
PK/PD of Positively Charged ADC in Mice



Abstract
1. Introduction
2. Materials and Methods
2.1. Production and Characterization of Wild-Type (WT) and Positively Charged mAb
2.2. Synthesis and Characterization of WT and Positively Charged ADCs
2.3. In Vitro Cytotoxicity of Positively Charged ADC
2.4. In Vivo Biodistribution Study of Positively Charged ADC
2.4.1. Development of Xenograft Mouse Model
2.4.2. Pharmacokinetics of Positively Charged ADC in Plasma, Tumor, and Tissues
2.5. Bioanalytical Methods
2.5.1. Sample Pretreatment
2.5.2. ELISA for Total mAb Quantification
2.5.3. LC–MS/MS for Unconjugated MMAE Quantification
2.5.4. Papain Deconjugation for Total MMAE Quantification
2.6. In Vivo Efficacy Study and Toxicity Assessment of Positively Charged ADC
3. Results
3.1. Synthesis and Characterization of Positively Charged ADCs
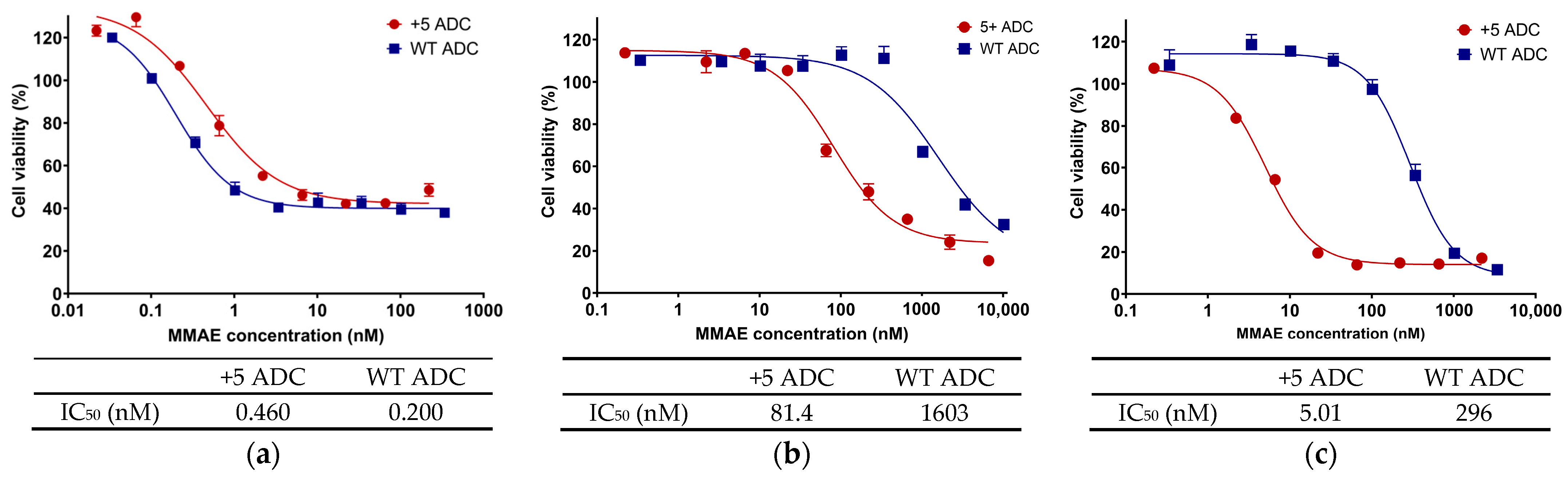
3.2. In Vitro Cytotoxicity of Positively Charged ADC
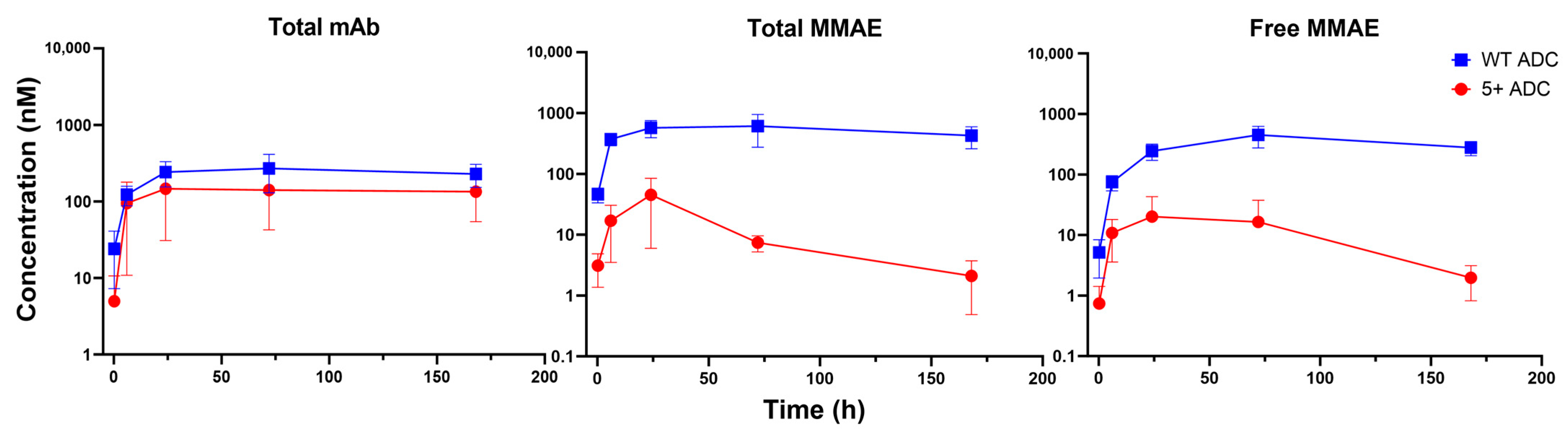
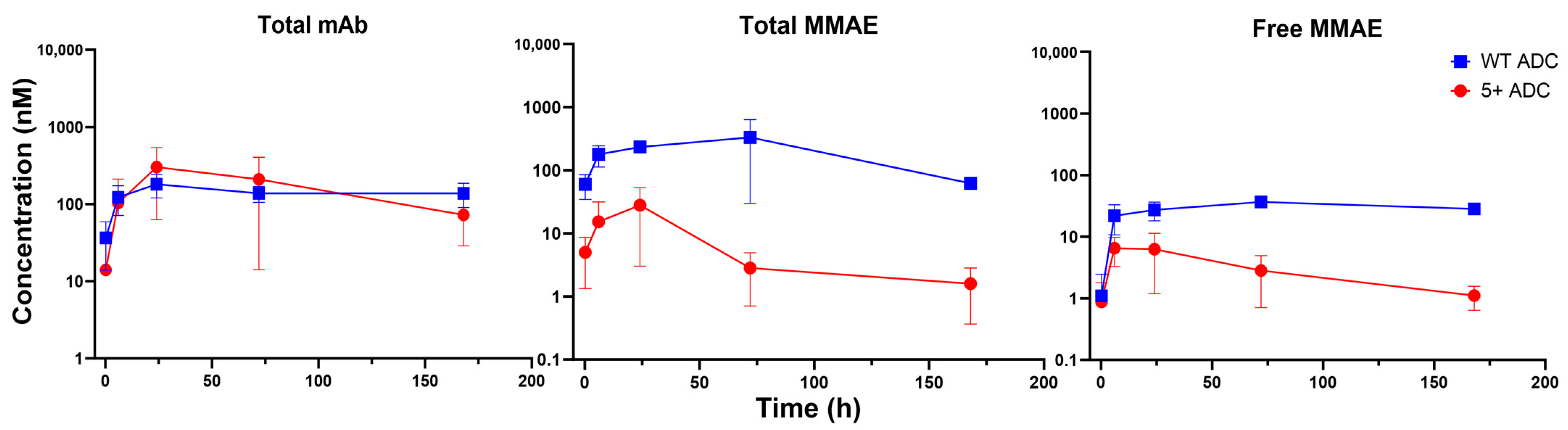
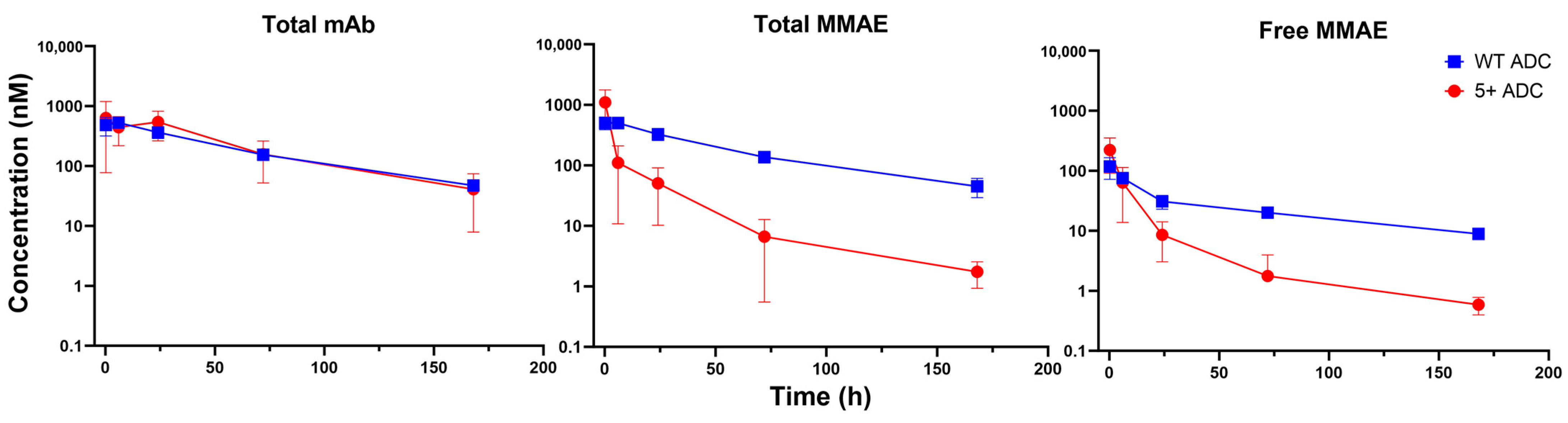
3.3. PK of Positively Charged ADC in Plasma
3.4. PK of Positively Charged ADC in Tumors
3.5. PK of Positively Charged ADC in Tissues
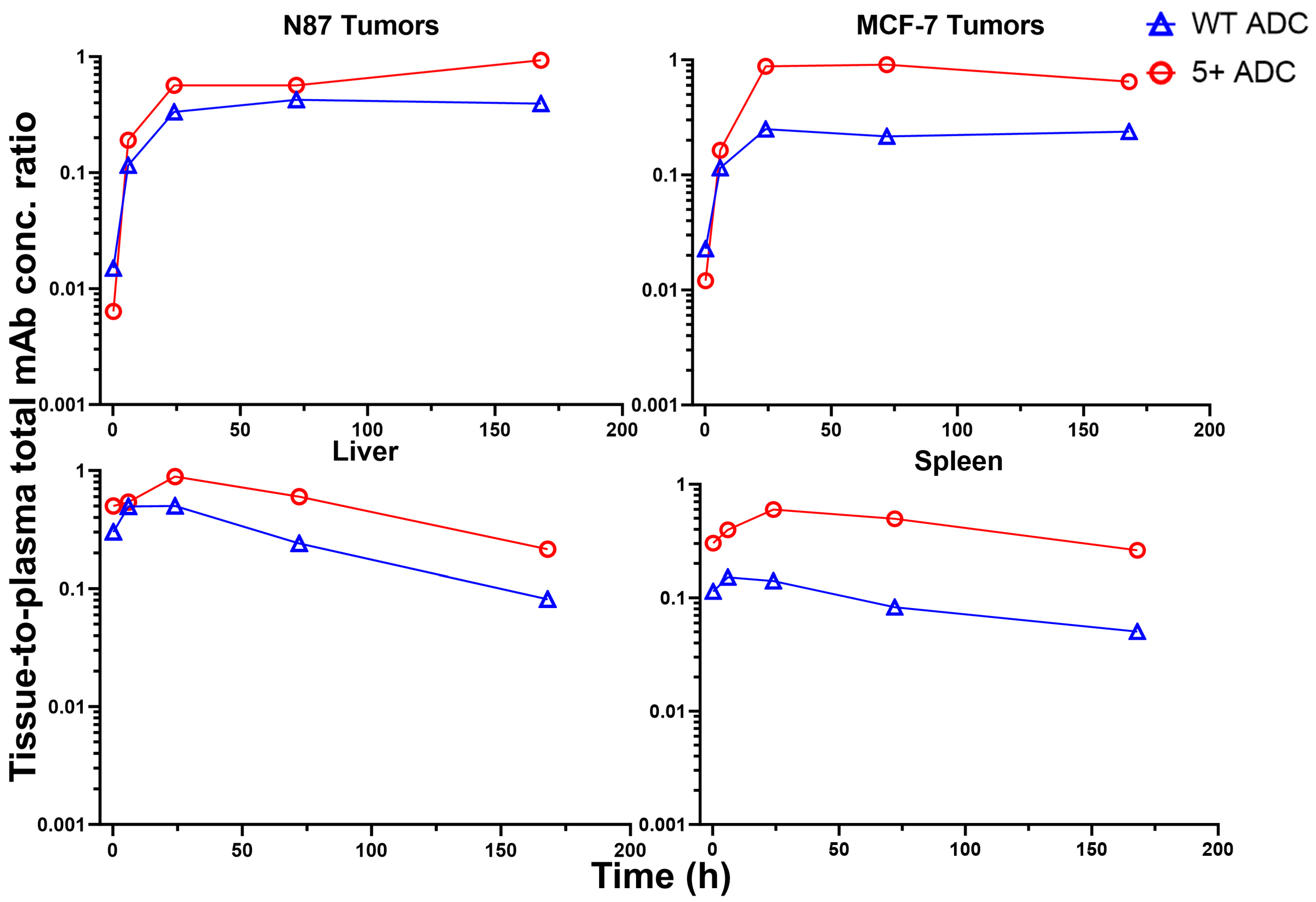
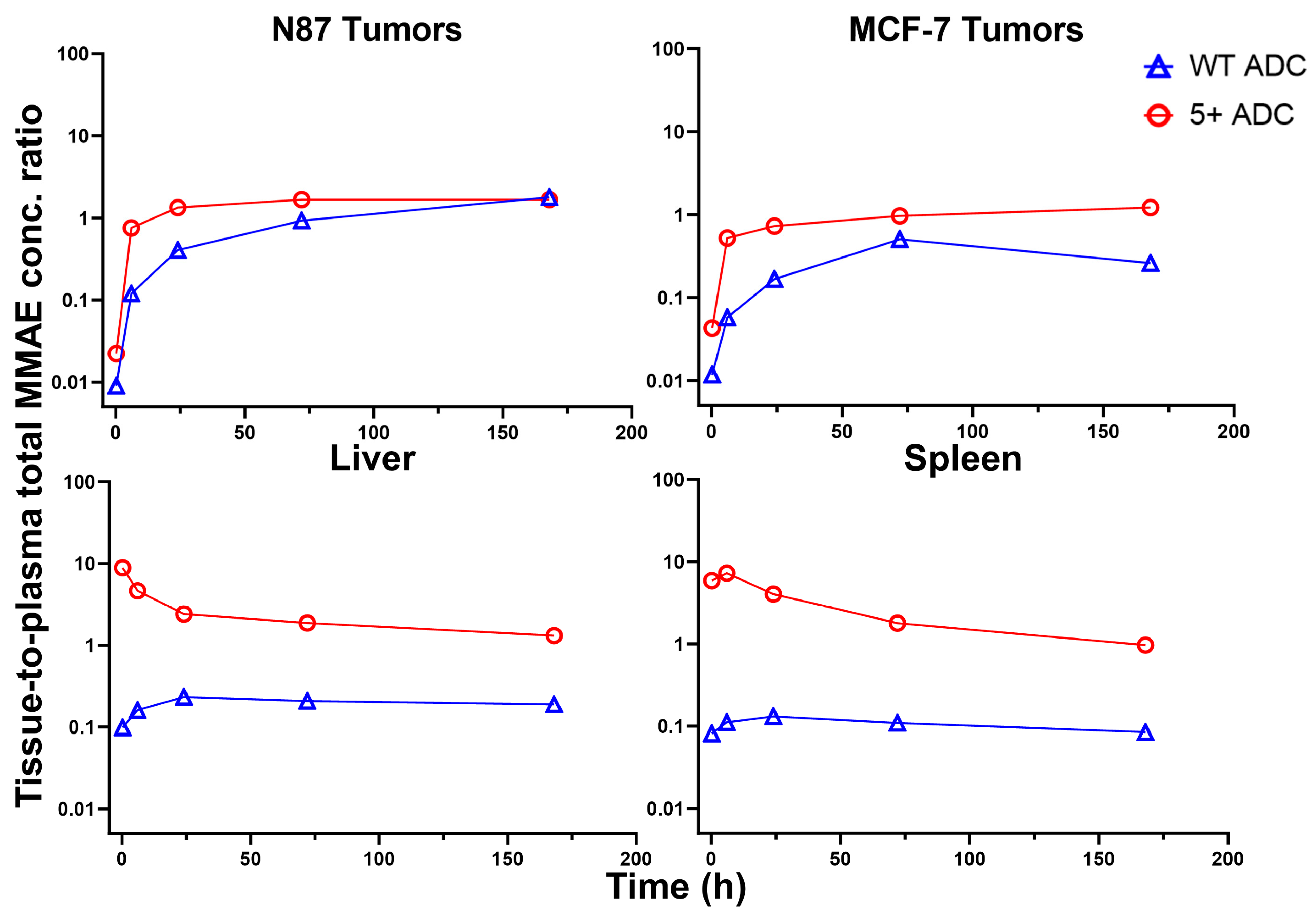
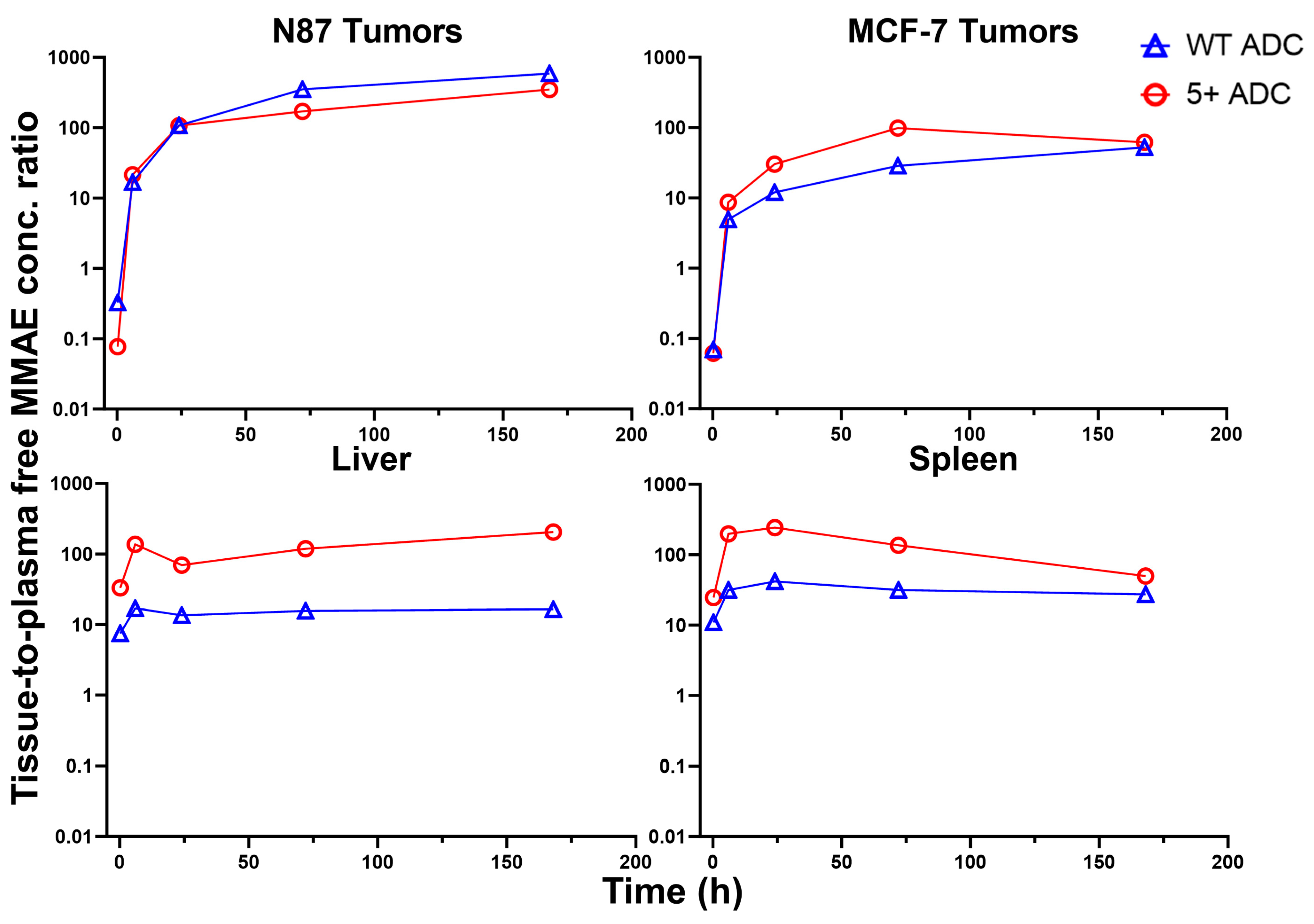
3.6. Tissue-to-Plasma Exposure Ratios
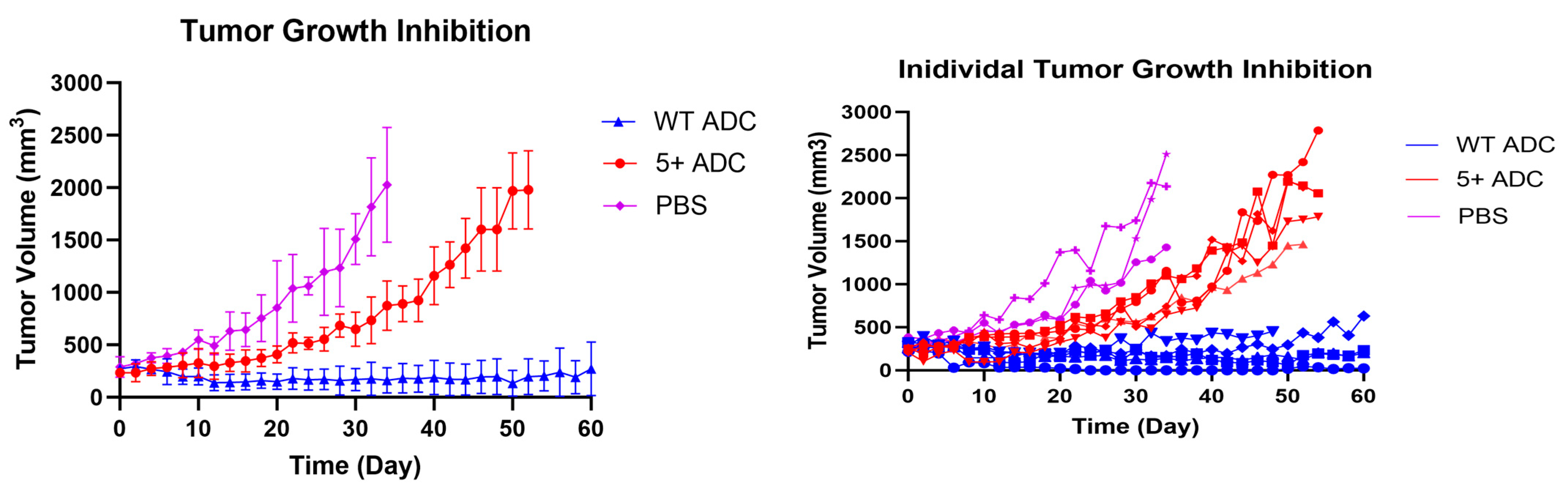
3.7. In Vivo Efficacy Study and Toxicity Assessment of Positively Charged ADC
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, W.Q.; Guo, H.F.; Li, L.Y.; Zhang, Y.F.; Cui, J.W. The promising role of antibody drug conjugate in cancer therapy: Combining targeting ability with cytotoxicity effectively. Cancer Med. 2021, 10, 4677–4696. [Google Scholar] [CrossRef]
- Liu, K.; Li, M.; Li, Y.; Li, Y.; Chen, Z.; Tang, Y.; Yang, M.; Deng, G.; Liu, H. A review of the clinical efficacy of FDA-approved antibody‒drug conjugates in human cancers. Mol. Cancer 2024, 23, 62. [Google Scholar] [CrossRef] [PubMed]
- McKertish, C.M.; Kayser, V. Advances and Limitations of Antibody Drug Conjugates for Cancer. Biomedicines 2021, 9, 872. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody drug conjugate: The “biological missile” for targeted cancer therapy. Signal Transduct. Target. Ther. 2022, 7, 93. [Google Scholar] [CrossRef] [PubMed]
- Ovacik, M.; Lin, K. Tutorial on Monoclonal Antibody Pharmacokinetics and Its Considerations in Early Development. Clin. Transl. Sci. 2018, 11, 540–552. [Google Scholar] [CrossRef]
- Boswell, C.A.; Tesar, D.B.; Mukhyala, K.; Theil, F.-P.; Fielder, P.J.; Khawli, L.A. Effects of Charge on Antibody Tissue Distribution and Pharmacokinetics. Bioconjugate Chem. 2010, 21, 2153–2163. [Google Scholar] [CrossRef]
- Liu, S.; Verma, A.; Kettenberger, H.; Richter, W.F.; Shah, D.K. Effect of variable domain charge on in vitro and in vivo disposition of monoclonal antibodies. MAbs 2021, 13, 1993769. [Google Scholar] [CrossRef]
- Stüber, J.C.; Rechberger, K.F.; Miladinović, S.M.; Pöschinger, T.; Zimmermann, T.; Villenave, R.; Eigenmann, M.J.; Kraft, T.E.; Shah, D.K.; Kettenberger, H.; et al. Impact of charge patches on tumor disposition and biodistribution of therapeutic antibodies. AAPS Open 2022, 8, 3. [Google Scholar] [CrossRef]
- Bumbaca Yadav, D.; Sharma, V.K.; Boswell, C.A.; Hotzel, I.; Tesar, D.; Shang, Y.; Ying, Y.; Fischer, S.K.; Grogan, J.L.; Chiang, E.Y.; et al. Evaluating the Use of Antibody Variable Region (Fv) Charge as a Risk Assessment Tool for Predicting Typical Cynomolgus Monkey Pharmacokinetics. J. Biol. Chem. 2015, 290, 29732–29741. [Google Scholar] [CrossRef]
- Singh, A.P.; Shah, D.K. Measurement and Mathematical Characterization of Cell-Level Pharmacokinetics of Antibody-Drug Conjugates: A Case Study with Trastuzumab-vc-MMAE. Drug Metab. Dispos. 2017, 45, 1120–1132. [Google Scholar] [CrossRef]
- Singh, A.P.; Sharma, S.; Shah, D.K. Quantitative characterization of in vitro bystander effect of antibody-drug conjugates. J. Pharmacokinet. Pharmacodyn. 2016, 43, 567–582. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.P.; Kim, S.J.; Shah, D.K. Whole-Body Pharmacokinetics of Antibody in Mice Determined using Enzyme-Linked Immunosorbent Assay and Derivation of Tissue Interstitial Concentrations. J. Pharm. Sci. 2021, 110, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.P.; Cheung, Y.K.; Shah, D.K. Whole-Body Pharmacokinetics and Physiologically Based Pharmacokinetic Model for Monomethyl Auristatin E (MMAE). J. Clin. Med. 2021, 10, 1332. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-P.; Cheung, Y.K.; Liu, S.; Shah, D.K. Development of a generalized pharmacokinetic model to characterize clinical pharmacokinetics of monomethyl auristatin E-based antibody–drug conjugates. Br. J. Clin. Pharmacol. 2024, 90, 1637–1655. [Google Scholar] [CrossRef]
- Liu, S.; Shah, D.K. Physiologically Based Pharmacokinetic Modeling to Characterize the Effect of Molecular Charge on Whole-Body Disposition of Monoclonal Antibodies. AAPS J. 2023, 25, 48. [Google Scholar] [CrossRef]
- Hu, X.Y.; Wang, R.; Jin, J.; Liu, X.J.; Cui, A.L.; Sun, L.Q.; Li, Y.P.; Li, Y.; Wang, Y.C.; Zhen, Y.S.; et al. An EGFR-targeting antibody-drug conjugate LR004-VC-MMAE: Potential in esophageal squamous cell carcinoma and other malignancies. Mol. Oncol. 2019, 13, 246–263. [Google Scholar] [CrossRef]
- Li, F.; Emmerton, K.K.; Jonas, M.; Zhang, X.; Miyamoto, J.B.; Setter, J.R.; Nicholas, N.D.; Okeley, N.M.; Lyon, R.P.; Benjamin, D.R.; et al. Intracellular Released Payload Influences Potency and Bystander-Killing Effects of Antibody-Drug Conjugates in Preclinical Models. Cancer Res. 2016, 76, 2710–2719. [Google Scholar] [CrossRef]
- Chang, H.P.; Li, Z.; Shah, D.K. Development of a Physiologically-Based Pharmacokinetic Model for Whole-Body Disposition of MMAE Containing Antibody-Drug Conjugate in Mice. Pharm. Res. 2022, 39, 1–24. [Google Scholar] [CrossRef]
- Anami, Y.; Yamazaki, C.M.; Xiong, W.; Gui, X.; Zhang, N.; An, Z.; Tsuchikama, K. Glutamic acid-valine-citrulline linkers ensure stability and efficacy of antibody-drug conjugates in mice. Nat. Commun. 2018, 9, 2512. [Google Scholar] [CrossRef]
- Jones, R.D.; Taylor, A.M.; Tong, E.Y.; Repa, J.J. Carboxylesterases are uniquely expressed among tissues and regulated by nuclear hormone receptors in the mouse. Drug Metab. Dispos. 2013, 41, 40–49. [Google Scholar] [CrossRef]
- Oda, S.; Fukami, T.; Yokoi, T.; Nakajima, M. A comprehensive review of UDP-glucuronosyltransferase and esterases for drug development. Drug Metab. Pharmacokinet. 2015, 30, 30–51. [Google Scholar] [CrossRef]
- Dorywalska, M.; Dushin, R.; Moine, L.; Farias, S.E.; Zhou, D.; Navaratnam, T.; Lui, V.; Hasa-Moreno, A.; Casas, M.G.; Tran, T.T.; et al. Molecular Basis of Valine-Citrulline-PABC Linker Instability in Site-Specific ADCs and Its Mitigation by Linker Design. Mol. Cancer Ther. 2016, 15, 958–970. [Google Scholar] [CrossRef]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable linkers in antibody-drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef] [PubMed]
- Doronina, S.O.; Toki, B.E.; Torgov, M.Y.; Mendelsohn, B.A.; Cerveny, C.G.; Chace, D.F.; DeBlanc, R.L.; Gearing, R.P.; Bovee, T.D.; Siegall, C.B.; et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotechnol. 2003, 21, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Atkinson, J.; Gulesserian, S.; Zeng, Z.; Nater, J.; Ou, J.; Yang, P.; Morrison, K.; Coleman, J.; Malik, F.; et al. Modulation of Macropinocytosis-Mediated Internalization Decreases Ocular Toxicity of Antibody-Drug Conjugates. Cancer Res. 2018, 78, 2115–2126. [Google Scholar] [CrossRef]
- Lyon, R.P.; Bovee, T.D.; Doronina, S.O.; Burke, P.J.; Hunter, J.H.; Neff-LaFord, H.D.; Jonas, M.; Anderson, M.E.; Setter, J.R.; Senter, P.D. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat. Biotechnol. 2015, 33, 733–735. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; D’Atri, V.; Ehkirch, A.; Fekete, S.; Hernandez-Alba, O.; Gahoual, R.; Leize-Wagner, E.; François, Y.; Guillarme, D.; Cianférani, S. Cutting-edge multi-level analytical and structural characterization of antibody-drug conjugates: Present and future. Expert Rev. Proteom. 2019, 16, 337–362. [Google Scholar] [CrossRef]
- Sun, X.; Ponte, J.F.; Yoder, N.C.; Laleau, R.; Coccia, J.; Lanieri, L.; Qiu, Q.; Wu, R.; Hong, E.; Bogalhas, M.; et al. Effects of Drug-Antibody Ratio on Pharmacokinetics, Biodistribution, Efficacy, and Tolerability of Antibody-Maytansinoid Conjugates. Bioconjug Chem. 2017, 28, 1371–1381. [Google Scholar] [CrossRef]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef]
- Vagenende, V.; Han, A.X.; Mueller, M.; Trout, B.L. Protein-Associated Cation Clusters in Aqueous Arginine Solutions and Their Effects on Protein Stability and Size. ACS Chem. Biol. 2013, 8, 416–422. [Google Scholar] [CrossRef]
- Kheddo, P.; Tracka, M.; Armer, J.; Dearman, R.J.; Uddin, S.; van der Walle, C.F.; Golovanov, A.P. The effect of arginine glutamate on the stability of monoclonal antibodies in solution. Int. J. Pharm. 2014, 473, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.K.; Betts, A.M. Antibody biodistribution coefficients: Inferring tissue concentrations of monoclonal antibodies based on the plasma concentrations in several preclinical species and human. MAbs 2013, 5, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.P.; Cheung, Y.K.; Shah, D.K. Chapter 7—Discovery and development of ADCs: Obstacles and opportunities. In Overcoming Obstacles in Drug Discovery and Development; He, K., Hollenberg, P.F., Wienkers, L.C., Eds.; Academic Press: Cambridge, MA, USA, 2023; pp. 75–106. [Google Scholar]
- Kellogg, B.A.; Garrett, L.; Kovtun, Y.; Lai, K.C.; Leece, B.; Miller, M.; Payne, G.; Steeves, R.; Whiteman, K.R.; Widdison, W.; et al. Disulfide-Linked Antibody−Maytansinoid Conjugates: Optimization of In Vivo Activity by Varying the Steric Hindrance at Carbon Atoms Adjacent to the Disulfide Linkage. Bioconjugate Chem. 2011, 22, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Sheyi, R.; de la Torre, B.G.; Albericio, F. Linkers: An Assurance for Controlled Delivery of Antibody-Drug Conjugate. Pharmaceutics 2022, 14, 396. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters (Unit) | Positively Charged ADC | WT ADC |
|---|---|---|
| Total mAb | ||
| (h) | 77.8 | 350 |
| CL (mL/h) | 0.0449 | 0.00518 |
| (mL) | 4.88 | 2.57 |
| (nM) | 716 ± 188 | 1600 ± 57.4 |
| (h∙nM) | 34,100 ± 5400 | 113,600 ± 5700 |
| (h∙nM) | 44,500 | 386,000 |
| Total MMAE | ||
| (h) | 41.7 | 57.5 |
| CL (mL/h) | 1.74 | 0.0452 |
| (mL) | 65.2 | 2.93 |
| (nM) | 172 ± 38.6 | 5080 ± 406 |
| (h∙nM) | 2200 ± 473 | 157,900 ± 7700 |
| (h∙nM) | 2300 | 177,000 |
| Unconjugated MMAE | ||
| (h) | 17.6 | 70.3 |
| (nM) | 8.72 ± 2.33 | 15.8 ± 3.66 |
| (h∙nM) | 23.3 | 278 |
| (h∙nM) | 23.9 | 331 |
| Parameters (Unit) | Positively Charged ADC | WT ADC |
|---|---|---|
| Total mAb | ||
| (h) | 24 | 24 |
| (nM) | 146 ± 51.6 | 364 ± 91.5 |
| (h∙nM) | 22,500 | 52,600 |
| Total MMAE | ||
| (h) | 24 | 72 |
| (nM) | 45.3 ± 17.6 | 610 ± 112 |
| (h∙nM) | 2057 | 87,300 |
| Unconjugated MMAE | ||
| (h) | 24 | 72 |
| (nM) | 20.1 ± 10.3 | 403 ± 71.2 |
| (h∙nM) | 1843 | 53,106 |
| Parameters (Unit) | Positively Charged ADC | WT ADC |
|---|---|---|
| Total mAb | ||
| (h) | 24 | 24 |
| (nM) | 301 ± 107 | 181 ± 35.1 |
| (h∙nM) | 28,500 | 24,000 |
| Total MMAE | ||
| (h) | 24 | 72 |
| (nM) | 28.1 ± 11.2 | 333 ± 175 |
| (h∙nM) | 1184 | 33,550 |
| Unconjugated MMAE | ||
| (h) | 6 | 72 |
| (nM) | 6.52 ± 1.45 | 36.7 ± 0.613 |
| (h∙nM) | 519 | 5148 |
| Parameters (Unit) | Positively Charged ADC | WT ADC |
|---|---|---|
| Total mAb | ||
| (h) | 24 | 6 |
| (nM) | 820 ± 300 | 527 ± 5.61 |
| (h∙nM) | 42,100 | 31,300 |
| Total MMAE | ||
| (h) | 0.167 | 0.167 |
| (nM) | 1100 ± 296 | 502 ± 46.6 |
| (h∙nM) | 5370 | 28,700 |
| Unconjugated MMAE | ||
| (h) | 0.167 | 0.167 |
| (nM) | 221 ± 58.1 | 118 ± 18.9 |
| (h∙nM) | 1540 | 3980 |
| Parameters (Unit) | Positively Charged ADC | WT ADC |
|---|---|---|
| Total mAb | ||
| (h) | 24 | 0.167 |
| (nM) | 387 ± 41.8 | 182 ± 10.5 |
| (h∙nM) | 26,700 | 10,800 |
| Total MMAE | ||
| (h) | 0.167 | 0.167 |
| (nM) | 750 ± 164 | 412 ± 23.3 |
| (h∙nM) | 4380 | 16,500 |
| Unconjugated MMAE | ||
| (h) | 0.167 | 0.167 |
| (nM) | 135 ± 41.1 | 173 ± 24.8 |
| (h∙nM) | 1970 | 8520 |
| Parameters (Unit) | Positively Charged ADC a | WT ADC a |
|---|---|---|
| Total mAb | ||
| Liver | 123 | 27.5 |
| Spleen | 78.3 | 9.47 |
| NCI-N87 | 66.1 | 46.3 |
| MCF7 | 83.6 | 21.2 |
| Total MMAE | ||
| Liver | 244 | 18.2 |
| Spleen | 199 | 10.4 |
| NCI-N87 | 93.6 | 55.3 |
| MCF7 | 53.9 | 21.3 |
| Unconjugated MMAE | ||
| Liver | 6600 | 1430 |
| Spleen | 8440 | 3070 |
| NCI-N87 | 7920 | 19,100 |
| MCF7 | 2230 | 1850 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, H.-P.; Le, H.K.; Liu, S.; Shah, D.K. PK/PD of Positively Charged ADC in Mice. Pharmaceutics 2025, 17, 377. https://doi.org/10.3390/pharmaceutics17030377
Chang H-P, Le HK, Liu S, Shah DK. PK/PD of Positively Charged ADC in Mice. Pharmaceutics. 2025; 17(3):377. https://doi.org/10.3390/pharmaceutics17030377
Chicago/Turabian StyleChang, Hsuan-Ping, Huyen Khanh Le, Shufang Liu, and Dhaval K. Shah. 2025. "PK/PD of Positively Charged ADC in Mice" Pharmaceutics 17, no. 3: 377. https://doi.org/10.3390/pharmaceutics17030377
APA StyleChang, H.-P., Le, H. K., Liu, S., & Shah, D. K. (2025). PK/PD of Positively Charged ADC in Mice. Pharmaceutics, 17(3), 377. https://doi.org/10.3390/pharmaceutics17030377