
Targeting Drug-Tolerant Persister Cancer Cells: Can Nanomaterial-Based Strategies Be Helpful for Anti-DTP Therapies?

 , , and
, , and 
Abstract
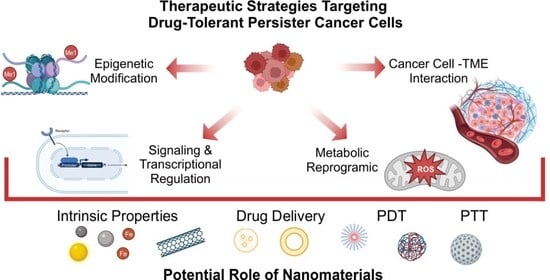
1. Introduction
2. Key Characteristics of Drug-Tolerant Persister (DTP) Cells
3. Therapeutic Strategies Targeting DTP Survival Mechanism
3.1. Targeting DTP Epigenetic Modifiers
3.2. Targeting DTP Cell Signaling and Transcriprional Regulation
3.3. Targeting DTP Metabolic Modifiers
3.4. Targeting DTP Cell–TME Interaction
4. Application of Nanotechnology for Cancer Treatment
4.1. Application of Nanotechnology to Combat Persister Cancer Cells
4.1.1. Nanotechnology in Targeting DTP Epigenetic Modifiers
4.1.2. Nanotechnology in Targeting DTP Cell Signaling and Transcriptional Regulation
4.1.3. Nanotechnology in Targeting DTP Metabolic Modifiers
4.1.4. Nanotechnology in Targeting DTP Cancer Cell–TME Interactions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nanomaterial/ Delivery System | Cancer Type/Model | Combination/ Co-Therapy | Key Findings/Mechanism | Ref. |
|---|---|---|---|---|
| Bovine serum albumin nanoparticles (BSA NPs) | Colorectal cancer (c-Met positive) | Crizotinib-IR808 conjugate | Enables NIR-II imaging-guided chemo-photo- therapy; targeted tumor delivery; synergistic chemo-phototherapy under laser irradiation | [129] |
| Mitochondria-targeting lipid-polymer hybrid nanoparticles PLGA/CPT/DSSP (poly(d,1-lactide-co-glycolide)/C18-PEG2000-TPP/DLPE-S-S-mPEG4000) | Breast cancer (MCF-7 cells) | RSL3 (GPX4 inhibitor) + Artemisinin | Synergistic apoptosis and ferroptosis; RSL3 enhances artemisinin-induced cell death via GPX4 inhibition | [130] |
| Mesoporous silica nanoparticles (MSN) with chitosan and lactobionic acid | Hepatocellular carcinoma (HCC) | Sorafenib + Ursolic acid | Enhanced tumor targeting, pH-responsive release, synergistic inhibition of growth and metastasis, increased apoptosis | [131] |
| Cancer cell–platelet hybrid membrane- camouflaged lipid nanoparticles | Hepatocellular carcinoma (HCC) | Sorafenib + Triptolide | Long circulation, tumor targeting, synergistic tumor inhibition, reduced sorafenib dose and toxicity | [132] |
| Ultra-small lipid nanoparticles (usLNPs) | Hepatocellular carcinoma (HCC) | Sorafenib + Midkine-siRNA | Overcomes sorafenib resistance, enhanced tumor accumulation, potent gene silencing, strong tumor eradication | [133] |
| Zwitterionic polymer-coated Fe3O4 nanoparticles | Colon cancer | Sorafenib + Fe3O4 | Induces ferroptosis, extended circulation, enhanced tumor accumulation, strong tumor inhibition | [134] |
| Bismuth-based mesoporous nanomaterial (NBOF) coated with polyethylene glycol and folic acid conjugates(P-FA) | Hepatocellular carcinoma (HCC) | Sorafenib + NBOF | Synergistic photothermal and molecular therapy, enhanced imaging, significant tumor growth reduction | [135] |
| HDL-like organic-core lipid nanoparticles | Prostate, AML, pancreatic cancer | Multi-kinase inhibitor (PIK-750 | SR-B1 targeted delivery, potent cell death, reduced tumor growth, and few side effects | [136] |
| Lipid nanoparticles (EGFR-PEG bispecific Ab) | Neuroblastoma | PLK1 siRNA (kinase silencing) | Enhanced targeting, gene silencing, and reduced tumor growth in vivo | [137] |
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| •OH | Hydroxyl Radicals |
| Ab | Antibody |
| AEW541 | Insulin-Like Growth Factor–Type 1 Receptor (IGF1-R) Inhibitor |
| AgNPs | Silver Nanoparticles |
| ALDH | Aldehyde Dehydrogenase |
| AML | Acute Myeloid Leukemia |
| Ang-1 | Angiopoietin 1 |
| Ang-2 | Angiopoietin 2 |
| AURKA | Aurora A Kinase |
| AXL | AXL Receptor Tyrosine Kinase |
| BBD | Box–Behnken design |
| BC | Bladder Cancer |
| BRAF | B-Raf Proto-Oncogene, Serine/threonine Kinase |
| Ca+2 | Calcium |
| CAFs | Cancer-Associated Fibroblasts |
| CAP | Cold Atmospheric Plasma |
| CDT | Chemodynamic Therapy |
| CDK7 | Cyclin-dependent Kinase 7 |
| Chl/Fe | Iron Chlorophyll |
| CL-LPHNPs | Crizotinib-Loaded Lipid-Polymer Hybrid Nanoparticles |
| CNPs | Cluster-Structured Nanoparticles |
| CNTs | Carbon Nanotubes |
| CPBA | Carboxyphenylboronic Acid |
| CPI-455 | Pan-KDM5 Inhibitor |
| CRIZ | Crizotinib |
| cSLNs | Cationic Solid Lipid Nanoparticles |
| DC | Dendritic Cell |
| DDAB | Dimethyl Di-octadecyl Ammonium Bromide |
| DDS | Drug Delivery System |
| DNA | Deoxyribonucleic Acid |
| DOTMA | 1,2-di-O-octadecenyl-3-trimethylammonium-propane |
| Dox | Doxorubicin |
| DSBs | DNA Double-Strand Breaks |
| DTP | Drug-Tolerant Persister |
| EGFR | Epidermal Growth Factor Receptor |
| EPR | Enhanced Permeability and Retention effect |
| ERK | Extracellular Signal-regulated Kinase |
| FA | Folic Acid |
| FAK | Focal Adhesion Kinase |
| FAO | Fatty Acid Oxidation |
| FDA | Food and Drug Administration |
| FGF | Fibroblast Growth Factor |
| FGFR | Fibroblast Growth Factor Receptor |
| FOSL1 | Fos-like Antigen 1 |
| FSHR | Follicle-Stimulating Hormone Receptor |
| GA | Gambogic Acid |
| GBC | Gallbladder Cancer |
| GO | Graphene Oxide |
| GPX4 | Glutathione Peroxidase 4 |
| H3K9me3 | Histone 3 Lysine 9 Trimethylation |
| HCC | Hepatocellular Carcinoma |
| HDAC | Histone Deacetylase |
| HGF | Hepatocyte Growth Factor |
| IC50 | Half-maximal Inhibitory Concentration |
| ICD | Immunogenic Cell Death |
| IDO-1 | Indoleamine-2,3-dioxygenase 1 |
| ICG-001 | Selective Wnt/β-Catenin Signaling Inhibitor |
| IGF-1 | Insulin-like Growth Factor |
| IGFR | Insulin-like Growth Factor Receptor |
| IHC | Immunohistochemistry |
| IL-6 | Interleukin 6 |
| JQ1 | Jun Qi 1 |
| JUN | Jun pProto-oncogene, AP-1 Transcription Factor Subunit |
| KDM5A | Lysine Demethylase 5A |
| LCN2 | Lipocalin 2 |
| MAPK | Mitogen-Activated Protein Kinase |
| MDR | Multi-Drug Resistance |
| MEK | Mitogen-Activated Protein Kinase |
| MET | Mesenchymal–Epithelial Transition Factor |
| miRNAs | Small Non-Coding MicroRNAs |
| MMPs | Matrix Metalloproteases |
| MPEG-PLA | Methoxy Poly(ethylene glycol)-poly(l-lactic acid) |
| MS023 | Human Type I Pprotein Arginine Methyltransferases (Prmts) Inhibitor |
| NGFR | Nerve Growth Factor Receptor |
| NPs | Nanoparticles |
| NPS1034 | AXL/MET Inhibitor |
| NSCLC | Non-Small Cell Lung Cancer |
| PD-L1 | Programmed Death-Ligand 1 |
| PDT | Photo-Dynamic Therapy |
| PDX | Patient-Derived Xenograft Model |
| PEG–PCL–PEG, PECE | Poly(ethylene glycol)–poly(e-caprolactone)–poly(ethylene glycol) |
| pGO | PEGylated Graphene Oxide |
| PLGA-PEG | Poly(ethylene glycol)-block-poly(lactide-co-glycolide) |
| RAF | Raf-1 Proto-Oncogene, Serine/threonine Kinase |
| RAS | Rat Sarcoma |
| RBP2 | Retinol-Binding Protein 2 |
| rGO-AgNPs | Reduced Graphene Oxide-silver Nanoparticles |
| RNA | Ribonucleic Acid |
| RNAPII | RNA Polymerase II |
| RONS | Reactive Oxygen and Nitrogen Species |
| ROS | Reactive Oxygen Species |
| RSL3 | Inhibitor of GPX4 |
| RTK | Receptor Tyrosine Kinase |
| siEphA2 | siRNA Silencing EphA2 |
| SLPs | Synthetic Lipoproteins |
| SMAD2 | Mothers Against Decapentaplegic Homolog 2 |
| SORA | Sorafenib |
| SPION | Superparamagnetic Iron Oxide Nanoparticles |
| TAM | Tumor-Associated Macrophages |
| TEAD | Transcriptional Enhanced Associate Domain Transcription Factors |
| TGF-β | Transforming Growth Factor Beta |
| Thio | Thioridazine |
| THZ1 | CDK7 Inhibitor |
| Tie2R | Angiopoietin Receptor |
| TKI | Tyrosine Kinase Inhibitor |
| TME | Tumor Micro Environment |
| TNF-α | Tumor Necrosis Factor-α |
| TSA | Trichostatin A |
| VEGF | Vascular Endothelial Growth Factor |
| VEGFR | Vascular Endothelial Growth Factor Receptor |
| YAP | Yes-associated Protein Transcriptional Coactivator |
| XL-880 | Foretinib |
References
- Blatter, S. S Rottenberg Minimal Residual Disease in Cancer Therapy–Small Things Make All the Difference. Drug Resist. Updates 2015, 21–22, 1–10. [Google Scholar] [CrossRef]
- Mordant, P.; Loriot, Y.; Lahon, B.; Castier, Y.; Lesèche, G.; Soria, J.C.; Massard, C.; Deutsch, E. Minimal Residual Disease in Solid Neoplasia: New Frontier or Red-Herring? Cancer Treat. Rev. 2012, 38, 101–110. [Google Scholar] [CrossRef]
- Cabanos, H.F.; Hata, A.N. Emerging Insights into Targeted Therapy-Tolerant Persister Cells in Cancer. Cancers 2021, 13, 2666. [Google Scholar] [CrossRef]
- Redmond, K.M.; Wilson, T.R.; Johnston, P.G.; Longley, D.B. Resistance Mechanisms to Cancer Chemotherapy. Front. Biosci. 2008, 13, 5138–5154. [Google Scholar] [CrossRef]
- Harms, A.; Maisonneuve, E.; Gerdes, K. Mechanisms of Bacterial Persistence during Stress and Antibiotic Exposure. Science 2016, 354, aaf4268. [Google Scholar] [CrossRef] [PubMed]
- Michiels, J.E.; Van den Bergh, B.; Verstraeten, N.; Michiels, J. Molecular Mechanisms and Clinical Implications of Bacterial Persistence. Drug Resist. Updates 2016, 29, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Helaine, S.; Kugelberg, E. Bacterial Persisters: Formation, Eradication, and Experimental Systems. Trends Microbiol. 2014, 22, 417–424. [Google Scholar] [CrossRef]
- Verstraeten, N.; Michiels, J. Bacterial Persistence: Methods and Protocols; Methods in Molecular Biology; Springer: New York, NY, USA, 2021; Volume 2357, ISBN 978-1-0716-1620-8. [Google Scholar]
- Zou, J.; Peng, B.; Qu, J.; Zheng, J. Are Bacterial Persisters Dormant Cells Only? Front. Microbiol. 2022, 12, 708580. [Google Scholar] [CrossRef] [PubMed]
- Decollogny, M.; Rottenberg, S. Persisting Cancer Cells Are Different from Bacterial Persisters. Trends Cancer 2024, 10, 393–406. [Google Scholar] [CrossRef]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations. Elsevier Cell 2010, 141, 69–80. [Google Scholar] [CrossRef]
- Vinogradova, M.; Gehling, V.S.; Gustafson, A.; Arora, S.; Tindell, C.A.; Wilson, C.; Williamson, K.E.; Guler, G.D.; Gangurde, P.; Manieri, W.; et al. An Inhibitor of KDM5 Demethylases Reduces Survival of Drug-Tolerant Cancer Cells. Nat. Chem. Biol. 2016, 12, 531–538. [Google Scholar] [CrossRef]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, M.J.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, S.L.; et al. Drug-Tolerant Persister Cancer Cells Are Vulnerable to GPX4 Inhibition. Nature 2017, 551, 247–250. [Google Scholar] [CrossRef]
- Menon, D.R.; Das, S.; Krepler, C.; Vultur, A.; Rinner, B.; Schauer, S.; Kashofer, K.; Wagner, K.; Zhang, G.; Bonyadi Rad, E.; et al. A Stress-Induced Early Innate Response Causes Multidrug Tolerance in Melanoma. Oncogene 2015, 34, 4448–4459. [Google Scholar] [CrossRef]
- Moody, S.; Schinzel, A.; Singh, S.; Izzo, F.; Strickland, M.R.; Luo, L.; Thomas, S.R.; Boehm, J.S.; Kim, S.Y.; Wang, Z.C.; et al. PRKACA Mediates Resistance to HER2-Targeted Therapy in Breast Cancer Cells and Restores Anti-Apoptotic Signaling. Oncogene 2015, 34, 2061–2071. [Google Scholar] [CrossRef]
- McDonald, P.; Dedhar, S. Persister Cell Plasticity in Tumour Drug Resistance. Semin. Cell Dev. Biol. 2024, 156, 1–10. [Google Scholar] [CrossRef]
- Liang, X.W.; Liu, B.; Chen, J.C.; Cao, Z.; Chu, F.R.; Lin, X.; Wang, S.Z.; Wu, J.C. Characteristics and Molecular Mechanism of Drug-Tolerant Cells in Cancer: A Review. Front Oncol 2023, 13, 1177466. [Google Scholar] [CrossRef]
- Pu, Y.; Li, L.; Peng, H.; Liu, L.; Heymann, D.; Robert, C.; Vallette, F.; Shen, S. Drug-Tolerant Persister Cells in Cancer: The Cutting Edges and Future Directions. Nat. Rev. Clin. Oncol. 2023, 20, 799–813. [Google Scholar] [CrossRef] [PubMed]
- Raha, D.; Wilson, T.R.; Peng, J.; Peterson, D.; Yue, P.; Evangelista, M.; Wilson, C.; Merchant, M.; Settleman, J. The Cancer Stem Cell Marker Aldehyde Dehydrogenase Is Required to Maintain a Drug-Tolerant Tumor Cell Subpopulation. Cancer Res. 2014, 74, 3579–3590. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare Cell Variability and Drug-Induced Reprogramming as a Mode of Cancer Drug Resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018, 174, 843–855.e19. [Google Scholar] [CrossRef]
- Álvarez-Varela, A.; Novellasdemunt, L.; Barriga, F.M.; Hernando-Momblona, X.; Cañellas-Socias, A.; Cano-Crespo, S.; Sevillano, M.; Cortina, C.; Stork, D.; Morral, C.; et al. Mex3a Marks Drug-Tolerant Persister Colorectal Cancer Cells That Mediate Relapse after Chemotherapy. Nat. Cancer 2022, 3, 1052–1070. [Google Scholar] [CrossRef]
- Gupta, P.B.; Fillmore, C.M.; Jiang, G.; Shapira, S.D.; Tao, K.; Kuperwasser, C.; Lander, E.S. Stochastic State Transitions Give Rise to Phenotypic Equilibrium in Populations of Cancer Cells. Cell 2011, 146, 633–644. [Google Scholar] [CrossRef]
- Russo, M.; Crisafulli, G.; Sogari, A.; Reilly, N.M.; Arena, S.; Lamba, S.; Bartolini, A.; Amodio, V.; Magrì, A.; Novara, L.; et al. Adaptive Mutability of Colorectal Cancers in Response to Targeted Therapies. Science 2019, 366, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Karki, P.; Angardi, V.; Mier, J.C.; Orman, M.A. A Transient Metabolic State in Melanoma Persister Cells Mediated by Chemotherapeutic Treatments. Front. Mol. Biosci. 2022, 8, 780192. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Pompei, S.; Sogari, A.; Corigliano, M.; Crisafulli, G.; Puliafito, A.; Lamba, S.; Erriquez, J.; Bertotti, A.; Gherardi, M.; et al. A Modified Fluctuation-Test Framework Characterizes the Population Dynamics and Mutation Rate of Colorectal Cancer Persister Cells. Nat. Genet. 2022, 54, 976–984. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Chen, M.; Mariella, E.; Peng, H.; Rehman, S.K.; Sancho, E.; Sogari, A.; Toh, T.S.; Balaban, N.Q.; Batlle, E.; et al. Cancer Drug-Tolerant Persister Cells: From Biological Questions to Clinical Opportunities. Nat. Rev. Cancer 2024, 24, 694–717. [Google Scholar] [CrossRef]
- Liu, S.; Jiang, A.; Tang, F.; Duan, M.; Li, B. Drug-Induced Tolerant Persisters in Tumor: Mechanism, Vulnerability and Perspective Implication for Clinical Treatment. Mol. Cancer 2025, 24, 150. [Google Scholar] [CrossRef]
- Giri, P.; Banerjee, A.; Layek, B. A Recent Review on Cancer Nanomedicine. Cancers 2023, 15, 2256. [Google Scholar] [CrossRef]
- Wang, B.; Hu, S.; Teng, Y.; Chen, J.; Wang, H.; Xu, Y.; Wang, K.; Xu, J.; Cheng, Y.; Gao, X. Current Advance of Nanotechnology in Diagnosis and Treatment for Malignant Tumors. Signal Transduct. Target. Ther. 2024, 9, 200. [Google Scholar] [CrossRef]
- Wang, S.; Cheng, K.; Chen, K.; Xu, C.; Ma, P.; Dang, G.; Yang, Y.; Lei, Q.; Huang, H.; Yu, Y.; et al. Nanoparticle-Based Medicines in Clinical Cancer Therapy. Nano Today 2022, 45, 101512. [Google Scholar] [CrossRef]
- Boumahdi, S.; de Sauvage, F.J. The Great Escape: Tumour Cell Plasticity in Resistance to Targeted Therapy. Nat. Rev. Drug Discov. 2020, 19, 39–56. [Google Scholar] [CrossRef]
- Oren, Y.; Tsabar, M.; Cuoco, M.; Amir-Zilberstein, L.; Cabanos, H.F.; Hütter, J.C.; Hu, B.; Thakore, P.I.; Tabaka, M.; Fulco, C.P.C. Cycling Cancer Persister Cells Arise from Lineages with Distinct Programs. Nature 2021, 596, 576–582. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G.J. Cancer Stem Cells in Solid Tumours: Accumulating Evidence and Unresolved Questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef]
- Moghal, N.; Li, Q.; Stewart, E.L.; Navab, R.; Mikubo, M.; D’Arcangelo, E.; Martins-Filho, S.N.; Raghavan, V.; Pham, N.A.; Li, M.; et al. Single-Cell Analysis Reveals Transcriptomic Features of Drug-Tolerant Persisters and Stromal Adaptation in a Patient-Derived EGFR-Mutated Lung Adenocarcinoma Xenograft Model. J. Thorac. Oncol. 2023, 18, 499–515. [Google Scholar] [CrossRef]
- Kobayashi, I.; Takahashi, F.; Nurwidya, F.; Nara, T.; Hashimoto, M.; Murakami, A.; Yagishita, S.; Tajima, K.; Hidayat, M.; Shimada, N.; et al. Oct4 Plays a Crucial Role in the Maintenance of Gefitinib-Resistant Lung Cancer Stem Cells. Biochem. Biophys. Res. Commun. 2016, 473, 125–132. [Google Scholar] [CrossRef]
- Liau, B.B.; Sievers, C.; Donohue, L.K.; Gillespie, S.M.; Flavahan, W.A.; Miller, T.E.; Venteicher, A.S.; Hebert, C.H.; Carey, C.D.; Rodig, S.J.; et al. Adaptive Chromatin Remodeling Drives Glioblastoma Stem Cell Plasticity and Drug Tolerance. Cell Stem Cell 2017, 20, 233–246.e7. [Google Scholar] [CrossRef]
- Vallette, F.M.; Olivier, C.; Lézot, F.; Oliver, L.; Cochonneau, D.; Lalier, L.; Cartron, P.F.; Heymann, D. Dormant, Quiescent, Tolerant and Persister Cells: Four Synonyms for the Same Target in Cancer. Biochem. Pharmacol. 2019, 162, 169–176. [Google Scholar] [CrossRef]
- Roesch, A.; Vultur, A.; Bogeski, I.; Wang, H.; Zimmermann, K.M.; Speicher, D.; Körbel, C.; Laschke, M.W.; Gimotty, P.A.; Philipp, S.E.; et al. Overcoming Intrinsic Multidrug Resistance in Melanoma by Blocking the Mitochondrial Respiratory Chain of Slow-Cycling JARID1Bhigh Cells. Cancer Cell 2013, 23, 811–825. [Google Scholar] [CrossRef]
- Tanei, T.; Morimoto, K.; Shimazu, K.; Seung, J.K.; Tanji, Y.; Taguchi, T.; Tamaki, Y.; Noguchi, S. Association of Breast Cancer Stem Cells Identified by Aldehyde Dehydrogenase 1 Expression with Resistance to Sequential Paclitaxel and Epirubicin-Based. Clin. Cancer Res. 2009, 15, 4234–4241. [Google Scholar] [CrossRef]
- Rusan, M.; Li, K.; Li, Y.; Christensen, C.; Abraham, B.J.; Kwiatkowski, N.; Buczkowski, K.A.; Bockorny, B.; Chen, T.; Li, S.; et al. Suppression of Adaptive Responses to Targeted Cancer Therapy by Transcriptional Repression. Cancer Discov. 2018, 8, 59–73. [Google Scholar] [CrossRef]
- Echeverria, G.V.; Ge, Z.; Seth, S.; Zhang, X.; Jeter-Jones, S.; Zhou, X.; Cai, S.; Tu, Y.; McCoy, A.; Peoples, M.; et al. Resistance to Neoadjuvant Chemotherapy in Triple-Negative Breast Cancer Mediated by a Reversible Drug-Tolerant State. Sci. Transl. Med. 2019, 11, eaav0936. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and Definitions for Research on Epithelial–Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef]
- Kurppa, K.; Liu, Y.; To, C.; Zhang, T.; Fan, M.; Vajdi, A.; Knelson, E.H.; Xie, Y.; Lim, K.; Cejas, P.; et al. Treatment-Induced Tumor Dormancy through YAP-Mediated Transcriptional Reprogramming of the Apoptotic Pathway. Cancer Cell 2020, 37, 104–122. [Google Scholar] [CrossRef]
- Raoof, S.; Mulford, I.; Frisco-Cabanos, H.; Nangia, V.; Timonina, D.; Labrot, E.; Hafeez, N.; Bilton, S.J.; Drier, Y.; Ji, F.; et al. Targeting FGFR Overcomes EMT-Mediated Resistance in EGFR Mutant Non-Small Cell Lung Cancer. Oncogene 2019, 38, 6399–6413. [Google Scholar] [CrossRef]
- Lotsberg, M.; Wnuk-Lipinska, K.; Terry, S.; Tan, T.Z.; Lu, N.; Trachsel-Moncho, L.; Røsland, G.V.; Siraji, M.I.; Hellesøy, M.; Rayford, A.; et al. AXL Targeting Abrogates Autophagic Flux and Induces Immunogenic Cell Death in Drug-Resistant Cancer Cells. J. Thorac. Oncol. 2020, 1, 973–999. [Google Scholar] [CrossRef]
- Böpple, K.; Oren, Y.; Henry, W.S.; Dong, M.; Weller, S.; Thiel, J.; Kleih, M.; Gaißler, A.; Zipperer, D.; Kopp, H.G.; et al. ATF3 Characterizes Aggressive Drug-Tolerant Persister Cells in HGSOC. Cell Death Dis. 2024, 15, 290. [Google Scholar] [CrossRef]
- Aldonza, M.B.D.; Ku, J.; Hong, J.Y.; Kim, D.; Yu, S.J.; Lee, M.S.; Prayogo, M.C.; Tan, S.; Kim, D.; Han, J.; et al. Prior Acquired Resistance to Paclitaxel Relays Diverse EGFR-Targeted Therapy Persistence Mechanisms. Sci. Adv. 2020, 6, eaav7416. [Google Scholar] [CrossRef]
- Risom, T.; Langer, E.; Chapman, M.; Rantala, J.; Fields, A.J.; Boniface, C.; Alvarez, M.J.; Kendsersky, N.D.; Pelz, C.R.; Johnson-Camacho, K.; et al. Differentiation-State Plasticity Is a Targetable Resistance Mechanism in Basal-like Breast Cancer. Nat. Commun. 2018, 9, 3815. [Google Scholar] [CrossRef]
- Lupo, B.; Sassi, F.; Pinnelli, M.; Galimi, F.; Zanella, E.R.; Vurchio, V.; Migliardi, G.; Gagliardi, P.A.; Puliafito, A.; Manganaro, D.; et al. Colorectal Cancer Residual Disease at Maximal Response to EGFR Blockade Displays a Druggable Paneth Cell–like Phenotype. Sci Transl Med 2020, 12, eaax8313. [Google Scholar] [CrossRef]
- Davies, A.H.; Beltran, H.; Zoubeidi, A. Cellular Plasticity and the Neuroendocrine Phenotype in Prostate Cancer. Nat. Rev. Urol. 2018, 15, 271–286. [Google Scholar] [CrossRef]
- Biehs, B.; Dijkgraaf, G.J.P.; Piskol, R.; Alicke, B.; Boumahdi, S.; Peale, F.; Gould, S.E.; de Sauvage, F.J. A Cell Identity Switch Allows Residual BCC to Survive Hedgehog Pathway Inhibition. Nature 2018, 562, 429–433. [Google Scholar] [CrossRef]
- Rehman, S.; Haynes, J.; Collignon, E.; Brown, K.R.; Wang, Y.; Nixon, A.M.; Bruce, J.P.; Wintersinger, J.A.; Mer, A.S.; Lo, E.B.; et al. Colorectal Cancer Cells Enter a Diapause-like DTP State to Survive Chemotherapy. Cell 2021, 184, 226–242. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Li, C.; Chen, L.; Liu, Y.; Hou, R.; Zhou, X. Research Advances on Embryonic Diapause in Mammals. Anim. Reprod. Sci. 2018, 198, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dhimolea, E.; de Matos Simoes, R.; Kansara, D.; Al’Khafaji, A.; Bouyssou, J.; Weng, X.; Sharma, S.; Raja, J.; Awate, P.; Shirasaki, R.; et al. An Embryonic Diapause-like Adaptation with Suppressed Myc Activity Enables Tumor Treatment Persistence. Cancer Cell 2021, 39, 240–256.e11. [Google Scholar] [CrossRef]
- Duy, C.; Li, M.; Teater, M.; Meydan, C.; Garrett-Bakelman, F.E.; Lee, T.C.; Chin, C.R.; Durmaz, C.; Kawabata, K.C.; Dhimolea, E.; et al. Chemotherapy Induces Senescence-like Resilient Cells Capable of Initiating AML Recurrence. Cancer Discov. 2021, 11, 1542–1561. [Google Scholar] [CrossRef]
- Chang, C.A.; Jen, J.; Jiang, S.; Sayad, A.; Mer, A.S.; Brown, K.R.; Nixon, A.M.L.; Dhabaria, A.; Tang, K.H.; Venet, D.; et al. Ontogeny and Vulnerabilities of Drug-Tolerant Persisters in HER2+ Breast Cancer. Cancer Discov. 2022, 12, 1022–1045. [Google Scholar] [CrossRef]
- Mani, N.; Daiya, A.; Chowdhury, R.; Mukherjee, S.; Chowdhury, S. Epigenetic Adaptations in Drug-Tolerant Tumor Cells. Elsevier 2023, 158, 293–335. [Google Scholar] [CrossRef]
- Bell, C.C.; Gilan, O. Principles and Mechanisms of Non-Genetic Resistance in Cancer. Br. J. Cancer 2020, 122, 465–472. [Google Scholar] [CrossRef]
- Aygün, O.; Mehta, S.; Grewal, S.I.S. HDAC-Mediated Suppression of Histone Turnover Promotes Epigenetic Stability of Heterochromatin. Nat. Struct. Mol. Biol. 2013, 20, 547–554. [Google Scholar] [CrossRef]
- Guler, G.D.; Tindell, C.A.; Pitti, R.; Wilson, C.; Nichols, K.; KaiWai Cheung, T.; Kim, H.J.; Wongchenko, M.; Yan, Y.; Haley, B.; et al. Repression of Stress-Induced LINE-1 Expression Protects Cancer Cell Subpopulations from Lethal Drug Exposure. Cancer Cell 2017, 32, 221–237.e13. [Google Scholar] [CrossRef]
- Song, X.; Lan, Y.; Zheng, X.; Zhu, Q.; Liao, X.; Liu, K.; Zhang, W.; Peng, Q.B.; Zhu, Y.; Zhao, L.; et al. Targeting Drug-tolerant Cells: A Promising Strategy for Overcoming Acquired Drug Resistance in Cancer Cells. ChenMedComm 2023, 4, e342. [Google Scholar] [CrossRef]
- Fujimura, T.; Furugaki, K.; Mizuta, H.; Muraoka, S.; Nishio, M.; Adachi, J.; Uchibori, K.; Miyauchi, E.; Hayashi, H.; Katayama, R.; et al. Targeting ErbB and Tankyrase1/2 Prevent the Emergence of Drug-Tolerant Persister Cells in ALK-Positive Lung Cancer. npj Precis. Oncol. 2024, 8, 264. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, R.; Mashima, T.; Kawata, N.; Kumagai, K.; Migita, T.; Sano, T.; Mizunuma, N.; Yamaguchi, K.; Seimiya, H. ALDH1A3-MTOR Axis as a Therapeutic Target for Anticancer Drug-Tolerant Persister Cells in Gastric Cancer. Cancer Sci. 2020, 111, 962–973. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.; Schreiber, S.L.; Conrad, M. Persister Cancer Cells: Iron Addiction and Vulnerability to Ferroptosis. Mol. Cell 2022, 82, 728–740. [Google Scholar] [CrossRef]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a Therapy-Resistant State of Cancer Cells on a Lipid Peroxidase Pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef]
- Zhang, Z.; Tan, Y.; Huang, C.; Wei, X. Redox Signaling in Drug-Tolerant Persister Cells as an Emerging Therapeutic Target. EBioMedicine 2023, 89, 104483. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Yu, W.; Liu, J.; Tang, D.; Yang, L.; Chen, X. Oxidative Cell Death in Cancer: Mechanisms and Therapeutic Opportunities. Cell Death Diseas 2024, 15, 556. [Google Scholar] [CrossRef]
- Mancini, C.; Lori, G.; Pranzini, E.; Taddei, M.L. Metabolic Challengers Selecting Tumor-Persistent Cells. Trends Endocrinol. Metab. 2024, 35, 263–276. [Google Scholar] [CrossRef]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Rong Qian, Z.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour Micro-Environment Elicits Innate Resistance to RAF Inhibitors through HGF Secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef]
- Sun, X.; Bieber, J.M.; Hammerlindl, H.; Chalkley, R.J.; Li, K.H.; Burlingame, A.L.; Jacobson, M.P.; Wu, L.F.; Altschuler, S.J. Modulating Environmental Signals to Reveal Mechanisms and Vulnerabilities of Cancer Persisters. Sci. Adv. 2022, 8, 7711. [Google Scholar] [CrossRef]
- Taniguchi, H.; Yamada, T.; Wang, R.; Tanimura, K.; Adachi, Y.; Nishiyama, A.; Tanimoto, A.; Takeuchi, S.; Araujo, L.H.; Boroni, M.; et al. AXL Confers Intrinsic Resistance to Osimertinib and Advances the Emergence of Tolerant Cells. Nat. Commun. 2019, 10, 259. [Google Scholar] [CrossRef]
- Li, Y.; Chen, H.; Xie, X.; Yang, B.; Wang, X.; Zhang, J.; Qiao, T.; Guan, J.; Qiu, Y.; Huang, Y.X.; et al. PINK1-Mediated Mitophagy Promotes Oxidative Phosphorylation and Redox Homeostasis to Induce Drug-Tolerant Persister Cancer Cells. Cancer Res. 2023, 83, 398–413. [Google Scholar] [CrossRef]
- Sánchez-Danés, A.; Larsimont, J.; Liagre, M.; Muñoz-Couselo, E.; Lapouge, G.; Brisebarre, A.; Dubois, C.; Suppa, M.; Sukumaran, V.; Del Marmol, V.; et al. A Slow-Cycling LGR5 Tumour Population Mediates Basal Cell Carcinoma Relapse after Therapy. Nature 2018, 562, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, B.K.; Singh, V.V.; Solanki, M.K.; Kumar, A.; Ruokolainen, J.; Kesari, K.K. Smart Nanomaterials in Cancer Theranostics: Challenges and Opportunities. ACS Omega 2023, 8, 14290–14320. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Li, M.; Dey, R.; Chen, Y. Nanomaterials for Cancer Therapy: Current Progress and Perspectives. J. Hematol. Oncol. 2021, 14, 85. [Google Scholar] [CrossRef]
- Banu, H.; Sethi, D.; Edgar, A.; Sheriff, A.; Rayees, N.; Renuka, N.; Faheem, S.M.; Premkumar, K.; Vasanthakumar, G. Doxorubicin Loaded Polymeric Gold Nanoparticles Targeted to Human Folate Receptor upon Laser Photothermal Therapy Potentiates Chemotherapy in Breast. J. Photochem. Photobiol. B 2015, 149, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Abbasi Kajani, A.; Haghjooy Javanmard, S.; Asadnia, M.; Razmjou, A. Recent Advances in Nanomaterials Development for Nanomedicine and Cancer. ACS Appl. Bio Mater. 2021, 4, 5908–5925. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, S. Advantages of Nanomedicine in Cancer Therapy: A Review. ACS Appl. Nano Mater. 2023, 6, 22594–22610. [Google Scholar] [CrossRef]
- Thanh, N.; Green, L.A. Functionalisation of Nanoparticles for Biomedical Applications. Nano Today 2010, 5, 213–230. [Google Scholar] [CrossRef]
- Su, G.; Zhou, H.; Mu, Q.; Zhang, Y.; Li, L.; Jiao, P.; Jiang, G.; Yan, B. Effective Surface Charge Density Determines the Electrostatic Attraction between Nanoparticles and Cells. J. Phys. Chem. C 2012, 116, 4993–4998. [Google Scholar] [CrossRef]
- Hühn, D.; Kantner, K.; Geidel, C.; Brandholt, S.; De Cock, I.; Soenen, S.J.H.; Riveragil, P.; Montenegro, J.M.; Braeckmans, K.; Müllen, K.; et al. Polymer-Coated Nanoparticles Interacting with Proteins and Cells: Focusing on the Sign of the Net Charge. ACS Nano 2013, 7, 3253–3263. [Google Scholar] [CrossRef]
- Irimie, A.I.; Braicu, C.; Cojocneanu-Petric, R.; Berindan-Neagoe, I.; Campian, R.S. Novel Technologies for Oral Squamous Carcinoma Biomarkers in Diagnostics and Prognostics. Acta Odontol. Scand. 2015, 73, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Baetke, S.C.; Lammers, T.; Kiessling, F. Applications of Nanoparticles for Diagnosis and Therapy of Cancer. Br. J. Radiol. 2015, 88, 20150207. [Google Scholar] [CrossRef]
- Zhu, R.; Zhang, F.; Peng, Y.; Xie, T.; Wang, Y.; Lan, Y. Current Progress in Cancer Treatment Using Nanomaterials. Front. Oncol. 2022, 12, 930125. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Wei, Y.; Zhang, X.; Lin, L.; Bao, Y.; Cao, H.; Chen, H.; Yu, J.; Yang, J.; Zhang, Y.; et al. Enhanced EPR Effects by Tumour Stromal Cell Mimicking Nanoplatform on Invasive Pituitary Adenoma. Mater. Today Bio 2024, 24, 100895. [Google Scholar] [CrossRef]
- Tomuleasa, C.; Braicu, C.; Irimie, A.; Craciun, L.; Berindan-Neagoe, I. Nanopharmacology in Translational Hematology and Oncology. Int. J. Nanomed. 2014, 9, 3465–3479. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Orza, A.; Soriţǎu, O.; Tomuleasa, C.; Olenic, L.; Florea, A.; Pana, O.; Bratu, I.; Pall, E.; Florian, S.; Casciano, D.; et al. Reversing Chemoresistance of Malignant Glioma Stem Cells Using Gold Nanoparticles. Int. J. Nanomed. 2013, 8, 689–702. [Google Scholar] [CrossRef]
- Tomuleasa, C.; Soritau, O.; Orza, A.; Dudea, M.; Petrushev, B.; Mosteanu, O.; Susman, S.; Florea, A.; Pall, E.; Aldea, M.; et al. Gold Nanoparticles Conjugated with Cisplatin/Doxorubicin/Capecitabine Lower the Chemoresistance of Hepatocellular Carcinoma-Derived Cancer Cells. J. Gastrointestin. Liver Dis. 2012, 21, 187–196. [Google Scholar]
- Liu, Y.; Yang, M.; Zhang, J.; Zhi, X.; Li, C.; Zhang, C.; Pan, F.; Wang, K.; Yang, Y.; Martinez De La Fuentea, J.; et al. Human Induced Pluripotent Stem Cells for Tumor Targeted Delivery of Gold Nanorods and Enhanced Photothermal Therapy. ACS Nano 2016, 10, 2375–2385. [Google Scholar] [CrossRef]
- Vangijzegem, T.; Lecomte, V.; Ternad, I.; Van Leuven, L.; Muller, R.N.; Stanicki, D.; Laurent, S. Superparamagnetic Iron Oxide Nanoparticles (SPION): From Fundamentals to State-of-the-Art Innovative Applications for Cancer Therapy. Pharmaceutics 2023, 15, 236. [Google Scholar] [CrossRef]
- Tao, L.; Faig, A.; Uhrich, K.E. Liposomal Stabilization Using a Sugar-Based, PEGylated Amphiphilic Macromolecule. J. Colloid. Interface Sci. 2014, 431, 112–116. [Google Scholar] [CrossRef]
- Akhtar, N.; Mohammed, S.A.A.; Singh, V.; Abdellatif, A.A.H.; Mohammad, H.A.; Ahad, A.; Yusuf, M.; Khadri, H.; Naz, M.; Khan, O.; et al. Liposome-Based Drug Delivery of Various Anticancer Agents of Synthetic and Natural Product Origin: A Patent Overview. Pharm. Pat. Anal. 2020, 9, 87–116. [Google Scholar] [CrossRef]
- Cheng, X.; Yan, H.; Pang, S.; Ya, M.; Qiu, F.; Qin, P.; Zeng, C.; Lu, Y. Liposomes as Multifunctional Nano-Carriers for Medicinal Natural Products. Front. Chem. 2022, 10, 963004. [Google Scholar] [CrossRef]
- Kulkarni, B.; Qutub, S.; Ladelta, V.; Khashab, N.M.; Hadjichristidis, N. AIE-Based Fluorescent Triblock Copolymer Micelles for Simultaneous Drug Delivery and Intracellular Imaging. Biomacromolecules 2021, 22, 5243–5255. [Google Scholar] [CrossRef]
- Kuperkar, K.; Patel, D.; Atanase, L.; Bahadur, P. Amphiphilic Block Copolymers: Their Structures, and Self-Assembly to Polymeric Micelles and Polymersomes as Drug Delivery Vehicles. Polym. 2022, 14, 4702. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, Z.; Zhang, Y. The Application of Carbon Nanotubes in Target Drug Delivery Systems for Cancer Therapies. Nanoscale Res. Lett. 2011, 6, 555. [Google Scholar] [CrossRef]
- Kang, B.; Chang, S.; Dai, Y.; Yu, D.; Chen, D. Cell Response to Carbon Nanotubes: Size-Dependent Intracellular Uptake Mechanism and Subcellular Fate. Small 2010, 6, 2362–2366. [Google Scholar] [CrossRef] [PubMed]
- Wong Shi Kam, N.; Liu, Z.; Dai, H.; Kam, W.S.; Liu, Z.; Dai, H. Carbon Nanotubes as Intracellular Transporters for Proteins and DNA: An Investigation of the Uptake Mechanism and Pathway. Angew. Chem. Int. Ed. 2005, 44, 1–6. [Google Scholar] [CrossRef]
- Gherman, C.; Tudo, M.; Constantin, B.; Flaviu, T.; Stefan, R.; Maria, B.; Chira, S.; Braicu, C.; Pop, L.; Petric, R.C.; et al. Pharmacokinetics Evaluation of Carbon Nanotubes Using FTIR Analysis and Histological Analysis. J. Nanosci. Nanotechnol. 2015, 15, 2865–2869. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xu, K.; Qin, L.; Zhao, D.; Yang, N.; Wang, D.; Yang, Y. Hollow Nanomaterials in Advanced Drug Delivery Systems: From Single- to Multiple Shells. Adv. Mater. 2023, 35, 2203890. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Liu, H.; Ye, Y.; Lei, Y.; Islam, R.; Tan, S.; Tong, R.; Miao, Y.B.; Cai, L. Smart Nanoparticles for Cancer Therapy. Signal Transduct. Target. Ther. 2023, 8, 418. [Google Scholar] [CrossRef]
- Tang, L.; He, S.; Yin, Y.; Liu, H.; Hu, J.; Cheng, J.; Wang, W. Combination of Nanomaterials in Cell-Based Drug Delivery Systems for Cancer Treatment. Pharmaceutics 2021, 13, 1888. [Google Scholar] [CrossRef]
- Tian, H.; Zhang, T.; Qin, S.; Huang, Z.; Zhou, L.; Shi, J.; Nice, E.C.; Xie, N.; Huang, C.; Shen, Z. Enhancing the Therapeutic Efficacy of Nanoparticles for Cancer Treatment Using Versatile Targeted Strategies. J. Hematol. Oncol. 2022, 15, 132. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Niu, M.; Yuan, X.; Wu, K.; Liu, A. CD44 as a Tumor Biomarker and Therapeutic Target. Exp. Hematol. Oncol. 2020, 9, 36. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Magid, M.S.; Kramer, E.E.; Chen, Y.-T. Follicle-Stimulating Hormone Receptor Is Expressed in Human Ovarian Surface Epithelium and Fallopian Tube. Am. J. Pathol. 1996, 14, 47–53. [Google Scholar]
- Igaz, N.; Kovács, D.; Rázga, Z.; Kónya, Z.; Boros, I.M.; Kiricsi, M. Modulating Chromatin Structure and DNA Accessibility by Deacetylase Inhibition Enhances the Anti-Cancer Activity of Silver Nanoparticles. Colloids Surf. B Biointerfaces 2016, 146, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, B.; Thankachan, S.; Suresh, P.S. Co-Delivery of JQ1 and Gambogic Acid through Tumor-Targeting PEGylated Nanographene Oxide with FSH Beta Peptide (33–53) Identifies SNHG7-Hsa-MiR-324-3p SMAD2 Axis as a Prognostic Biomarker in Ovarian Cancer. J. Drug Deliv. Sci. Technol. 2025, 104, 106537. [Google Scholar] [CrossRef]
- Masoudi, E.; Soleimani, M.; Zarinfard, G.; Homayoun, M.; Bakhtiari, M. The Effects of Chitosan-Loaded JQ1 Nanoparticles on OVCAR-3 Cell Cycle and Apoptosis-Related Gene Expression. Res. Pharm. Sci. 2024, 19, 53–63. [Google Scholar] [CrossRef]
- Zhang, X.F.; Huang, F.H.; Zhang, G.L.; Bai, D.P.; de Felici, M.; Huang, Y.F.; Gurunathan, S. Novel Biomolecule Lycopene-Reduced Graphene Oxide-Silver Nanoparticle Enhances Apoptotic Potential of Trichostatin A in Human Ovarian Cancer Cells (SKOV3). Int. J. Nanomed. 2017, 12, 7551–7575. [Google Scholar] [CrossRef]
- Oner, E.; Kotmakci, M.; Baird, A.M.; Gray, S.G.; Debelec Butuner, B.; Bozkurt, E.; Kantarci, A.G.; Finn, S.P. Development of EphA2 SiRNA-Loaded Lipid Nanoparticles and Combination with a Small-molecule Histone Demethylase Inhibitor in Prostate Cancer Cells and Tumor. J. Nanobiotechnology 2021, 19, 71. [Google Scholar] [CrossRef]
- Zhong, T.; Liu, X.; Li, H.; Zhang, J. Co-Delivery of Sorafenib and Crizotinib Encapsulated with Polymeric Nanoparticles for the Treatment of in Vivo Lung Cancer Animal Model. Drug Deliv. 2021, 28, 2108–2118. [Google Scholar] [CrossRef]
- Korucu Aktas, P.; Baysal, I.; Yabanoglu-Ciftci, S.; Arica, B. Development and In Vitro Evaluation of Crizotinib-Loaded Lipid–Polymer Hybrid Nanoparticles Using Box–Behnken Design in Non-Small Cell Lung Cancer. AAPS PharmSciTech 2023, 24, 178. [Google Scholar] [CrossRef]
- Huang, C.S.; Xu, Q.C.; Dai, C.; Wang, L.; Tien, Y.C.; Li, F.; Su, Q.; Huang, X.T.; Wu, J.; Zhao, W.; et al. Nanomaterial-Facilitated Cyclin-Dependent Kinase 7 Inhibition Suppresses Gallbladder Cancer Progression via Targeting Transcriptional Addiction. ACS Nano 2021, 15, 14744–14755. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Zou, B.; Luo, L.; Zhong, C.; Zhang, P.; Cheng, H.; Guo, Y.; Gou, M. Codelivery of Thioridazine and Doxorubicin Using Nanoparticles for Effective Breast Cancer Therapy. Int. J. Nanomed. 2016, 11, 4545–4552. [Google Scholar] [CrossRef] [PubMed]
- Mohiuddin, S.; Ngo, H.; Orman, M.A. Unveiling the Critical Roles of Cellular Metabolism Suppression in Antibiotic Tolerance. npj Antimicrob. Resist. 2024, 2, 17. [Google Scholar] [CrossRef]
- Rivankar, S. An Overview of Doxorubicin Formulations in Cancer Therapy. J. Cancer Res. Ther. 2014, 10, 853–858. [Google Scholar] [CrossRef]
- An, J.; Li, H.; Wen, W.; Liu, G.; Huang, Z.; Zhao, X.; Liang, G. Dual Inhibition of GPX4 and LCN2 via MiR-214-3p Loaded Iron Oxide Nanoparticles for Enhancing Ferroptosis in Liver Cancer. Colloids Surf. A Physicochem. Eng. Asp. 2025, 719, 137049. [Google Scholar] [CrossRef]
- Gonzalez, T.; Muminovic, M.; Nano, O.; Vulfovich, M. Folate Receptor Alpha—A Novel Approach to Cancer Therapy. Int. J. Mol. Sci. 2024, 25, 1046. [Google Scholar] [CrossRef]
- Chaudhary, N.; Choudhary, B.S.; Shah, S.G.; Khapare, N.; Dwivedi, N.; Gaikwad, A.; Joshi, N.; Raichanna, J.; Basu, S.; Gurjar, M.; et al. Lipocalin 2 Expression Promotes Tumor Progression and Therapy Resistance by Inhibiting Ferroptosis in Colorectal Cancer. Int. J. Cancer 2021, 149, 1495–1511. [Google Scholar] [CrossRef]
- Yang, W.; SriRamaratnam, R.; Welsch, M.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wirz, R. Cold Atmospheric Plasma (CAP) Technology and Applications; Morgan and Claypool: San Rafael, CA, USA, 2021; ISBN 9781636391816. [Google Scholar]
- Cao, X.; Chen, M.; Fang, T.; Deng, Y.; Wang, L.; Wang, H.; Chen, Z.; Chen, G. RSL3-Loaded Nanoparticles Amplify the Therapeutic Potential of Cold Atmospheric Plasma. J. Nanobiotechnology 2025, 23, 136. [Google Scholar] [CrossRef]
- Gurunathan, S.; Jeyaraj, M.; Kang, M.H.; Kim, J.H. Tangeretin-Assisted Platinum Nanoparticles Enhance the Apoptotic Properties of Doxorubicin: Combination Therapy for Osteosarcoma Treatment. Nanomaterials 2019, 9, 1089. [Google Scholar] [CrossRef]
- Genc, S.; Taghizadehghalehjoughi, A.; Yeni, Y.; Jafarizad, A.; Hacimuftuoglu, A.; Nikitovic, D.; Docea, A.O.; Mezhuev, Y.; Tsatsakis, A. Fe3O4 Nanoparticles in Combination with 5-FU Exert Antitumor Effects Superior to Those of the Active Drug in a Colon Cancer Cell Model. Pharmaceutics 2023, 15, 245. [Google Scholar] [CrossRef] [PubMed]
- Chin, Y.C.; Yang, L.X.; Hsu, F.T.; Hsu, C.W.; Chang, T.W.; Chen, H.Y.; Chen, L.Y.C.; Chia, Z.C.; Hung, C.H.; Su, W.C.; et al. Iron Oxide@ Chlorophyll Clustered Nanoparticles Eliminate Bladder Cancer by Photodynamic Immunotherapy-Initiated Ferroptosis and Immunostimulation. J. Nanobiotechnology 2022, 20, 373. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.A.; Horvat, N.K.; Marques, O.; Chocarro, S.; Mertens, C.; Colucci, S.; Schmitt, S.; Carrella, L.M.; Morsbach, S.; Koynov, K.; et al. Core Cross-Linked Polymeric Micelles for Specific Iron Delivery: Inducing Sterile Inflammation in Macrophages. Adv. Heal. Mater. 2021, 10, e2100385. [Google Scholar] [CrossRef]
- Horvat, N.K.; Chocarro, S.; Marques, O.; Bauer, T.A.; Qiu, R.; Diaz-Jimenez, A.; Helm, B.; Chen, Y.; Sawall, S.; Sparla, R.; et al. Superparamagnetic Iron Oxide Nanoparticles Reprogram the Tumor Microenvironment and Reduce Lung Cancer Regrowth after Crizotinib Treatment. ACS Nano 2024, 18, 11025–11041. [Google Scholar] [CrossRef]
- Hu, Z.; Li, R.; Cui, X.; Chen, Z. Albumin-Based Cyanine Crizotinib Conjugate Nanoparticles for NIR-II Imaging-Guided Synergistic Chemophototherapy. ACS Appl. Mater. Interfaces 2023, 15, 33890–33902. [Google Scholar] [CrossRef]
- Yu, H.; Li, J.; Deng, K.; Zhou, W.; Li, K.; Wang, C.X.; Wang, Q.; Wu, M.; Huang, S.W. GPX4 Inhibition Synergistically Boosts Mitochondria Targeting Nanoartemisinin-Induced Apoptosis/Ferroptosis Combination Cancer Therapy. Biomater. Sci. 2023, 11, 5831–5845. [Google Scholar] [CrossRef]
- Zhao, R.; Li, T.; Zheng, G.; Jiang, K.; Fan, L.; Shao, J. Simultaneous Inhibition of Growth and Metastasis of Hepatocellular Carcinoma by Co-Delivery of Ursolic Acid and Sorafenib Using Lactobionic Acid Modified and PH-Sensitive Chitosan-Conjugated Mesoporous Silica Nanocomplex. Biomaterials 2017, 143, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yang, G.; Han, L.; Wang, R.; Gong, C.; Yuan, Y. Sorafenib and Triptolide Loaded Cancer Cell-Platelet Hybrid Membrane-Camouflaged Liquid Crystalline Lipid Nanoparticles for the Treatment of Hepatocellular Carcinoma. J. Nanobiotechnology 2021, 19, 360. [Google Scholar] [CrossRef]
- Younis, M.; Khalil, I.; Elewa, Y.; Kon, Y.; Harashima, H. Ultra-Small Lipid Nanoparticles Encapsulating Sorafenib and Midkine-SiRNA Selectively-Eradicate Sorafenib-Resistant Hepatocellular Carcinoma in Vivo. J. Control. Release 2021, 331, 335–349. [Google Scholar] [CrossRef]
- Lin, J.; Zhang, J.; Wang, K.; Guo, S.; Yang, W. Zwitterionic Polymer Coated Sorafenib-Loaded Fe 3 O 4 Composite Nanoparticles Induced Ferroptosis for Cancer Therapy. J. Mater. Chem. B 2022, 10, 5784–5795. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Song, K.; Wang, X.; He, Z.; Du, J.; Sun, J.L.; Xu, R.C.; Liu, Z.Y.; Qi, Z.R.; Wang, F.; et al. Bismuth-Based Mesoporous Nanoball Carrying Sorafenib for Synergistic Photothermal and Molecularly-Targeted Therapy in an Orthotopic Hepatocellular Carcinoma Xenograft Mouse Model. Colloids Surf. B Biointerfaces 2025, 245, 114279. [Google Scholar] [CrossRef] [PubMed]
- Rink, J.; Calvert, A.; Kwon, D.; Zhang, X.; Yin, H.H.; Horne, D.; Nguyen, S.T.; Lin, A.Y.; Rosen, S.T.; Gordon, L.I.; et al. Receptor Targeted Delivery of the Multi-Kinase Inhibitor F7/PIK-75 by Organic-Core Templated Lipid Nanoparticles as Cancer Therapy. Cancer Res. 2025, 85, 4467. [Google Scholar] [CrossRef]
- Logan, A.; Howard, C.; Huda, P.; Kimpton, K.; Ma, Z.; Thurecht, K.J.; McCarroll, J.A.; Moles, E.; Kavallaris, M. Targeted Delivery of Polo-like Kinase 1 SiRNA Nanoparticles Using an EGFR-PEG Bispecific Antibody Inhibits Proliferation of High-Risk Neuroblastoma. J. Control Release 2024, 367, 806–820. [Google Scholar] [CrossRef]



| Cancer Types | Exp. Model | Treatment | Mechanism of DTP | Target of Therapy | Proposed Drug to Target DTPs | Ref. |
|---|---|---|---|---|---|---|
| NCLC | Cell lines; PC9, HCC4006 | Osimertinib + trametinib | Transcriptional regulation | YAP-TEAD pathway | TEAD-inhibitor (MYF-01-37) | [44] |
| Cell lines; PC9 | EGFR TKI Erlotinib | Epigenetic modification and transcription regulation | H3K9 methylation over LINE-1 elements | HDAC inhibitor (MS275 or TSA) | [61] | |
| Cell lines; PC9, H1975, HCC827, HCC4006 | EGFR TKI Erlotinib | Signaling pathway | FGFR3 signaling | Pan-FGFR inhibitor (infigratinib) | [45] | |
| Cell lines; PC9, HCC4011 | EGFR TKI Osimertinib | Signaling pathway | AXL signaling | AXL inhibitor (NPS1034) | [72] | |
| NCLC, GC | Cell lines; PC9, MKN45 | EGFR TKI Erlotinib, crizotinib | Metabolic changes | ALDH | ALDH inhibitor (Disulfiram) | [19] |
| LUAD | Cell lines; A549, H460 and PDO | MEK inhibitor Trametinib | Metabolic changes | Mitophagy | Chloroquine | [73] |
| Melanoma | Cell lines; SK-Mel-5, SK-Mel-28, G-361 | Vemurafenib | Tumor microenvironment | HGF/MET paracrine pathway | MET inhibitor crizotinib | [70] |
| BBC | PDX | Hedgehog inhibitor Vismodegib | Signaling pathway | Wnt signaling | Wnt signal inhibitor (LGK-974) | [74] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghoderao, P.; Kwiatkowska-Borowczyk, E.; Sahare, S.; Dams-Kozlowska, H. Targeting Drug-Tolerant Persister Cancer Cells: Can Nanomaterial-Based Strategies Be Helpful for Anti-DTP Therapies? Pharmaceutics 2025, 17, 1428. https://doi.org/10.3390/pharmaceutics17111428
Ghoderao P, Kwiatkowska-Borowczyk E, Sahare S, Dams-Kozlowska H. Targeting Drug-Tolerant Persister Cancer Cells: Can Nanomaterial-Based Strategies Be Helpful for Anti-DTP Therapies? Pharmaceutics. 2025; 17(11):1428. https://doi.org/10.3390/pharmaceutics17111428
Chicago/Turabian StyleGhoderao, Prachi, Eliza Kwiatkowska-Borowczyk, Sanjay Sahare, and Hanna Dams-Kozlowska. 2025. "Targeting Drug-Tolerant Persister Cancer Cells: Can Nanomaterial-Based Strategies Be Helpful for Anti-DTP Therapies?" Pharmaceutics 17, no. 11: 1428. https://doi.org/10.3390/pharmaceutics17111428
APA StyleGhoderao, P., Kwiatkowska-Borowczyk, E., Sahare, S., & Dams-Kozlowska, H. (2025). Targeting Drug-Tolerant Persister Cancer Cells: Can Nanomaterial-Based Strategies Be Helpful for Anti-DTP Therapies? Pharmaceutics, 17(11), 1428. https://doi.org/10.3390/pharmaceutics17111428