
Recent Advancements in Development and Therapeutic Applications of Genome-Targeting Triplex-Forming Oligonucleotides and Peptide Nucleic Acids



Abstract
1. Introduction
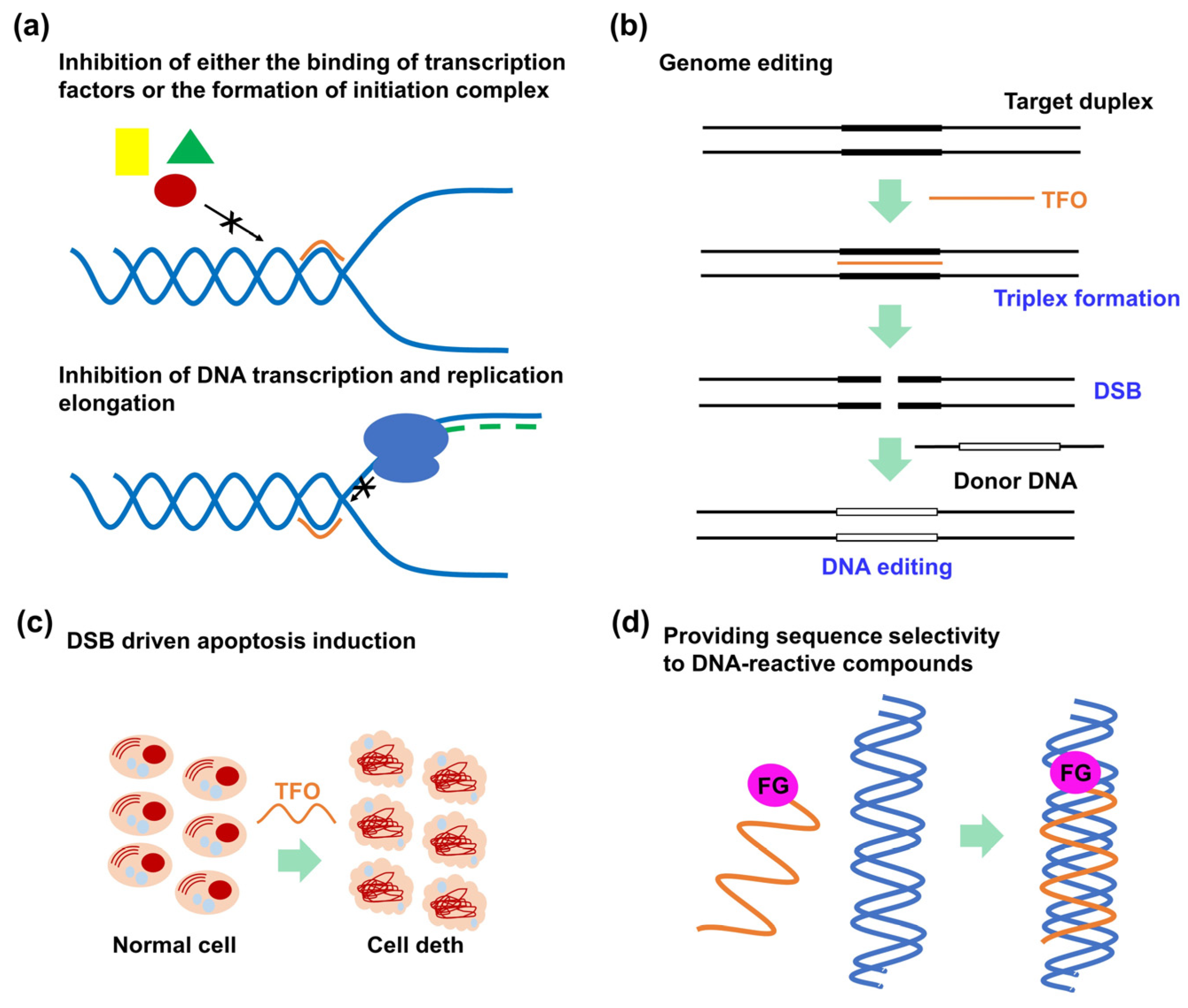
2. Triplex-Forming Oligonucleotides
2.1. Outline of the Triplex-Forming Oligonucleotide Technology
2.2. Advancements in the Engineering of Triplex-Forming Oligonucleotides
2.2.1. Triplex-Stabilization Technology
Artificial Nucleobases Recognizing Pyrimidine Base Interruption in the Target Double-Stranded DNA
- Recognition of the thymine–adenine base pair in a parallel motif
- Recognition of the guanine–cytosine base pair in a parallel motif
- Recognition of the thymine–adenine base pair in an antiparallel motif
- Recognition of the cytosine–guanine base pair in the antiparallel motif
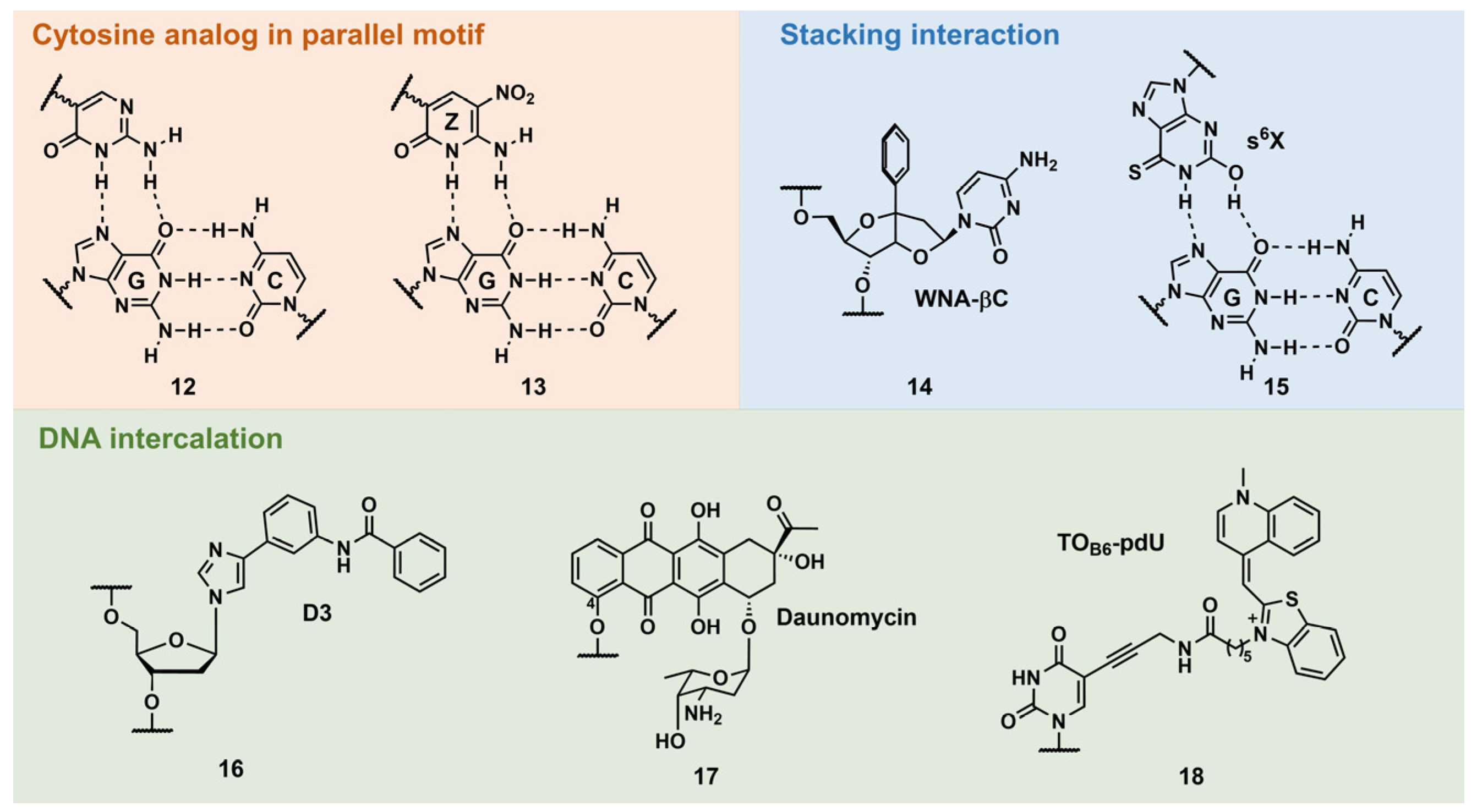
Other Triplex-Stabilization Techniques Using Artificial Nucleobases
- Cytosine mimic that can form hydrogen bonds with the guanine–cytosine base pair in a parallel motif
- Stacking interactions
- DNA intercalation
2.2.2. Recent Advancements in Functional Triplex-Forming Oligonucleotides for DNA Targeting
Targeted Crosslinking Using Psoralen-Conjugated Triplex-Forming Oligonucleotide
Targeted Crosslinking Using Platinum-Conjugated Triplex-Forming Oligonucleotides
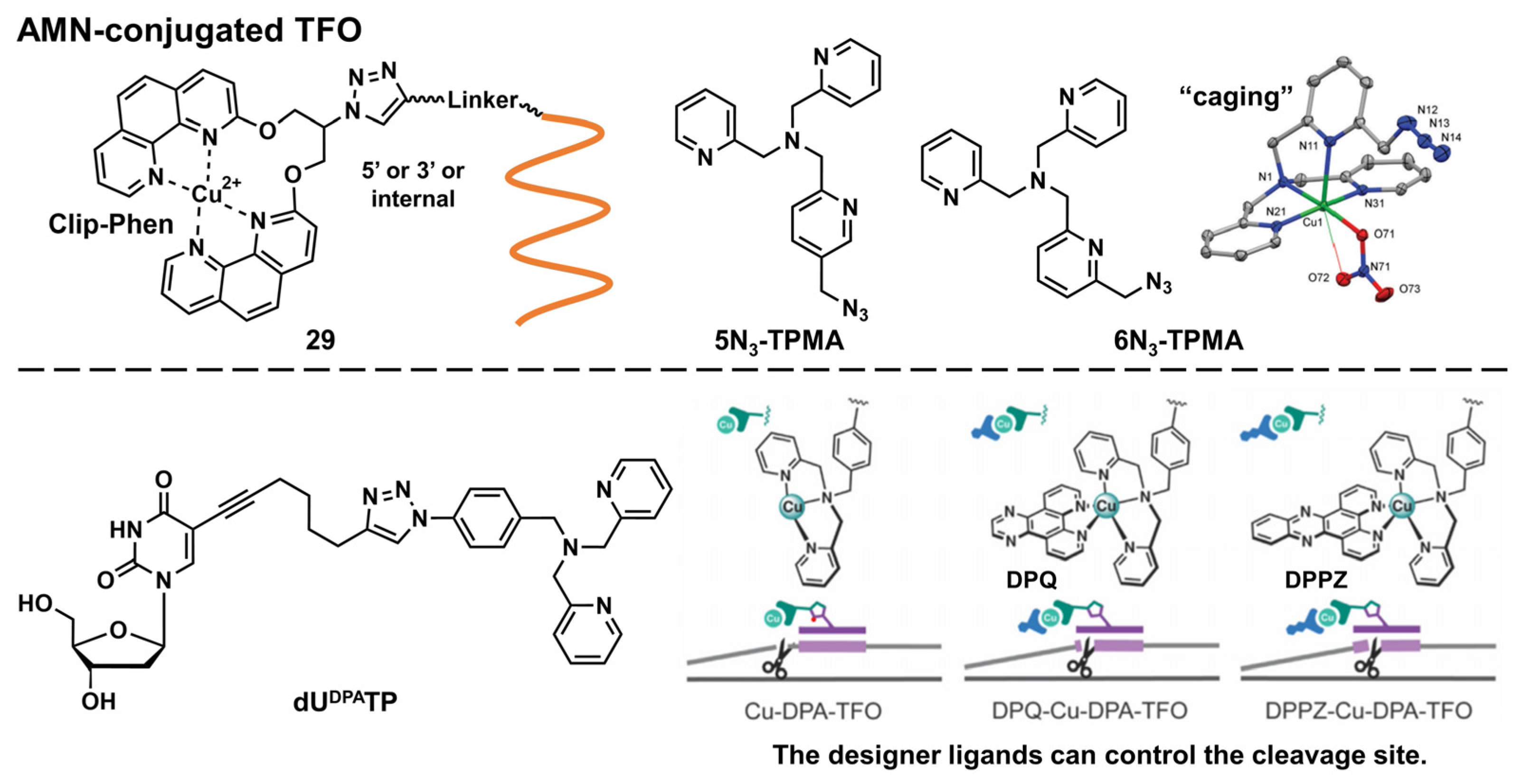
Artificial Metallonuclease-Conjugated Triplex-Forming Oligonucleotides
2.3. Recent Advancements in Therapeutic Applications of Triplex-Forming Oligonucleotides
3. Peptide Nucleic Acid
3.1. Outline of the Peptide Nucleic Acid Technology
3.2. Advancements in Peptic Nucleic Acid Engineering
3.2.1. Modification of the Peptic Nucleic Acid Backbone
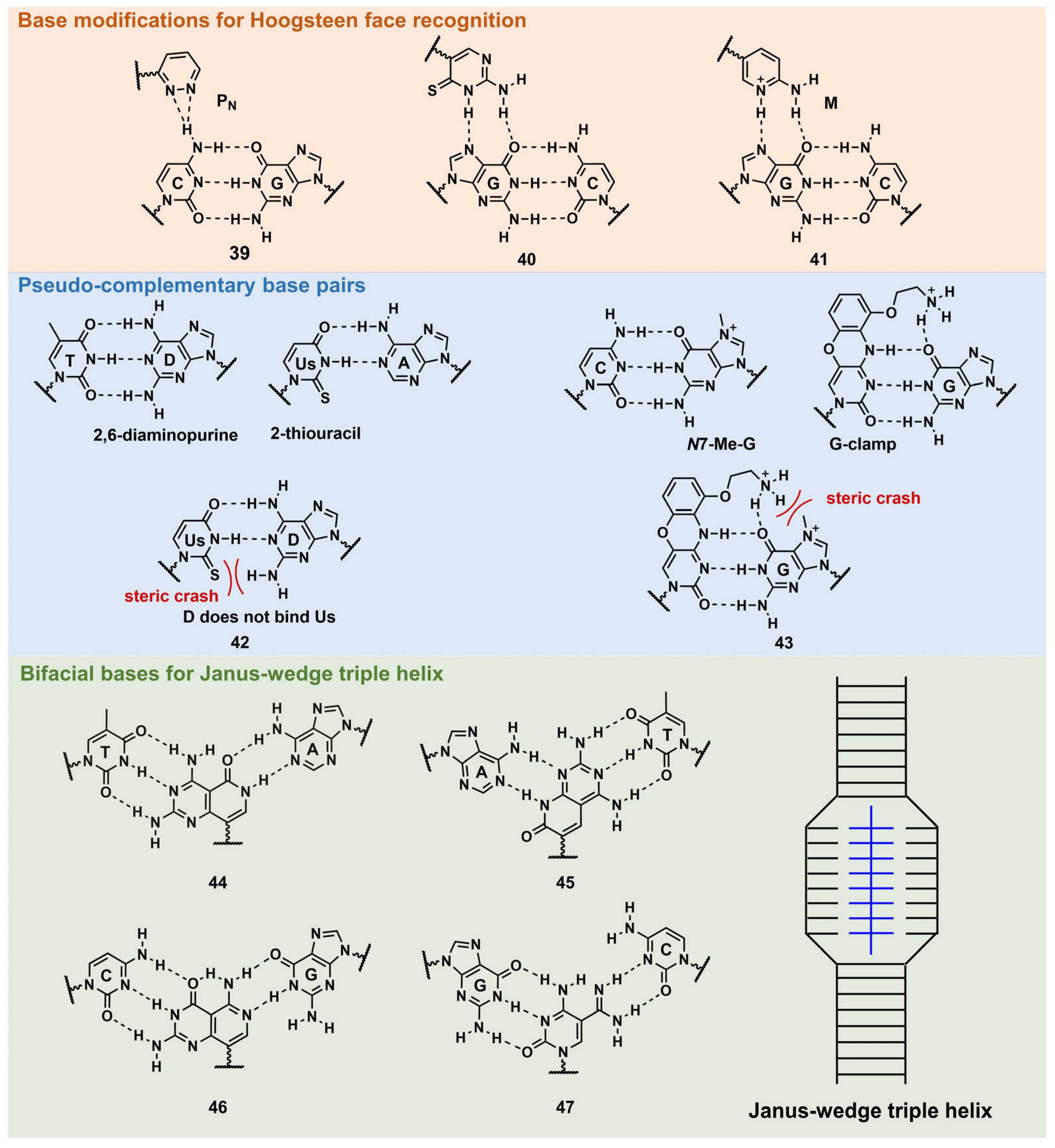
3.2.2. Base Modification of Peptide Nucleic Acid
Base Modifications for Enhanced Triplex Formation
Base Modifications for Peptide Nucleic Acid Functionalization
3.2.3. Recent Advancements in Crosslinkable Peptide Nucleic Acid for DNA Targeting
3.3. Recent Advancements in the Therapeutic Applications of Peptide Nucleic Acids
3.3.1. Modulation of DNA Expression
3.3.2. Genome Editing
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Terada, C.; Kawamoto, S.; Yamayoshi, A.; Yamamoto, T. Chemistry of Therapeutic Oligonucleotides That Drives Interactions with Biomolecules. Pharmaceutics 2022, 14, 2647. [Google Scholar] [CrossRef]
- Oyama, S.; Yamamoto, T.; Yamayoshi, A. Recent Advances in the Delivery Carriers and Chemical Conjugation Strategies for Nucleic Acid Drugs. Cancers 2021, 13, 3881. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Modell, A.E.; Lim, D.; Nguyen, T.M.; Sreekanth, V.; Choudhary, A. CRISPR-Based Therapeutics: Current Challenges and Future Applications. Trends Pharmacol. Sci. 2022, 43, 151–161. [Google Scholar] [CrossRef]
- Breier, D.; Peer, D. Genome Editing in Cancer: Challenges and Potential Opportunities. Bioact. Mater. 2023, 21, 394–402. [Google Scholar] [CrossRef]
- Zhang, C.C. Cas9-induced immune response: A potential caution for human genome editing. Biochem. Biophys. Res. Commun. 2019, 520, 706–707. [Google Scholar] [CrossRef]
- Duca, M.; Vekhoff, P.; Oussedik, K.; Halby, L.; Arimondo, P.B. The triple helix: 50 years later, the outcome. Nucleic Acids Res. 2008, 36, 5123–5138. [Google Scholar] [CrossRef]
- Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991, 254, 1497–1500. [Google Scholar] [CrossRef]
- Moser, H.E.; Dervan, P.B. Sequence-Specific Cleavage of Double Helical DNA by Triple Helix Formation. Science 1987, 238, 645–650. [Google Scholar] [CrossRef]
- Doan, T.L.; Perrouault, L.; Praseuth, D.; Habhoub, N.; Decout, J.L.; Thuong, N.T.; Lhomme, J.; Hélène, C. Sequence-specific recognition, photocrosslinking and cleavage of the DNA double helix by an oligo-[α]-thymidylate covalently linked to an azidoproflavine derivative. Nucleic Acids Res. 1987, 15, 7749–7760. [Google Scholar] [CrossRef]
- Jain, A.; Wang, G.; Vasquez, K.M. DNA Triple Helices: Biological Consequences and Therapeutic Potential. Biochimie 2008, 90, 1117–1130. [Google Scholar] [CrossRef] [PubMed]
- Goni, J.R.; Vaquerizas, J.M.; Dopazo, J.; Orozco, M. Exploring the Reasons for the Large Density of Triplex-Forming Oligonucleotide Target Sequences in the Human Regulatory Regions. BMC Genom. 2006, 7, 63. [Google Scholar] [CrossRef] [PubMed]
- Gaddis, S.S.; Wu, Q.; Thames, H.D.; Digiovanni, J.; Walborg, E.F.; Macleod, M.C.; Vasquez, K.M. A Web-Based Search Engine for Triplex-Forming Oligonucleotide Target Sequences. Oligonucleotides 2006, 16, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Jenjaroenpun, P.; Kuznetsov, V.A. TTS Mapping: Integrative WEB Tool for Analysis of Triplex Formation Target DNA Sequences, G-quadruplets and Non-Protein Coding Regulatory DNA Elements in the Human Genome. BMC Genom. 2009, 10, S9. [Google Scholar] [CrossRef]
- Jenjaroenpun, P.; Chew, C.S.; Yong, T.P.; Choowongkomon, K.; Thammasorn, W.; Kuznetsov, V.A. The TTSMI Database: A Catalog of Triplex Target DNA Sites Associated with Genes and Regulatory Elements in the Human Genome. Nucleic Acids Res. 2015, 43, D110–D116. [Google Scholar] [CrossRef]
- Li, C.; Zhou, Z.; Ren, C.; Deng, Y.; Peng, F.; Wang, Q.; Zhang, H.; Jiang, Y. Triplex-Forming Oligonucleotides as An Anti-Gene Technique for Cancer Therapy. Front. Pharmacol. 2022, 13, 1007723. [Google Scholar] [CrossRef]
- Hewett, P.W.; Daft, E.L.; Laughton, C.A.; Ahmad, S.; Ahmed, A.; Murray, J.C. Selective Inhibition of the Human Tie-1 Promoter with Triplex-Forming Oligonucleotides Targeted to Ets Binding Sites. Mol. Med. 2006, 12, 8–16. [Google Scholar] [CrossRef]
- Karympalis, V.; Kalopita, K.; Zarros, A.; Carageorgiou, H. Regulation of Gene Expression via Triple Helical Formations. Biochemistry 2004, 69, 855–860. [Google Scholar] [CrossRef]
- Young, S.L.; Krawczyk, S.H.; Matteucci, M.D.; Toole, J.J. Triple Helix Formation Inhibits Transcription Elongation in Vitro. Proc. Natl. Acad. Sci. USA 1991, 88, 10023–10026. [Google Scholar] [CrossRef]
- Song, J.; Intody, Z.; Li, M.; Wilson, J.H. Activation of gene expression by triplex-directed psoralen crosslink. Gene 2004, 324, 183–190. [Google Scholar] [CrossRef]
- Ghosh, M.K.; Katyal, A.; Chandra, R.; Brahmachari, V. Targeted activation of transcription in vivo through hairpin-triplex forming oligonucleotide in Saccharomyces cerevisiae. Mol. Cell. Biochem. 2005, 278, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, S.; Ait-Si-Ali, S.; Nagibneva, I.; Troalen, F.; Le Villain, J.P.; Harel-Bellan, A.; Svinarchuk, F. Gene activation by triplex-forming oligonucleotide coupled to the activation domain of protein VP16. Nucleic Acids Res. 1999, 27, 3995–4000. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cecconello, A.; Magro, M.; Vianello, F.; Simmel, F.C. Rational design of hybrid DNA-RNA triplex structures as modulators of transcription activity in vitro. Nucleic Acids Res. 2022, 50, 13172–13182. [Google Scholar] [CrossRef] [PubMed]
- Faruqi, A.F.; Seidman, M.M.; Segal, D.J.; Carroll, D.; Glazer, P.M. Recombination Induced by Triple-Helix-Targeted DNA Damage in Mammalian Cells. Mol. Cell. Biol. 1996, 16, 6820–6828. [Google Scholar] [CrossRef]
- Kaushik Tiwari, M.; Rogers, F.A. XPD-dependent activation of apoptosis in response to triplex-induced DNA damage. Nucleic Acids Res. 2013, 41, 8979–8994. [Google Scholar] [CrossRef][Green Version]
- Kaushik Tiwari, M.; Colon-Rios, D.A.; Rao Tumu, H.C.; Liu, Y.; Quijano, E.; Krysztofiak, A.; Chan, C.; Song, E.; Braddock, D.T.; Suh, H.-W.; et al. Direct Targeting of Amplified Gene Loci for Proapoptotic Anticancer Therapy. Nat. Biotechnol. 2022, 40, 325–334. [Google Scholar] [CrossRef]
- Kaushik Tiwari, M.; Adaku, N.; Peart, N.; Rogers, F.A. Triplex Structures Induce DNA Double Strand Breaks via Replication Fork Collapse in NER Deficient Cells. Nucleic Acids Res. 2016, 44, 7742–7754. [Google Scholar] [CrossRef]
- Faria, M.; Wood, C.D.; Perrouault, L.; Nelson, J.S.; Winter, A.; White, M.R.H.; Hélène, C.; Giovannangeli, C. Targeted inhibition of transcription elongation in cells mediated by triplex-forming oligonucleotides. Proc. Natl. Acad. Sci. USA 2000, 97, 3862–3867. [Google Scholar] [CrossRef]
- Semenyuk, A.; Darian, E.; Liu, J.; Majumdar, A.; Cuenoud, B.; Miller, P.S.; MacKerell, A.D., Jr.; Seidman, M.M. Targeting of an interrupted polypurine:polypyrimidine sequence in mammalian cells by a triplex-forming oligonucleotide containing a novel base analogue. Biochemistry 2010, 49, 7867–7878. [Google Scholar] [CrossRef]
- Hari, Y.; Obika, S.; Imanishi, T. Towards the Sequence-Selective Recognition of Double-Stranded DNA Containing Pyrimidine-Purine Interruptions by Triplex-Forming Oligonucleotides. Eur. J. Org. Chem. 2012, 2012, 2875–2887. [Google Scholar] [CrossRef]
- Griffin, L.C.; Dervan, P.B. Recognition of thymine adenine. base pairs by guanine in a pyrimidine triple helix motif. Science 1989, 245, 967–971. [Google Scholar] [CrossRef] [PubMed]
- Yoon, K.; Hobbs, C.A.; Koch, J.; Sardaro, M.; Kutny, R.; Weis, A.L. Elucidation of the sequence-specific third-strand recognition of four Watson-Crick base pairs in a pyrimidine triple-helix: T.AT, C.GC, T.CG, and Gn.TA. Proc. Natl. Acad. Sci. USA 1992, 89, 3840–3844. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Miller, P.S. Interactions of Cytosine Derivatives with T·A Interruptions in Pyrimidine·Purine·Pyrimidine DNA Triplex. Bioconjugate Chem. 1996, 7, 600–605. [Google Scholar] [CrossRef]
- Guianvarc’h, D.; Benhida, R.; Fourrey, J.L.; Maurisse, R.; Sun, J.S. Incorporation of a novel nucleobase allows stable oligonucleotide-directed triple helix formation at the target sequence containing a purine·pyrimidine interruption. Chem. Commun. 2001, 1814–1815. [Google Scholar] [CrossRef] [PubMed]
- Ohkubo, A.; Ohnishi, T.; Nishizawa, S.; Nishimura, Y.; Hisamatsu, S. The ability of a triplex-forming oligonucleotide to recognize T-A and C-G base pairs in a DNA duplex is enhanced by incorporating N-acetyl-2,7-diaminoquinoline. Bioorg. Med. Chem. 2020, 28, 115350. [Google Scholar] [CrossRef] [PubMed]
- Gerrard, S.R.; Edrees, M.M.; Bouamaied, I.; Fox, K.R.; Brown, T. CG base pair recognition within DNA triple helices by modified N-methylpyrrolo-dC nucleosides. Org. Biomol. Chem. 2010, 8, 5087–5096. [Google Scholar] [CrossRef]
- Hari, Y.; Akabane, M.; Obika, S. 2′-4′-BNA bearing a chiral guanidinopyrrolidine-containing nucleobase with potent ability to recognize the C:G base pair in a parallel-motif DNA triplex. Chem. Commun. 2013, 49, 7421–7423. [Google Scholar] [CrossRef]
- Obika, S.; Uneda, T.; Sugimoto, T.; Nanbu, D.; Minami, T.; Doi, T.; Imanishi, T. 2′-O,4′-C-methylene bridge nucleic acid (2′,4′-BNA): Synthesis and triplex-forming properties. Bioorg. Med. Chem. 2001, 9, 1001–1011. [Google Scholar] [CrossRef]
- Taniguchi, Y.; Magata, Y.; Osuki, T.; Notomi, R.; Wang, L.; Okamura, H.; Sasaki, S. Development of novel C-nucleoside analogues for the formation of antiparallel-type triplex DNA with duplex DNA that includes TA and dUA base pairs. Org. Biomol. Chem. 2020, 18, 2845–2851. [Google Scholar] [CrossRef]
- Nishizawa, S.; Tu, G.; Ogata, D.; Miyauchi, K.; Ohkubu, A. Development of antiparallel-type triplex-forming oligonucleotides containing quinoline derivatives capable of recognizing a T-A base pair in a DNA duplex. Bioorg. Med. Chem. 2022, 71, 116934. [Google Scholar] [CrossRef]
- Wang, L.; Taniguchi, Y.; Okamura, H.; Sasaki, S. Modification of the aminopyridine unit of 2′-deoxyaminopyridinyl-pseudocytidine allowing triplex formation at CG interruptions in homopurine sequences. Nucleic Acids Res. 2018, 46, 8679–8688. [Google Scholar] [CrossRef] [PubMed]
- Notomi, R.; Wang, L.; Osuki, T.; Okamura, H.; Sasaki, S.; Taniguchi, Y. Synthesis of C-nucleoside analogues based on the pyrimidine skeleton for the formation of anti-parallel-type triplex DNA with a CG mismatch site. Bioorg. Med. Chem. 2020, 28, 115782. [Google Scholar] [CrossRef] [PubMed]
- Notomi, R.; Wang, L.; Sasaki, S.; Taniguchi, Y. Design and synthesis of purine nucleoside analogues for the formation of stable anti-parallel-type triplex DNA with duplex DNA bearing the 5mCG base pair. RSC Adv. 2021, 11, 21390–21396. [Google Scholar] [CrossRef] [PubMed]
- Notomi, R.; Sasaki, S.; Taniguchi, Y. Recognition of 5-methyl-CG and CG base pairs in duplex DNA with high stability using antiparallel-type triplex-forming oligonucleotides with 2-guanidinoethyl-2′-deoxynebularine. Nucleic Acids Res. 2022, 50, 12071–12081. [Google Scholar] [CrossRef] [PubMed]
- Hartono, Y.D.; Pabon-Martinez, Y.V.; Uyar, A.; Wengel, J.K.; Lundin, K.E.; Zain, R.; Smith, C.I.E.; Nilsson, L.; Villa, A. Role of pseudoisocytosine tautomerization in triplex-forming oligonucleotides: In silico and in vitro studies. ACS Omega 2017, 2, 2165–2177. [Google Scholar] [CrossRef]
- Rusling, D.A. Triplex-forming properties and enzymatic incorporation of a base-modified nucleotide capable of duplex DNA recognition at neutral pH. Nucleic Acids Res. 2021, 49, 7256–7266. [Google Scholar] [CrossRef]
- Sasaki, S.; Taniguchi, Y.; Takahashi, R.; Senko, Y.; Kodama, K.; Nagatsugi, F.; Maeda, M. Selective Formation of Stable Triplexes Including a TA or a CG Interrupting Site with New Bicyclic Nucleoside Analogue (WNA). J. Am. Chem. Soc. 2004, 126, 516–528. [Google Scholar] [CrossRef]
- Inde, T.; Nishizawa, S.; Hattori, Y.; Kanamori, T.; Yuasa, H.; Seio, K.; Sekine, M.; Ohkubo, A. Synthesis of and triplex formation in oligonucleotides containing 2′-deoxy-6-thioxanthosine. Bioorg. Med. Chem. 2018, 26, 3785–3790. [Google Scholar] [CrossRef]
- Wang, E.; Koshlap, K.M.; Gillespie, P.; Dervan, P.B.; Feigon, J. Solution Structure of a Pyrimidine·Purine·Pyrimidine Triplex Containing the Sequence-specific Intercalating Non-natural Base D3. J. Mol. Biol. 1996, 257, 1052–1069. [Google Scholar] [CrossRef]
- Carbone, G.M.; McGuffie, E.; Napoli, S.; Flanagan, C.E.; Dembech, C.; Negri, U.; Arcamone, F.; Capobianco, M.L.; Catapano, C.V. DNA binding and antigene activity of a daunomycin-conjugated triplex-forming oligonucleotide targeting the P2 promoter of the human c-myc gene. Nucleic Acids Res. 2004, 32, 2396–2410. [Google Scholar] [CrossRef]
- Fox, K.R.; Brown, T.; Rusling, D.A. DNA recognition by parallel triplex formation. In DNA-Targeting Molecules as Therapeutic Agents; Waring, M.J., Ed.; RSC Publishing: Cambridge, UK, 2018; pp. 1–32. [Google Scholar]
- Walsh, S.; El-Sagheer, A.H.; Brown, T. Fluorogenic thiazole orange TOTFO probes stabilize parallel DNA triplexes at pH 7 and above. Chem. Sci. 2018, 9, 7681–7687. [Google Scholar] [CrossRef] [PubMed]
- Phan, K.; Ramachandran, V.; Fassihi, H.; Sebaratnam, F.D. Comparison of Narrowband UV-B With Psoralen-UV-A Phototherapy for Patients With Early-Stage Mycosis Fungoides: A Systematic Review and Meta-analysis. JAMA Dermatol. 2019, 155, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Buhimschi, D.A.; Gooden, M.D.; Jing, H.; Fels, R.D.; Hansen, S.K.; Beyer, F.W.; Dewhirst, W.M.; Walder, H.; Gasparro, P.F. Psoralen Derivatives with Enhanced Potency. Photochem. Photobiol. 2020, 96, 1014–1031. [Google Scholar] [CrossRef] [PubMed]
- Duval-Valentin, G.; Thuong, N.T.; Hélène, C. Specific Inhibition of Transcription by Triple Helix-Forming Oligonucleotide. Proc. Natl. Acad. Sci. USA 1992, 89, 504–508. [Google Scholar] [CrossRef]
- Havre, P.A.; Gunther, E.; Gasparro, F.; Glazer, P.M. Targeted Mutagenesis of DNA Using Triple Helix-Forming Oligonucleotides Linked to Psoralen. Proc. Natl. Acad. Sci. USA 1993, 90, 7879–7883. [Google Scholar] [CrossRef]
- Nakao, J.; Mikame, Y.; Eshima, H.; Yamamoto, T.; Dohno, C.; Wada, T.; Yamayoshi, A. Unique Crosslinking Properties of Psoralen-Conjugated Oligonucleotides Developed by Novel Psoralen N-Hydroxysuccinimide Esters. ChemBioChem 2023, 24, e202200789. [Google Scholar] [CrossRef]
- Raha, M.; Lacroix, L.; Glazer, P.M. Mutagenesis Mediated by Triple Helix-Forming Oligonucleotides Conjugated to Psoralen: Effects of Linker Arm Length and Sequence Context. Photochem. Photobiol. 1998, 67, 289–294. [Google Scholar] [CrossRef]
- Majumdar, A.; Muniandy, P.A.; Liu, J.; Liu, J.-I.; Liu, S.-T.; Cuenoud, B.; Seidman, M.M. Targeted Gene Knock-In and Sequence Modulation Mediated by a Psoralen-Linked Triplex-Forming Oligonucleotide. J. Biol. Chem. 2008, 283, 11244–11252. [Google Scholar] [CrossRef]
- Liu, J.; Majumdar, A.; Liu, J.; Thompson, L.H.; Seidman, M.M. Sequence Conversion by Single Strand Oligonucleotide Donors via Non-homologous End Joining in Mammalian Cells. J. Biol. Chem. 2010, 285, 23198–23207. [Google Scholar] [CrossRef]
- Kojima, A.; Nakao, J.; Shimada, N.; Yoshida, N.; Abe, Y.; Mikame, Y.; Yamamoto, T.; Wada, T.; Maruyama, A.; Yamayoshi, A. Selective Photo-Crosslinking Detection of Methylated Cytosine in DNA Duplex Aided by a Cationic Comb-Type Copolymer. ACS. Biomater. Sci. Eng. 2022, 8, 1799–1805. [Google Scholar] [CrossRef]
- Mikame, Y.; Eshima, H.; Toyama, H.; Nakao, J.; Matsuo, M.; Yamamoto, T.; Hari, Y.; Komano, J.A.; Yamayoshi, A. Development and Crosslinking Properties of Psoralen-Conjugated Triplex-Forming Oligonucleotides as Antigene Tools Targeting Genome DNA. ChemMedChem 2023, 13, e202300348. [Google Scholar] [CrossRef] [PubMed]
- Nakao, J.; Yamamoto, T.; Yamayoshi, A. Therapeutic Application of Sequence-Specific Binding Molecules for Novel Genome Editing Tools. Drug Metab. Pharmacokinet. 2022, 42, 100427. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.M.; Lippard, S.J. Cisplatin: From DNA Damage to Cancer Chemotherapy. Prog. Nucleic Acid Res. Mol. Biol. 2001, 67, 93–130. [Google Scholar] [PubMed]
- Sharma, S.K.; McLaughlin, L.W. Cross-Linking of a DNA Conjugate Tethering a cis-Bifunctional Platinated Complex to a Target DNA Duplex. J. Am. Chem. Soc. 2002, 124, 9658–9659. [Google Scholar] [CrossRef]
- Graham, M.K.; Miller, P.S. Inhibition of Transcription by Platinated Triplex-Forming Oligonucleotides. J. Biol. Inorg. Chem. 2012, 17, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Graham, M.K.; Brown, T.R.; Miller, P.S. Targeting the Human Androgen Receptor Gene with Platinated Triplex-Forming Oligonucleotides. Biochemistry 2015, 54, 2270–2282. [Google Scholar] [CrossRef]
- Hennessy, J.; McGorman, B.; Molphy, Z.; Farrell, N.P.; Singleton, D.; Brown, T.; Kellett, A. A Click Chemistry Approach to Targeted DNA Crosslinking with cis-Platinum(II)-Modified Triplex-Forming Oligonucleotides. Angew. Chem. Int. Ed. 2021, 60, 2–9. [Google Scholar]
- Fantoni, N.Z.; Brown, T.; Kellett, A. DNA-Targeted Metallodrugs: An Untapped Source of Artificial Gene Editing Technology. ChemBioChem 2021, 22, 2184–2205. [Google Scholar] [CrossRef]
- Panattoni, A.; El-Sagheer, A.H.; Kellett, A.; Hocek, M. Oxidative DNA Cleavage with Clip-Phenanthroline Triplex-Forming Oligonucleotide Hybrids. ChemBioChem 2020, 21, 991–1000. [Google Scholar] [CrossRef]
- Lauria, T.; Slator, C.; McKee, V.; Müller, M.; Stazzoni, S.; Crisp, A.L.; Carell, T.; Kellett, A. A Click Chemistry Approach to Developing Molecularly Targeted DNA Scissors. Chem. Eur. J. 2020, 26, 16782–16792. [Google Scholar] [CrossRef]
- Fantoni, N.Z.; McGorman, B.; Molphy, Z.; Singleton, D.; Walsh, S.; El-Sagheer, A.H.; McKee, V.; Brown, T.; Kellett, A. Development of Gene-Targeted Polypyridyl Triplex-Forming Oligonucleotide Hybrids. ChemBioChem 2020, 21, 3563–3574. [Google Scholar] [CrossRef] [PubMed]
- McGorman, B.; Fantoni, N.Z.; O’Carroll, S.; Ziemele, A.; El-Sagheer, A.H.; Brown, T.; Kellett, A. Enzymatic Synthesis of Chemical Nucleoase Triplex-Forming Oligonucleotides with Gene-Silencing Application. Nucleic Acids Res. 2022, 50, 5467–5481. [Google Scholar] [CrossRef] [PubMed]
- Paquet, J.; Henrionnet, C.; Pinzano, A.; Vincourt, J.B.; Gillet, P.; Netter, P.; Chary-Valckenaere, I.; Loeuille, D.; Pourel, J.; Grossin, L. Alternative for Anti-TNF Antibodies for Arthritis Treatment. Mol. Ther. 2011, 19, 1887–1895. [Google Scholar] [CrossRef]
- Huo, S.; Gong, N.; Jiang, Y.; Chen, F.; Guo, H.; Gan, Y.; Wang, Z.; Herrmann, A.; Liang, X.J. Gold-DNA nanosunflowers for efficient gene silencing with controllable transformation. Sci. Adv. 2019, 5, eaaw6264. [Google Scholar] [CrossRef]
- Yang, X.; Xu, Y.; Fu, J.; Shen, Z. Nanoparticle delivery of TFOs is a novel targeted therapy for HER2 amplified breast cancer. BMC Cancer 2023, 23, 680. [Google Scholar] [CrossRef]
- Das, A.; Pradhan, B. Evolution of peptide nucleic acid with modifications of its backbone and application in biotechnology. Chem. Biol. Drug Des. 2021, 97, 865–892. [Google Scholar] [CrossRef] [PubMed]
- Lonkar, P.; Kim, K.H.; Kuan, J.Y.; Chin, J.Y.; Rogers, F.A.; Knauert, M.P.; Kole, P.; Nielsen, P.E.; Glazer, P.M. Targeted correction of a thalassemia-associated β-globin mutation induced by pseudo-complementary peptide nucleic acids. Nucleic Acids Res. 2009, 37, 3635–3644. [Google Scholar] [CrossRef][Green Version]
- Rogers, F.A.; Vasquez, K.M.; Egholm, M.; Glazer, P.M. Site-directed recombination via bifunctional PNA-DNA conjugates. Proc. Natl. Acad. Sci. USA 2002, 99, 16695–16700. [Google Scholar] [CrossRef]
- Schleifman, E.B.; Bindra, R.; Leif, J.; del Campo, J.; Rogers, F.A.; Uchil, P.; Kutsch, O.; Shultz, L.D.; Kumar, P.; Greiner, D.L.; et al. Targeted Disruption of the CCR5 Gene in Human Hematopoietic Stem Cells Stimulated by Peptide Nucleic Acids. Chem. Biol. 2011, 18, 1189–1198. [Google Scholar] [CrossRef]
- Perera, J.D.R.; Carufe, K.E.W.; Glazer, P.M. Peptide nucleic acids and their role in gene regulation and editing. Biopolymers 2021, 112, e23460. [Google Scholar] [CrossRef]
- Swenson, C.S.; Heemstra, J.M. Peptide nucleic acids harness dual information codes in a single molecule. Chem. Commun. 2020, 56, 1926–1935. [Google Scholar] [CrossRef] [PubMed]
- Suparpprom, C.; Vilaivan, T. Perspectives on conformationally constrained peptide nucleic acid (PNA): Insights into the structural design, properties and applications. RSC Chem. Biol. 2022, 3, 648–697. [Google Scholar] [CrossRef] [PubMed]
- Brodyagin, N.; Katkevics, M.; Kotikam, V.; Ryan, C.A.; Rozners, E. Chemical approaches to discover the full potential of peptide nucleic acids in biomedical applications. Beilstein. J. Org. Chem. 2021, 17, 1641–1688. [Google Scholar] [CrossRef] [PubMed]
- Dragulescu-Andrasi, A.; Rapireddy, S.; Frezza, B.M.; Gayathri, C.; Gil, R.R.; Ly, D.H. A Simple γ-Backbone Modification Preorganizes Peptide Nucleic Acid into a Helical Structure. J. Am. Chem. Soc. 2006, 128, 10258–10267. [Google Scholar] [CrossRef] [PubMed]
- Sahu, B.; Chenna, V.; Lathrop, K.L.; Thomas, S.M.; Zon, G.; Livak, K.J.; Ly, D.H. Synthesis of Conformationally Preorganized and Cell-Permeable Guanidine-Based γ-Peptide Nucleic Acids (γ-GPNAs). J. Org. Chem. 2009, 74, 1509–1516. [Google Scholar] [CrossRef]
- Bahal, R.; Sahu, B.; Rapireddy, S.; Lee, C.M.; Ly, D.H. Sequence-Unrestricted, Watson-Crick Recognition of Double Helical B-DNA by (R)-MiniPEG-γPNAs. ChemBioChem 2012, 13, 56–60. [Google Scholar] [CrossRef]
- Tähtinen, V.; Verhassel, A.; Tuomela, J.; Virta, P. γ-(S)-Guanidinylmethyl-Modified Triplex-Forming Peptide Nucleic Acids Increase Hoogsteen-Face Affinity for a MicroRNA and Enhanced Cellular Uptake. ChemBioChem 2019, 20, 3041–3051. [Google Scholar] [CrossRef]
- Zheng, H.; Saha, M.; Appella, D.H. Synthesis of Fmoc-Protected (S,S)-trans-Cyclopentane Diamine Monomers Enables the Preparation and Study of Conformationally Restricted Peptide Nucleic Acids. Org. Lett. 2018, 20, 7637–7640. [Google Scholar] [CrossRef]
- Zheng, H.; Botos, I.; Clausse, V.; Nikolayevskiy, H.; Rastede, E.E.; Fouz, M.F.; Mazur, S.J.; Appella, D.H. Conformational constraints of cyclopentane peptide nucleic acids facilitate tunable binding to DNA. Nucleic Acids Res. 2021, 49, 713–725. [Google Scholar] [CrossRef]
- Kulkarni, P.; Datta, D.; Ramabhadran, R.O.; Ganesh, K.N. Gem-dimethyl peptide nucleic acid (α/β/γ-gdm-PNA) monomers: Synthesis and the role of gdm-substituents in preferential stabilization of Z/E-rotamers. Org. Biomol. Chem. 2021, 19, 6534–6545. [Google Scholar] [CrossRef]
- Kulkarni, P.; Datta, D.; Ramabhadran, R.O.; Ganesh, K.N. Gemdimethyl Peptide Nucleic Acids (α/β/γ-gdm-PNA): E/Z-Rotamers Influence the Selectivity in the Formation of Parallel/Antiparallel gdm-PNA:DNA/RNA Duplexes. ACS Omega 2022, 7, 40558–40568. [Google Scholar] [CrossRef] [PubMed]
- Shiraj, A.; Ramabhadran, R.O.; Ganesh, K.N. Aza-PNA: Engineering E-Rotamer Selectivity Directed by Intramolecular H-Bonding. Org. Lett. 2022, 24, 7421–7427. [Google Scholar] [CrossRef] [PubMed]
- Bhingardeve, P.; Madhanagopal, B.R.; Ganesh, K.N. Cγ-(S/R)-Bimodal Peptide Nucleic Acids (Cγ-bm-PNA) Form Coupled Double Duplexes by Synchronous Binding to Two Complementary DNA Strands. J. Org. Chem. 2020, 85, 13680–13693. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.K.; Madhanagopal, B.R.; Datta, D.; Ganesh, K.N. Structural Design and Synthesis of Bimodal PNA That Simultaneously Binds Two Complementary DNAs To Form Fused Double Duplexes. Org. Lett. 2020, 22, 5255–5260. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.K.; Madhanagopal, B.R.; Ganesh, K.N. Peptide Nucleic Acid with Double Face: Homothymine-Homocytosine Bimodal Cα-PNA (bm-Cα-PNA) Forms a Double Duplex of the bm-PNA2:DNA Tripolex. J. Org. Chem. 2021, 86, 414–428. [Google Scholar] [CrossRef]
- Bhingardeve, P.; Jain, P.; Ganesh, K.N. Molecular Assembly of Triplex of Duplexes from Homothyminyl-Homocytosinyl Cγ-(S/R)-Bimodal Peptide Nucleic Acids with dA/dG and the Cell Permeability of Bimodal Peptide Nucleic Acids. ACS Omega 2021, 6, 19757–19770. [Google Scholar] [CrossRef]
- Ong, A.A.L.; Toh, D.F.K.; Patil, K.M.; Meng, Z.; Yuan, Z.; Krishna, M.S.; Devi, G.; Haruehanroengra, P.; Lu, Y.; Xia, K.; et al. General Recognition of U-G, U-A, and C-G Pairs by Double-Stranded RNA-Binding PNAs Incorporated with an Artificial Nucleobase. Biochemistry 2019, 58, 1319–1331. [Google Scholar] [CrossRef]
- Toh, D.F.K.; Devi, G.; Patil, K.M.; Qu, Q.; Maraswami, M.; Xiao, Y.; Loh, T.P.; Zhao, Y.; Chen, G. Incorporating a guanidine-modified cytosine base into triplex-forming PNAs for the recognition of a C-G pyrimidine-purine inversion site of an RNA duplex. Nucleic Acids Res. 2016, 44, 9071–9082. [Google Scholar] [CrossRef]
- Egholm, M.; Christensen, L.; Deuholm, K.L.; Buchardt, O.; Coull, J.; Nielsen, P.E. Efficient pH-independent sequence-specific DNA binding by pseudoisocytosine-containing bis-PNA. Nucleic Acids Res. 1995, 23, 217–222. [Google Scholar] [CrossRef]
- Kumpina, I.; Brodyagin, N.; MacKay, J.A.; Kennedy, S.D.; Katkevics, M.; Rozners, E. Synthesis and RNA-Binding Properties of Extended Nucleobases for Triplex-Forming Peptide Nucleic Acids. J. Org. Chem. 2019, 84, 13276–13298. [Google Scholar] [CrossRef]
- Brodyagin, N.; Kumpina, I.; Applegate, J.; Katkevics, M.; Rozners, E. Pyridazine Nucleobase in Triplex-Forming PNA Improves Recognition of Cytosine Interruptions of Polypurine Tracts in RNA. ACS Chem. Biol. 2021, 16, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Devi, G.; Yuan, Z.; Lu, Y.; Zhao, Y.; Chen, G. Incorporation of thio-pseudoisocytosine into triplex-forming peptide nucleic acids for enhanced recognition of RNA duplexes. Nucleic Acids Res. 2014, 42, 4008–4018. [Google Scholar] [CrossRef] [PubMed]
- Kotikam, V.; Kennedy, S.; MacKay, J.A.; Rozners, E. Synthetic, Structural, and RNA Binding Studies on 2-Aminopyridine-Modified Triplex-Forming Peptide Nucleic Acids. Chem. Eur. J. 2019, 25, 4367–4372. [Google Scholar] [CrossRef] [PubMed]
- Ryan, C.A.; Brodyagin, N.; Lok, J.; Rozners, E. The 2-Aminopyridine Nucleobase Improves Triple-Helical Recognition of RNA and DNA When Used Instead of Pseudoisocytosine in Peptide Nucleic Acids. Biochemistry 2021, 60, 1919–1925. [Google Scholar] [CrossRef]
- Haaima, G.; Hansen, H.F.; Christensen, L.; Dahl, O.; Nielsen, P.E. Increased DNA binding and sequence discrimination of PNA oligomers containing 2,6-diaminopurine. Nucleic Acids Res. 1997, 25, 4639–4643. [Google Scholar] [CrossRef]
- Hudson, R.H.E.; Heidari, A.; Martin-Chan, T.; Park, G.; Wisner, J.A. On the Necessity of Nucleobase Protection for 2-Thiouracil for Fmoc-Based Pseudo-Complementary Peptide Nucleic Acid Oligomer Synthesis. J. Org. Chem. 2019, 84, 13252–13261. [Google Scholar] [CrossRef]
- López-Tena, M.; Farrera-Soler, L.; Barluenga, S.; Winssinger, N. Pseudo-Complementary G:C Base Pair for Mixed Sequence dsDNA Invasion and Its Applications in Diagnostics (SARS-CoV-2 Detection). JACS Au 2023, 3, 449–458. [Google Scholar] [CrossRef]
- Lin, K.Y.; Matteucci, M.D. A Cytosine Analogue Capable of Clamp-Like Binding to a Guanine in Helical Nucleic Acids. J. Am. Chem. Soc. 1998, 120, 8531–8532. [Google Scholar] [CrossRef]
- Rajeev, K.G.; Maier, M.A.; Lesnik, E.A. High-Affinity Peptide Nucleic Acid Oligomers Containing Tricyclic Cytosine Analogues. Org. Lett. 2002, 4, 4395–4398. [Google Scholar] [CrossRef]
- Chen, D.; Meena; Sharma, S.K.; McLaughlin, L.W. Formation and Stability of a Janus-Wedge Type of DNA Triple. J. Am. Chem. Soc. 2004, 126, 70–71. [Google Scholar] [CrossRef]
- Thadke, S.A.; Perera, J.D.R.; Hridya, V.M.; Bhatt, K.; Shaikh, A.Y.; Hsieh, W.C.; Chen, M.; Gayathri, C.; Gil, R.R.; Rule, G.S.; et al. Design of Bivalent Nucleic Acid Ligands for Recognition of RNA-Repeated Expansion Associated with Huntington’s Disease. Biochemistry 2018, 57, 2094–2108. [Google Scholar] [CrossRef] [PubMed]
- Thadke, S.A.; Hridya, V.M.; Perera, J.D.R.; Gil, R.R.; Mukherjee, A.; Ly, D.H. Shape selective bifacial recognition of double helical DNA. Commun. Chem. 2018, 1, 79. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Fan, X.J.; Nielsen, P.E. Efficient Sequence-Directed Psoralen Targeting Using Pseudocomplementary Peptide Nucleic Acids. Bioconjugate Chem. 2007, 18, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Nielsen, P.E.; Glazer, P.M. Site-directed gene mutation at mixed sequence targets by psoralen-conjugated pseudo-complementary peptide nucleic acids. Nucleic Acids Res. 2007, 35, 7604–7613. [Google Scholar] [CrossRef]
- Manicardi, A.; Gyssels, E.; Corradini, R.; Madder, A. Furan-PNA: A mildly inducible irreversible interstrand crosslinking system targeting single and double-stranded DNA. Chem. Commun. 2016, 52, 6930–6933. [Google Scholar] [CrossRef]
- Elskens, J.; Manicardi, A.; Costi, V.; Madder, A.; Corradini, R. Synthesis and improved cross-linking properties of C5-modified furan bearing PNAs. Molecules 2017, 22, 2010. [Google Scholar] [CrossRef]
- Muangkaew, P.; Vilaivan, T. Pyrrolidinyl Peptide Nucleic Acid Probes Capable of Crosslinking with DNA: Effects of Terminal and Internal Modifications on Crosslink Efficiency. ChemBioChem 2021, 22, 241–252. [Google Scholar] [CrossRef]
- Montemurro, L.; Raieli, S.; Angelucci, S.; Bartolucci, D.; Amadesi, C.; Lampis, S.; Scardovi, A.L.; Venturelli, L.; Nieddu, G.; Cerisoli, L.; et al. A Novel MYCN-Specific Antigene Oligonucleotide Deregulates Mitochondria and Inhibits Tumor Growth in MYCN-Amplified Neuroblastome. Cancer Res. 2019, 79, 6166–6177. [Google Scholar] [CrossRef]
- Lampis, S.; Raieli, S.; Montemurro, L.; Bartolucci, D.; Amadesi, C.; Bortolotti, S.; Angelucci, S.; Scardovi, A.L.; Nieddu, G.; Cerisoli, L.; et al. The MYCN inhibitor BGA002 restores the retinoic acid response leading to differentiation or apoptosis by the mTOR block in MYCN-amplified neuroblastoma. J. Exp. Clin. Cancer Res. 2022, 42, 160. [Google Scholar] [CrossRef]
- Tonelli, R.; Mclntyre, A.; Camerin, C.; Walters, Z.S.; Leo, K.D.; Selfe, J.; Purgato, S.; Missiaglia, E.; Tortori, A.; Renshaw, J.; et al. Antitumor Activity of Sustauned N-Myc Reduction in Rhabdomyosarcomas and Transcriptional Block by Antigene Therapy. Clin. Cancer Res. 2012, 18, 796–807. [Google Scholar] [CrossRef]
- Bahal, R.; Ali McNeer, N.; Quijano, E.; Liu, Y.; Sulkowski, P.; Turchick, A.; Lu, Y.C.; Bhunia, D.C.; Manna, A.; Greiner, D.L.; et al. In vivo correction of anaemia in β-thalassemic mice by γPNA-mediated gene editing with nanoparticle delivery. Nat. Commun. 2016, 7, 13304. [Google Scholar] [CrossRef] [PubMed]
- Ricciardi, A.S.; Bahal, R.; Farrelly, J.S.; Quijano, E.; Bianchi, A.H.; Luks, V.L.; Putman, R.; López-Giráldez, F.; Coskun, S.; Song, E.; et al. In utero nanoparticle delivery for site-specific genome editing. Nat. Commun. 2018, 9, 2481. [Google Scholar] [CrossRef] [PubMed]
- Chin, J.Y.; Glazer, P.M. Repair of DNA lesions associated with triplex-forming oligonucleotides. Mol. Carcinog. 2009, 48, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Piotrowski-Daspit, A.S.; Barone, C.; Lin, C.Y.; Deng, Y.; Wu, D.; Binns, T.C.; Xu, E.; Ricciardi, A.S.; Putman, R.; Garrison, A.; et al. In vivo correction of cystic fibrosis mediated by PNA nanoparticles. Sci. Adv. 2022, 8, eabo0522. [Google Scholar] [CrossRef] [PubMed]
- Putman, R.; Ricciardi, A.S.; Carufe, K.E.W.; Quijano, E.; Bahal, R.; Glazer, P.M.; Saltzman, W.M. Nanoparticle-mediated genome editing in single-cell embryos via peptide nucleic acids. Bioeng. Transl. Med. 2023, 8, e10458. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikame, Y.; Yamayoshi, A. Recent Advancements in Development and Therapeutic Applications of Genome-Targeting Triplex-Forming Oligonucleotides and Peptide Nucleic Acids. Pharmaceutics 2023, 15, 2515. https://doi.org/10.3390/pharmaceutics15102515
Mikame Y, Yamayoshi A. Recent Advancements in Development and Therapeutic Applications of Genome-Targeting Triplex-Forming Oligonucleotides and Peptide Nucleic Acids. Pharmaceutics. 2023; 15(10):2515. https://doi.org/10.3390/pharmaceutics15102515
Chicago/Turabian StyleMikame, Yu, and Asako Yamayoshi. 2023. "Recent Advancements in Development and Therapeutic Applications of Genome-Targeting Triplex-Forming Oligonucleotides and Peptide Nucleic Acids" Pharmaceutics 15, no. 10: 2515. https://doi.org/10.3390/pharmaceutics15102515
APA StyleMikame, Y., & Yamayoshi, A. (2023). Recent Advancements in Development and Therapeutic Applications of Genome-Targeting Triplex-Forming Oligonucleotides and Peptide Nucleic Acids. Pharmaceutics, 15(10), 2515. https://doi.org/10.3390/pharmaceutics15102515