

Membrane-Fusing Vehicles for Re-Sensitizing Transporter-Mediated Multiple-Drug Resistance in Cancer



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation and Characterization of MFVs
2.3. Cell Culture Studies
2.3.1. Confocal Laser Scanning Microscopy (CLSM)
2.3.2. Substrate Accumulation Assay
2.3.3. MTT Cytotoxicity Assay
2.3.4. Cell Toxicity of the Cytotoxic Drugs
2.4. Statistical Analysis
3. Results
3.1. Physicochemical Characterization of MFV
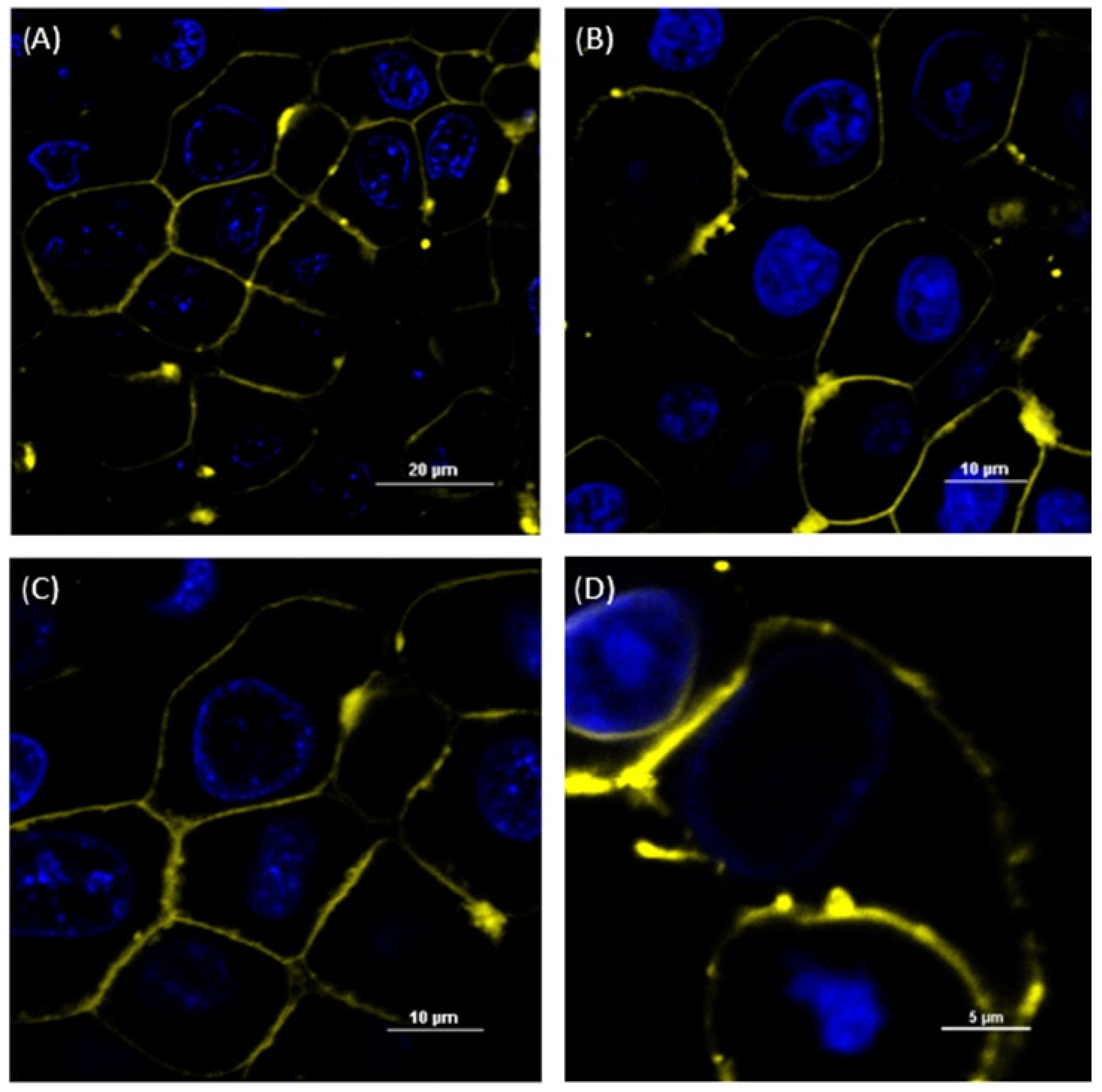
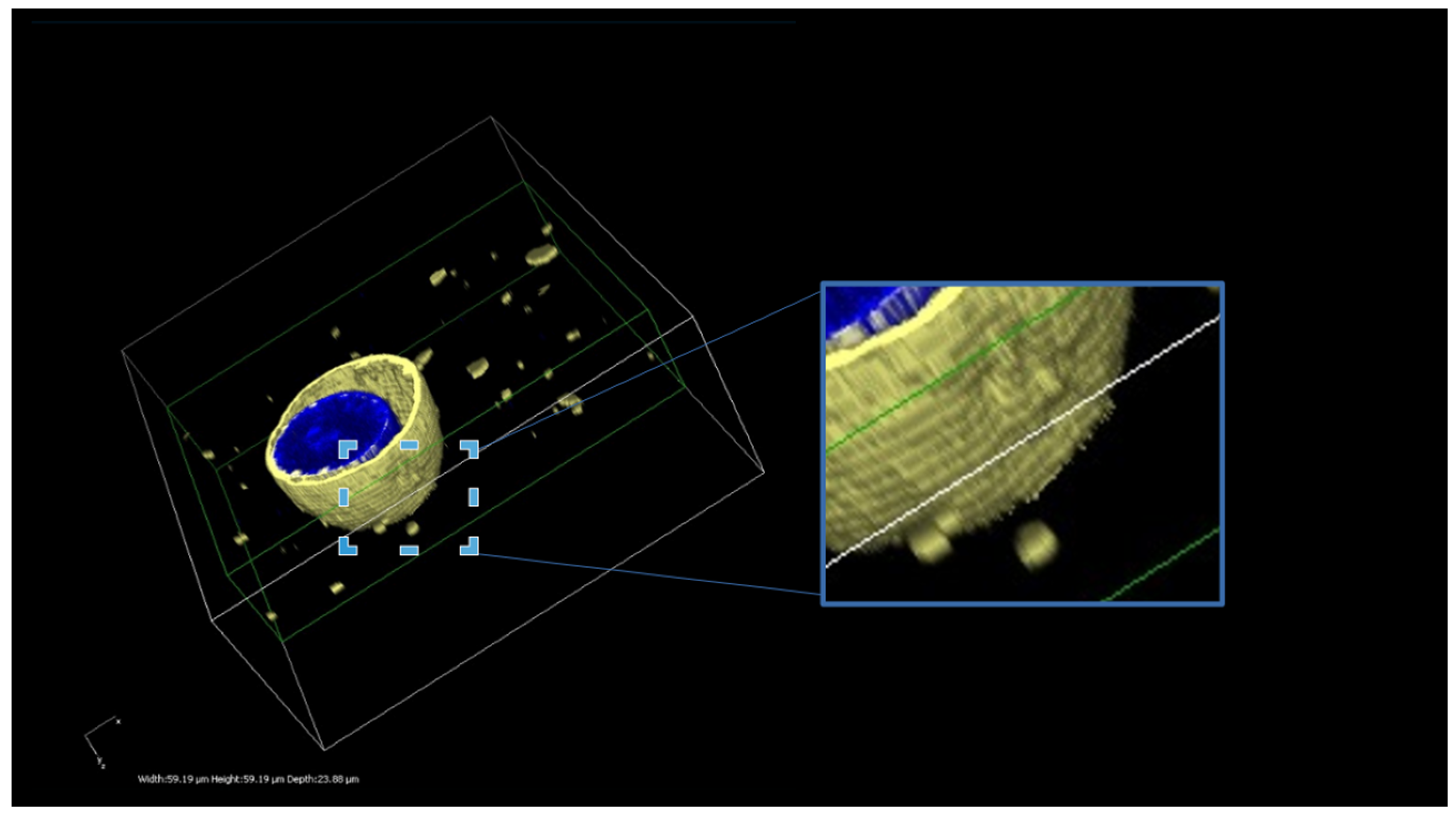
3.2. Imaging of Membrane-Fusing Vehicles
3.3. Analysis of In Vitro Experiment Results: Key Observations
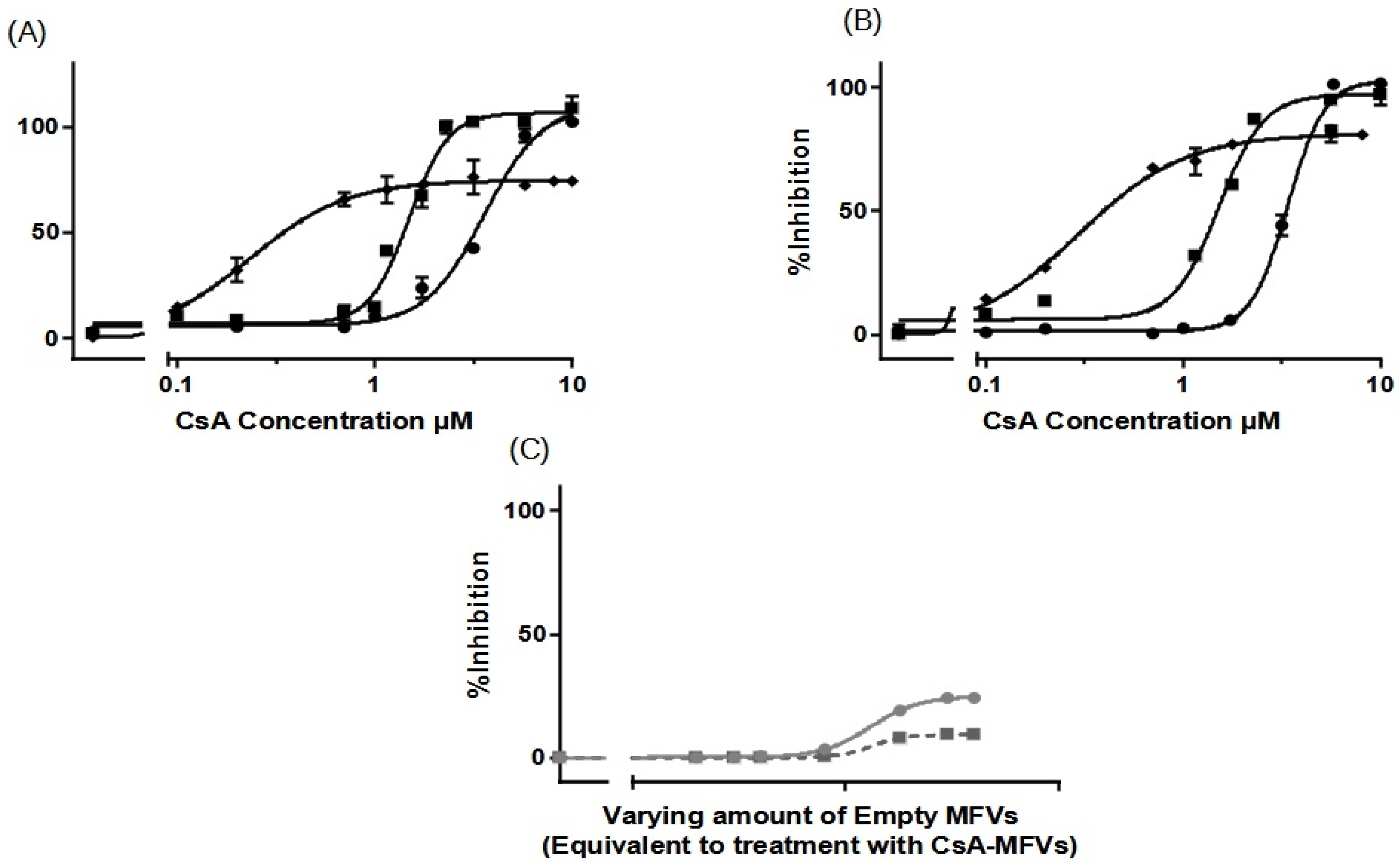
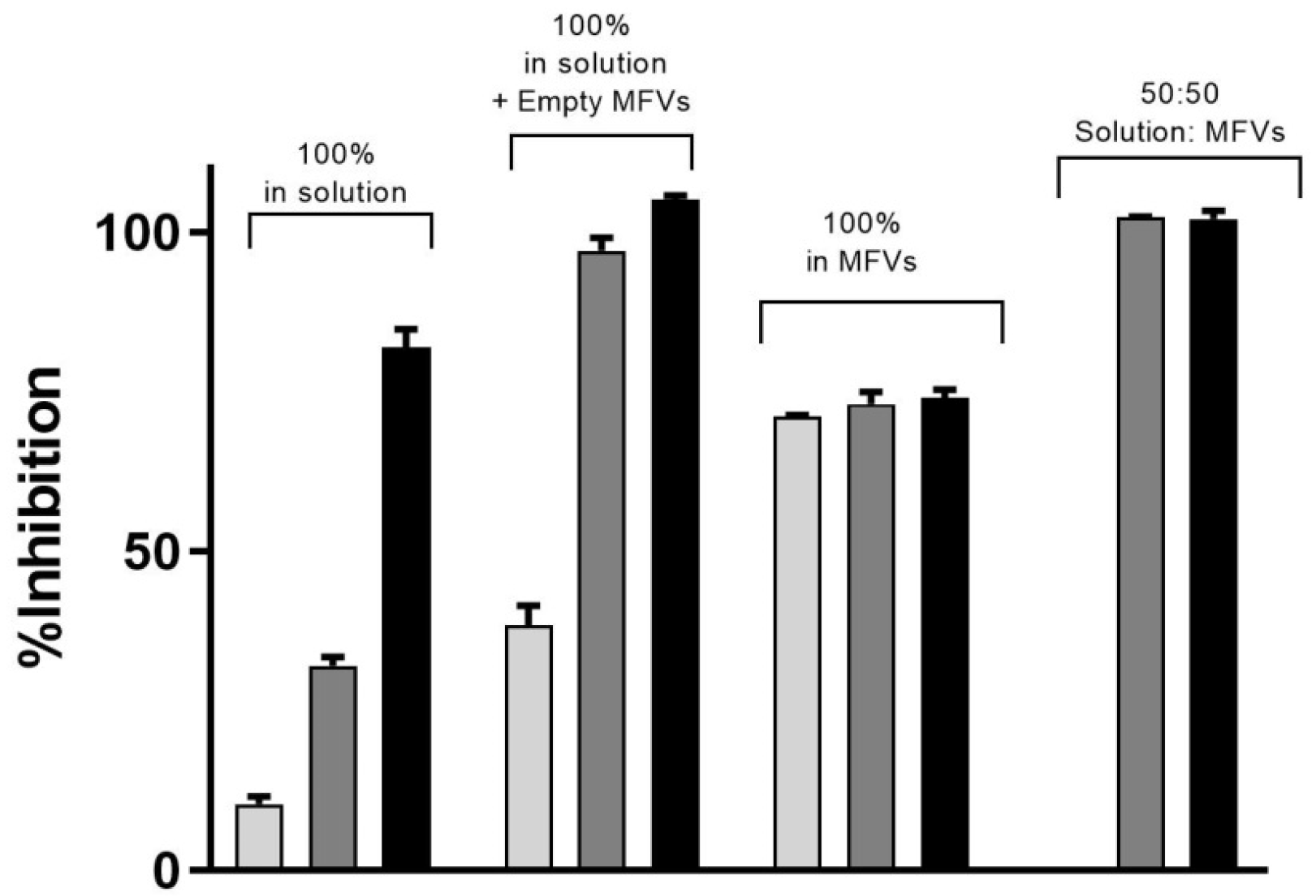
3.3.1. Substrate Accumulation Assay
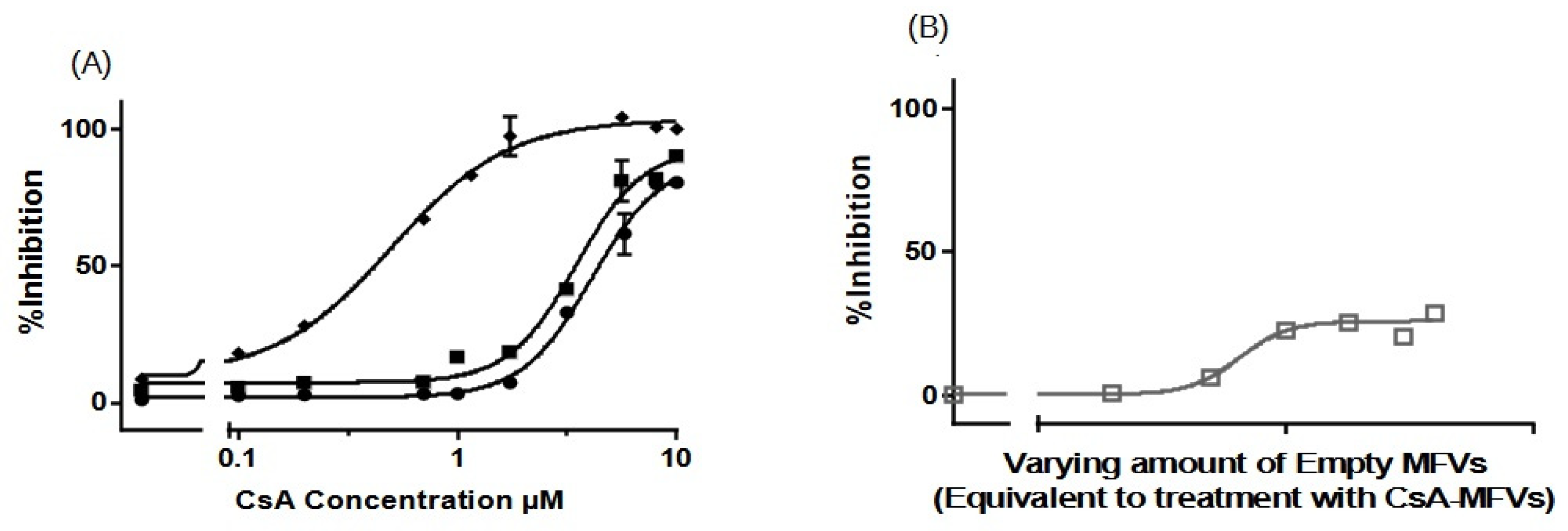
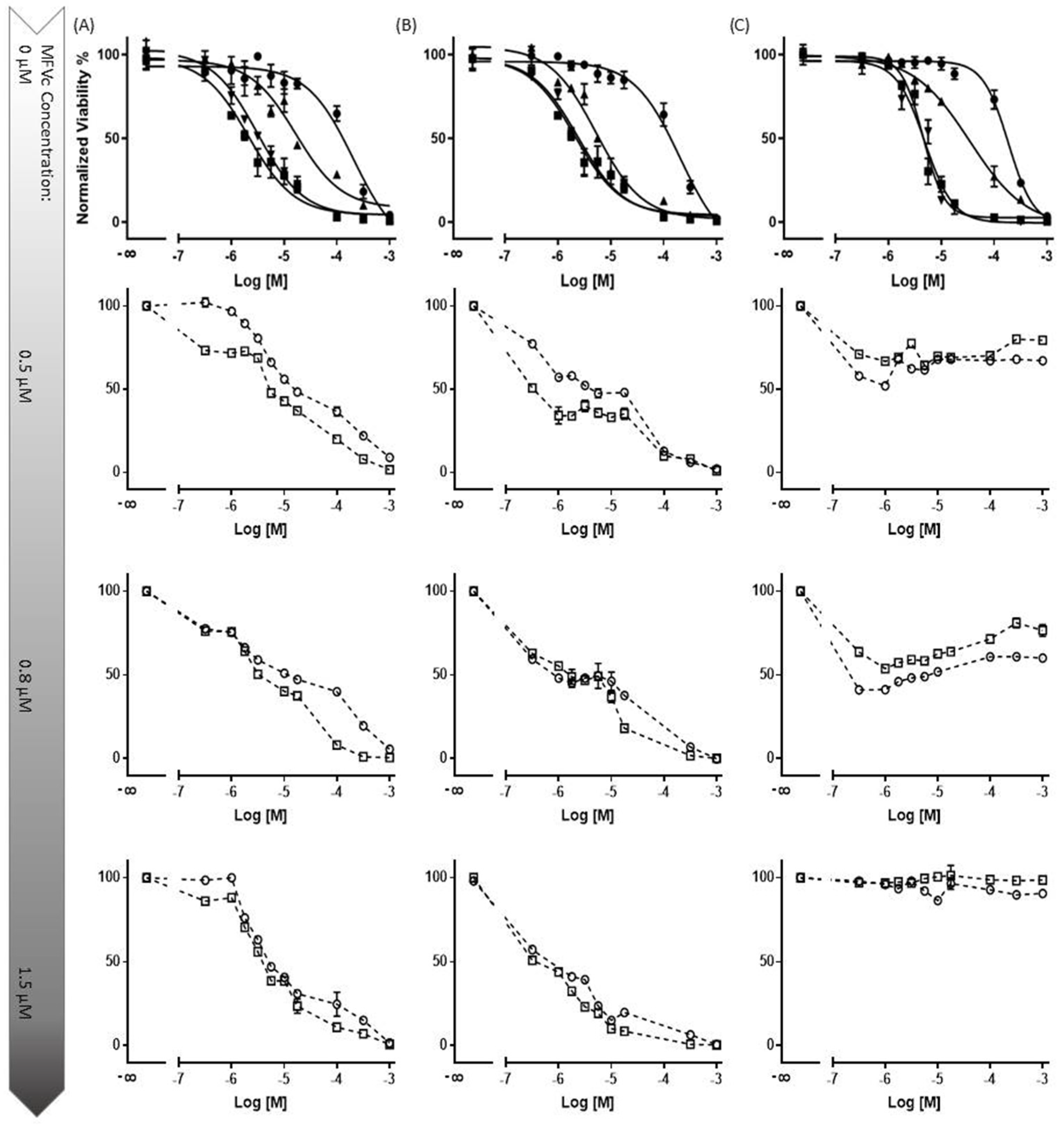
3.3.2. Concentration-Dependent Drug Delivery Using MFVs

3.3.3. Cell Toxicity of the Cytotoxic Drugs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef] [PubMed]
- Khunweeraphong, N.; Stockner, T.; Kuchler, K. The structure of the human ABC transporter ABCG2 reveals a novel mechanism for drug extrusion. Sci. Rep. 2017, 7, 13767. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Ni, D.; Kowal, J.; Manolaridis, I.; Jackson, S.M.; Stahlberg, H.; Locher, K.P. Structures of ABCG2 under turnover conditions reveal a key step in the drug transport mechanism. Nat. Commun. 2021, 12, 4376. [Google Scholar] [CrossRef]
- Choi, Y.H.; Yu, A.-M. ABC Transporters in Multidrug Resistance and Pharmacokinetics, and Strategies for Drug Development. Curr. Pharm. Des. 2014, 20, 793–807. [Google Scholar] [CrossRef] [PubMed]
- Sharom, F.J. The P-glycoprotein multidrug transporter. Essays Biochem. 2011, 50, 161–178. [Google Scholar] [CrossRef] [PubMed]
- Antoni, F. Zur Erlangung des Doktorgrades der Naturwissenschaften (Dr. Rer. Nat.) an der Fakultät Chemie S Pharmazie der Universität Regensburg. 291. Available online: https://epub.uni-regensburg.de/34630/1/Pompe_Dissertation%202016.pdf (accessed on 24 March 2024).
- Jackson, S.M.; Manolaridis, I.; Kowal, J.; Zechner, M.; Taylor, N.M.I.; Bause, M.; Bauer, S.; Bartholomaeus, R.; Bernhardt, G.; Koenig, B.; et al. Structural basis of small-molecule inhibition of human multidrug transporter ABCG2. Nat. Struct. Mol. Biol. 2018, 25, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Lei, T.; Wang, Z.; Shen, C.; Cao, D.; Hou, T. ADMET evaluation in drug discovery. 20. Prediction of breast cancer resistance protein inhibition through machine learning. J. Cheminform. 2020, 12, 16. [Google Scholar] [CrossRef]
- Rehman, S.; Nabi, B.; Fazil, M.; Khan, S.; Bari, N.K.; Singh, R.; Ahmad, S.; Kumar, V.; Baboota, S.; Ali, J. Role of P-Glycoprotein Inhibitors in the Bioavailability Enhancement of Solid Dispersion of Darunavir. BioMed Res. Int. 2017, 2017, e8274927. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.S.; Tammam, S.N.; Shetab Boushehri, M.A.; Lamprecht, A. MDR in cancer: Addressing the underlying cellular alterations with the use of nanocarriers. Pharmacol. Res. 2017, 126, 2–30. [Google Scholar] [CrossRef]
- Knight, V.; Koshkina, N.V.; Golunski, E.; Roberts, L.E.; Gilbert, B.E. Cyclosporin A aerosol improves the anticancer effect of paclitaxel aerosol in mice. Trans. Am. Clin. Climatol. Assoc. 2004, 115, 395–404; discussion 404. [Google Scholar]
- Gao, W.; Lin, Z.; Chen, M.; Yang, X.; Cui, Z.; Zhang, X.; Yuan, L.; Zhang, Q. The co-delivery of a low-dose P-glycoprotein inhibitor with doxorubicin sterically stabilized liposomes against breast cancer with low P-glycoprotein expression. Int. J. Nanomed. 2014, 9, 3425–3437. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zhang, H.; Jiang, L.; Wei, W.; Liu, C.; Cang, S. Enhancing the cytotoxic efficacy of combined effect of doxorubicin and Cyclosporin encapsulated photoluminescent graphene dotted mesoporous nanoparticles against lung cancer cell-specific drug targeting for the nursing care of cancer patients. J. Photochem. Photobiol. B Biol. 2019, 198, 111578. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Shi, L.; Ren, L.; Zhou, L.; Li, T.; Qiao, Y.; Wang, H. A nanomedicine approach enables co-delivery of cyclosporin A and gefitinib to potentiate the therapeutic efficacy in drug-resistant lung cancer. Signal Transduct. Target. Ther. 2018, 3, 16. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Liu, X.; Wang, H.; Liu, X.; Zhang, Q.; Li, K.; Chen, Y.; Zhu, Q.; Shen, Y.; Sui, M. A nanotherapeutic strategy to overcome chemoresistance to irinotecan/7-ethyl-10-hydroxy-camptothecin in colorectal cancer. Acta Biomater. 2021, 137, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Zhou, Y.; Liu, L.; Xu, Y.; Chen, Q.; Wang, Y.; Wu, S.; Deng, Y.; Zhang, J.; Shao, A. Nanoparticle-Based Drug Delivery in Cancer Therapy and Its Role in Overcoming Drug Resistance. Front. Mol. Biosci. 2020, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Soma, C.E.; Dubernet, C.; Bentolila, D.; Benita, S.; Couvreur, P. Reversion of multidrug resistance by co-encapsulation of doxorubicin and cyclosporin A in polyalkylcyanoacrylate nanoparticles. Biomaterials 2000, 21, 1–7. [Google Scholar] [CrossRef]
- Farid, M.; Faber, T.; Dietrich, D.; Lamprecht, A. Cell membrane fusing liposomes for cytoplasmic delivery in brain endothelial cells. Colloids Surf. B Biointerfaces 2020, 194, 111193. [Google Scholar] [CrossRef] [PubMed]
- Czogalla, A. Oral cyclosporine A—The current picture of its liposomal and other delivery systems. Cell. Mol. Biol. Lett. 2009, 14, 139–152. [Google Scholar] [CrossRef]
- Bangham, A.D.; Horne, R.W. Negative staining of phospholipids and their structural modification by surface-active agents as observed in the electron microscope. J. Mol. Biol. 1964, 8, 660–668. [Google Scholar] [CrossRef]
- Hupfeld, S.; Holsaeter, A.M.; Skar, M.; Frantzen, C.B.; Brandl, M. Liposome size analysis by dynamic/static light scattering upon size exclusion-/field flow-fractionation. J. Nanosci. Nanotechnol. 2006, 6, 3025–3031. [Google Scholar] [CrossRef]
- Aljohani, B.; Alotaibi, F.F.; Ghazaly, E.; Jaber, J.A.; Perrett, D.; Johnston, A. Development and Validation of A HPLC-UV Method for Dissolution Testing of Ciclosporin: Its Application to The Measurement of Brand and Generic Versions from Different Countries. J. Bioequiv. Availab. 2017, 9, 509–519. [Google Scholar] [CrossRef]
- Strouse, J.J.; Ivnitski-Steele, I.; Waller, A.; Young, S.M.; Perez, D.; Evangelisti, A.M.; Ursu, O.; Bologa, C.G.; Carter, M.B.; Salas, V.M.; et al. Fluorescent substrates for flow cytometric evaluation of efflux inhibition in ABCB1, ABCC1, and ABCG2 transporters. Anal. Biochem. 2013, 437, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Twentyman, P.R.; Rhodes, T.; Rayner, S. A comparison of rhodamine 123 accumulation and efflux in cells with P-glycoprotein-mediated and MRP-associated multidrug resistance phenotypes. Eur. J. Cancer 1994, 30A, 1360–1369. [Google Scholar] [CrossRef] [PubMed]
- Olson, D.P.; Taylor, B.J.; Ivy, S.P. Detection of MRP functional activity: Calcein AM but not BCECF AM as a multidrug resistance-related protein (MRP1) substrate. Cytometry 2001, 46, 105–113. [Google Scholar] [CrossRef]
- Robey, R.W.; Steadman, K.; Polgar, O.; Morisaki, K.; Blayney, M.; Mistry, P.; Bates, S.E. Pheophorbide a Is a Specific Probe for ABCG2 Function and Inhibition. Cancer Res. 2004, 64, 1242–1246. [Google Scholar] [CrossRef] [PubMed]
- Dakhlaoui, I.; Vahdati, S.; Maalej, E.; Chabchoub, F.; Wiese, M.; Marco-Contelles, J.; Ismaili, L. Synthesis and biological assessment of new pyrimidopyrimidines as inhibitors of breast cancer resistance protein (ABCG2). Bioorg. Chem. 2021, 116, 105326. [Google Scholar] [CrossRef] [PubMed]
- Krapf, M.K.; Gallus, J.; Vahdati, S.; Wiese, M. New Inhibitors of Breast Cancer Resistance Protein (ABCG2) Containing a 2,4-Disubstituted Pyridopyrimidine Scaffold. J. Med. Chem. 2018, 61, 3389–3408. [Google Scholar] [CrossRef] [PubMed]
- Brgles, M.; Šantak, M.; Halassy, B.; Forcic, D.; Tomašić, J. Influence of charge ratio of liposome/DNA complexes on their size after extrusion and transfection efficiency. Int. J. Nanomed. 2012, 7, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.J.; Ferreira, M.-J.U.; dos Santos, D.J.V.A. Molecular Docking Characterizes Substrate-Binding Sites and Efflux Modulation Mechanisms within P-Glycoprotein. J. Chem. Inf. Model. 2013, 53, 1747–1760. [Google Scholar] [CrossRef]
- Ferreira, R.J.; Bonito, C.A.; Cordeiro, M.N.D.S.; Ferreira, M.-J.U.; dos Santos, D.J.V.A. Structure-function relationships in ABCG2: Insights from molecular dynamics simulations and molecular docking studies. Sci. Rep. 2017, 7, 15534. [Google Scholar] [CrossRef]
- Storch, C.H.; Ehehalt, R.; Haefeli, W.E.; Weiss, J. Localization of the human breast cancer resistance protein (BCRP/ABCG2) in lipid rafts/caveolae and modulation of its activity by cholesterol in vitro. J. Pharmacol. Exp. Ther. 2007, 323, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Nosol, K.; Romane, K.; Irobalieva, R.N.; Alam, A.; Kowal, J.; Fujita, N.; Locher, K.P. Cryo-EM structures reveal distinct mechanisms of inhibition of the human multidrug transporter ABCB1. Proc. Natl. Acad. Sci. USA 2020, 117, 26245–26253. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transporter vs. Substrate | Hoechst 33324 | Rhodamine 123 | Pheophorbide A | Calcein |
|---|---|---|---|---|
| ABCB1 | + | + | ||
| ABCG2 | + | − | + | − |
| ABCC1 | − | + | − | + |
| MFVs | Particle Size (nm) | Zeta Potential (mV) | Drug Load (%) | Entrapment Efficiency (%) |
|---|---|---|---|---|
| Drug Free | 110 ± 4 | 42 ± 5 | - | - |
| With DiI | 112 ± 7 | 40 ± 3 | - | - |
| With Nile-red | 127 ± 0 | 47 ± 6 | - | - |
| With CsA (0.25 mM) | 104 ± 0 | 41 ± 3 | 9 ± 1% | 98 ± 1% |
| With CsA (0.50 mM) | 103 ± 0 | 41 ± 0 | 16 ± 2% | 97 ± 3% |
| With CsA (0.75 mM) | 104 ± 0 | 40 ± 0 | 21 ± 1% | 88 ± 1% |
| With CsA (1.00 mM) | 105 ± 1 | 39 ± 5 | 23 ± 4% | 74 ± 6% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vahdati, S.; Lamprecht, A. Membrane-Fusing Vehicles for Re-Sensitizing Transporter-Mediated Multiple-Drug Resistance in Cancer. Pharmaceutics 2024, 16, 493. https://doi.org/10.3390/pharmaceutics16040493
Vahdati S, Lamprecht A. Membrane-Fusing Vehicles for Re-Sensitizing Transporter-Mediated Multiple-Drug Resistance in Cancer. Pharmaceutics. 2024; 16(4):493. https://doi.org/10.3390/pharmaceutics16040493
Chicago/Turabian StyleVahdati, Sahel, and Alf Lamprecht. 2024. "Membrane-Fusing Vehicles for Re-Sensitizing Transporter-Mediated Multiple-Drug Resistance in Cancer" Pharmaceutics 16, no. 4: 493. https://doi.org/10.3390/pharmaceutics16040493
APA StyleVahdati, S., & Lamprecht, A. (2024). Membrane-Fusing Vehicles for Re-Sensitizing Transporter-Mediated Multiple-Drug Resistance in Cancer. Pharmaceutics, 16(4), 493. https://doi.org/10.3390/pharmaceutics16040493