | 101.0 | 101.2 | |
| Content uniformity (10 tablets, %) | Average | 101.2 | 102.0 | 101.6 | 101.0 | 99.5 | 102.5 | 102.5 | 103.1 | 100.9 | 100.5 | 101.5 |
| SD | 0.5 | 1.0 | 0.6 | 0.7 | 0.5 | 0.9 | 0.7 | 1.0 | 0.9 | 1.3 | 0.7 | |
| Acceptance value | 1.2 | 2.9 | 1.5 | 1.7 | 1.2 | 3.2 | 2.7 | 4.0 | 2.2 | 3.1 | 1.7 | |
| Batch | III-1 | III-2 | III-3 |
|---|---|---|---|
| Blending speed (rpm) | 17 | 17 | 17 |
| Blending time (min) | 9 | 11 | 13 |
| Blending revolution number (rev.) * | 153 | 187 | 221 |
| Batch | I-1 | I-2 | I-3 | |
|---|---|---|---|---|
| Blend uniformity (10 locations, %) | Average | 99.8 | 99.4 | 99.1 |
| RSD | 1.2 | 0.7 | 0.8 | |
| Min | 98.0 | 98.1 | 97.7 | |
| Max | 101.2 | 100.5 | 99.9 | |
| Batch | IV-1 | IV-2 | IV-3 |
|---|---|---|---|
| Lubrication speed (rpm) | 17 | 17 | 17 |
| Lubrication time (min) | 3 | 4 | 5 |
| Lubrication revolution number (rev.) * | 51 | 68 | 85 |
| Batch | V-1 | V-2 | V-3 | V-4 | V-5 |
|---|---|---|---|---|---|
| Turret speed (rpm) | 15 | 25 | 35 | 15 | 35 |
| Feeder speed (rpm) | 60 | 80 | 100 | 100 | 60 |
| Batch | V-1 | V-2 | V-3 | V-4 | V-5 |
|---|---|---|---|---|---|
| Tablet weight (n = 20, mg) | 303.3 ±2.7 | 305.1 ±3.7 | 304.7 ±3.7 | 307.5 ±2.1 | 305.3 ±4.1 |
| Assay (n = 3, %) | 99.9 ±0.5 | 100.0 ±0.3 | 98.6 ±0.3 | 99.4 ±0.2 | 98.5 ±0.4 |
| Content uniformity (n = 10, %) | 100.5 ±1.7 | 99.5 ±1.5 | 99.4 ±1.2 | 99.2 ±1.7 | 98.9 ±1.0 |
| Content uniformity acceptance value (%) | 4.1 | 3.6 | 2.9 | 4.1 | 4.1 |
| Tablet hardness (n = 10, KP) | 9.7 ±1.0 | 9.9 ±1.3 | 10.0 ±1.4 | 9.8 ±0.9 | 10.4 ±1.4 |
| Tablet friability (n = 3, %) | 0.18 ±0.01 | 0.19 ±0.02 | 0.20 ±0.01 | 0.20 ±0.01 | 0.18 ±0.02 |
| Process Step | Critical Process Parameter (CPP) | Proven Acceptance Range (PAR) | Set Point |
|---|---|---|---|
| Pre-blending | Blending time | 9–13 min | 11 min |
| Roller compaction | Roller speed | 20–30 rpm | 25 rpm |
| Roller pressure | 7–9 MPa | 8 MPa | |
| Roller gap | 2.0–3.0 mm | 2.5 mm | |
| Post-blending | Blending time | 9–13 min | 11 min |
| Lubrication | Lubrication time | 3–5 min | 4 min |
| Compression | Turret speed | 15–35 rpm | 25 rpm |
| Feeder speed | 60–100 rpm | 80 rpm |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bak, Y.W.; Woo, M.R.; Cho, H.J.; Kwon, T.K.; Im, H.T.; Cho, J.H.; Choi, H.-G. Advanced QbD-Based Process Optimization of Clopidogrel Tablets with Insights into Industrial Manufacturing Design. Pharmaceutics 2025, 17, 659. https://doi.org/10.3390/pharmaceutics17050659
Bak YW, Woo MR, Cho HJ, Kwon TK, Im HT, Cho JH, Choi H-G. Advanced QbD-Based Process Optimization of Clopidogrel Tablets with Insights into Industrial Manufacturing Design. Pharmaceutics. 2025; 17(5):659. https://doi.org/10.3390/pharmaceutics17050659
Chicago/Turabian StyleBak, Young Woo, Mi Ran Woo, Hyuk Jun Cho, Taek Kwan Kwon, Ho Teak Im, Jung Hyun Cho, and Han-Gon Choi. 2025. "Advanced QbD-Based Process Optimization of Clopidogrel Tablets with Insights into Industrial Manufacturing Design" Pharmaceutics 17, no. 5: 659. https://doi.org/10.3390/pharmaceutics17050659
APA StyleBak, Y. W., Woo, M. R., Cho, H. J., Kwon, T. K., Im, H. T., Cho, J. H., & Choi, H.-G. (2025). Advanced QbD-Based Process Optimization of Clopidogrel Tablets with Insights into Industrial Manufacturing Design. Pharmaceutics, 17(5), 659. https://doi.org/10.3390/pharmaceutics17050659