
Targeting Protein Kinases and Epigenetic Control as Combinatorial Therapy Options for Advanced Prostate Cancer Treatment


 ,
,  ,
,  and
and 
Abstract
1. Introduction
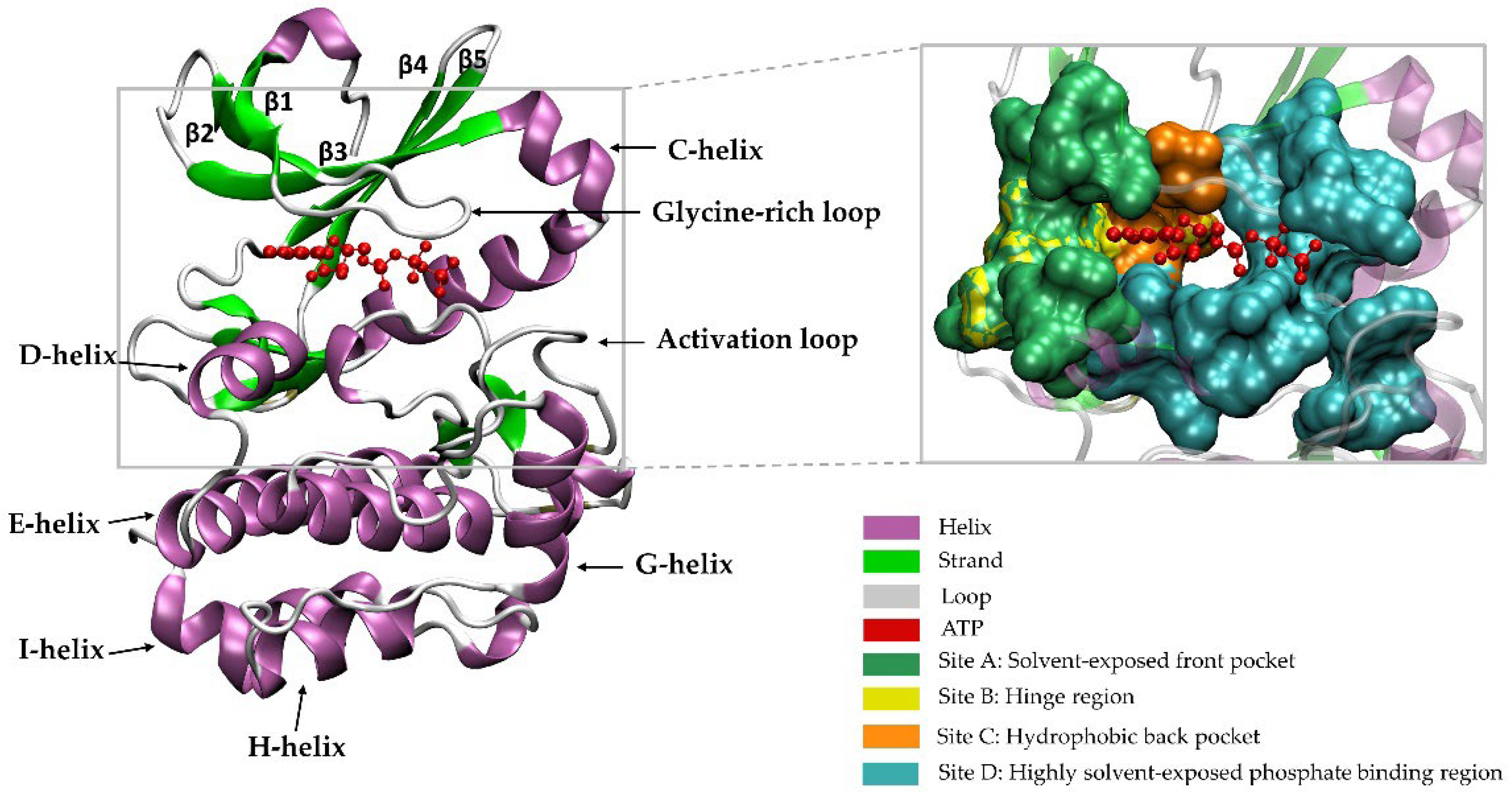
2. A Brief Introduction to the Family of PKs
Catalytic and Non-Catalytic Activities of PKs
3. PK Targeting Tools
4. PKs and PC Progression
4.1. AMP-Activated PK (AMPK)
4.2. Protein Kinase A (PKA)
4.3. Protein Kinase B (PKB)
4.4. Protein Kinase C (PKC)
4.5. Protein Kinase D (PKD)
4.6. DNA-Dependent Protein Kinase (DNA-PK)
4.7. CDC2-Like Protein Kinase (CLKs)
4.8. Serine-Argnine Protein Kinase 1 (SRPK1)
4.9. Pyruvate Kinase M2 (PKM2)
4.10. T lAK Cell Originated PK (TOPK)
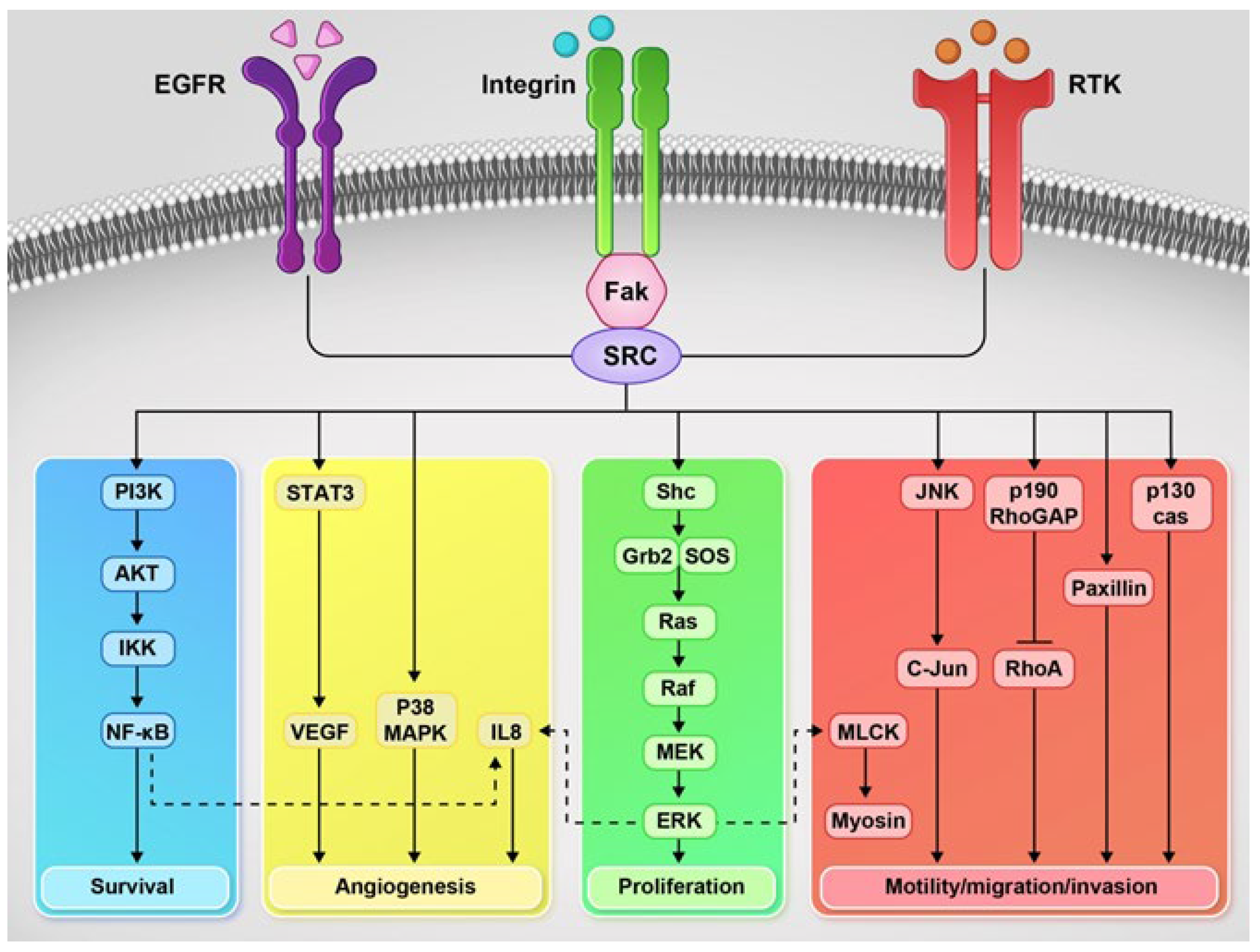
4.11. Src Family Kinases (SFKs)
4.12. Focal Adhesion Kinase (FAK)
4.13. Cyclin G-Associated Kinase (GAK)
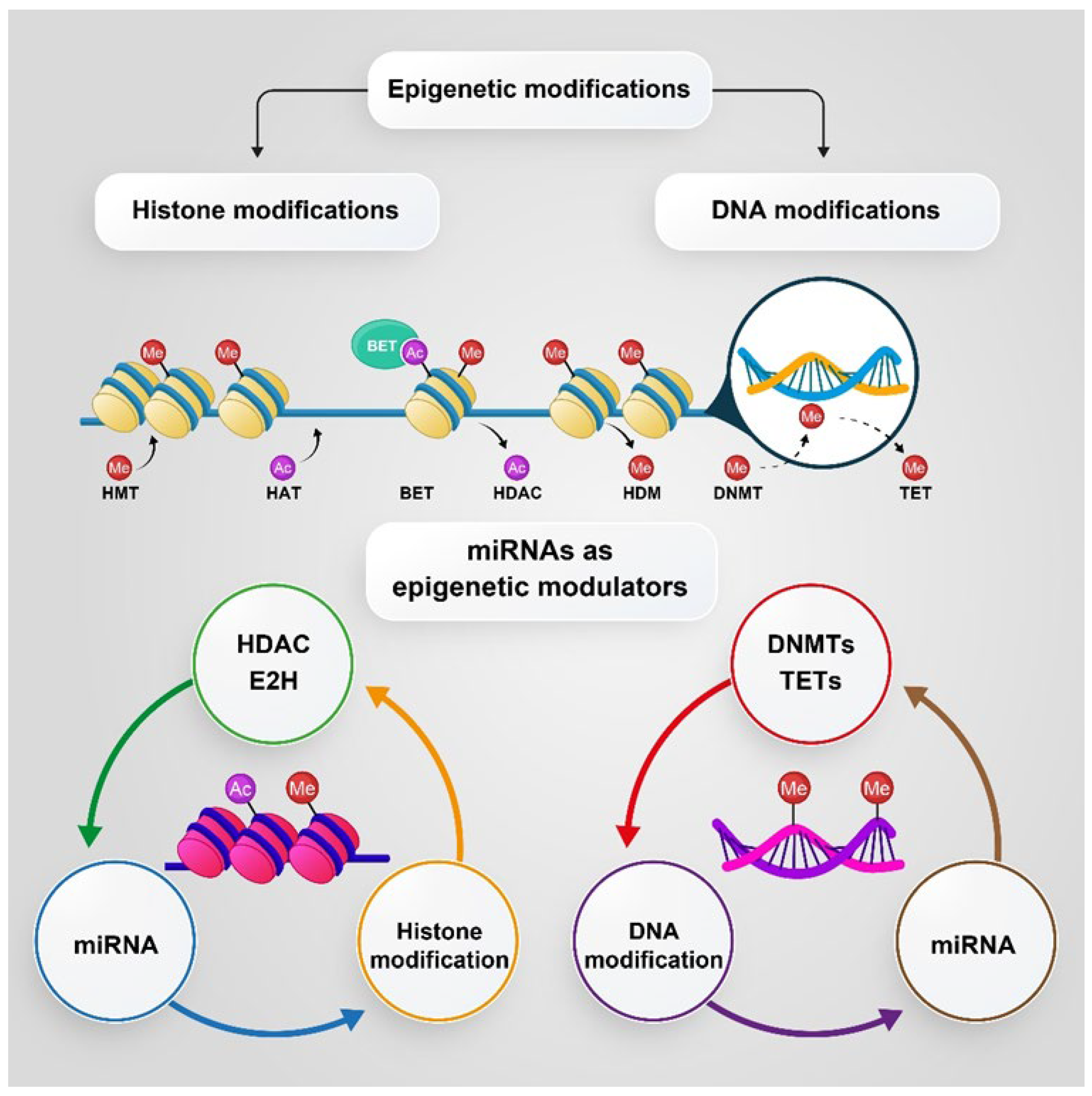
5. The Role of PKs in Epigenetic Changes and Progression of PC
5.1. DNA Methylation and Histone Modification
5.2. MicroRNAs (miRNAs), as Epigenetic Modulators
6. PC Treatment and Management
6.1. PK Inhibitors in PC
6.2. Epigenetic Targeting as a Therapeutic Strategy for Advanced PC
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2022 update. Pharmacol. Res. 2022, 175, 106037. [Google Scholar] [CrossRef]
- Fabian, M.A.; Biggs, W.H.; Treiber, D.K.; Atteridge, C.E.; Azimioara, M.D.; Benedetti, M.G.; Carter, T.A.; Ciceri, P.; Edeen, P.T.; Floyd, M.; et al. A small molecule–kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005, 23, 329–336. [Google Scholar] [CrossRef]
- Wells, C.I.; Al-Ali, H.; Andrews, D.M.; Asquith, C.R.M.; Axtman, A.D.; Dikic, I.; Ebner, D.; Ettmayer, P.; Fischer, C.; Frederiksen, M.; et al. The Kinase Chemogenomic Set (KCGS): An Open Science Resource for Kinase Vulnerability Identification. Int. J. Mol. Sci. 2021, 22, 566. [Google Scholar] [CrossRef]
- Ferguson, F.M.; Gray, N.S. Kinase inhibitors: The road ahead. Nat. Rev. Drug Discov. 2018, 17, 353–377. [Google Scholar] [CrossRef]
- Klaeger, S.; Heinzlmeir, S.; Wilhelm, M.; Polzer, H.; Vick, B.; Koenig, P.-A.; Reinecke, M.; Ruprecht, B.; Petzoldt, S.; Meng, C.; et al. The target landscape of clinical kinase drugs. Science 2017, 358, eaan4368. [Google Scholar] [CrossRef]
- Anastassiadis, T.; Deacon, S.W.; Devarajan, K.; Ma, H.; Peterson, J.R. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1039–1045. [Google Scholar] [CrossRef]
- Carles, F.; Bourg, S.; Meyer, C.; Bonnet, P. PKIDB: A Curated, Annotated and Updated Database of Protein Kinase Inhibitors in Clinical Trials. Molecules 2018, 23, 908. [Google Scholar] [CrossRef]
- Corti, M.; Lorenzetti, S.; Ubaldi, A.; Zilli, R.; Marcoccia, D. Endocrine Disruptors and Prostate Cancer. Int. J. Mol. Sci. 2022, 23, 1216. [Google Scholar] [CrossRef]
- Wang, X.; Wei, L.; Xiao, J.; Shan, K.; He, Q.; Huang, F.; Ge, X.; Gao, X.; Feng, N.; Chen, Y.Q. Cholesterol and saturated fatty acids synergistically promote the malignant progression of prostate cancer. Neoplasia 2022, 24, 86–97. [Google Scholar] [CrossRef]
- Zhang, H.; Spencer, K.; Burley, S.K.; Zheng, X.S. Toward improving androgen receptor-targeted therapies in male-dominant hepatocellular carcinoma. Drug Discov. Today 2021, 26, 1539–1546. [Google Scholar] [CrossRef]
- Lv, S.; Pu, X.; Luo, M.; Wen, H.; Xu, Z.; Wei, Q.; Dang, Q. Long noncoding RNA GAS5 interacts and suppresses androgen receptor activity in prostate cancer cells. Prostate 2021, 81, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Thienger, P.; Rubin, M.A. SETting Up for Epigenetic Regulation of Advanced Prostate Cancer. Cancer Cell 2020, 38, 309–311. [Google Scholar] [CrossRef] [PubMed]
- Chau, V.; Madan, R.A.; Aragon-Ching, J.B. Protein kinase inhibitors for the treatment of prostate cancer. Expert Opin. Pharmacother. 2021, 22, 1889–1899. [Google Scholar] [CrossRef]
- Gioeli, D.; Mandell, J.W.; Petroni, G.R.; Frierson, H.F.; Weber, M.J. Activation of mitogen-activated protein kinase associated with prostate cancer progression. Cancer Res. 1999, 59, 279–284. [Google Scholar]
- Xu, R.; Hu, J. The role of JNK in prostate cancer progression and therapeutic strategies. Biomed. Pharmacother. 2020, 121, 109679. [Google Scholar] [CrossRef]
- Ge, R.; Wang, Z.; Montironi, R.; Jiang, Z.; Cheng, M.; Santoni, M.; Huang, K.; Massari, F.; Lu, X.; Cimadamore, A. Epigenetic modulations and lineage plasticity in advanced prostate cancer. Ann. Oncol. 2020, 31, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Conteduca, V.; Hess, J.; Yamada, Y.; Ku, S.-Y.; Beltran, H. Epigenetics in prostate cancer: Clinical implications. Transl. Androl. Urol. 2021, 10, 3104–3116. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.S.; Li, J.; Stockert, J.A.; O’Connor, J.; Herzog, B.; Elaiho, C.; Galsky, M.D.; Tewari, A.K.; Yadav, K.K. Combination effect of therapies targeting the PI3K- and AR-signaling pathways in prostate cancer. Oncotarget 2016, 7, 76181–76196. [Google Scholar] [CrossRef]
- Haubrich, B.A.; Swinney, D.C. Enzyme Activity Assays for Protein Kinases: Strategies to Identify Active Substrates. Curr. Drug Discov. Technol. 2016, 13, 2–15. [Google Scholar] [CrossRef]
- Han, K.-H.E.; McGonigal, T. Role of Focal Adhesion Kinase in Human Cancer: A Potential Target for Drug Discovery. Anti-Cancer Agents Med. Chem. 2007, 7, 681–684. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef]
- Martin, J.; Anamika, K.; Srinivasan, N. Classification of Protein Kinases on the Basis of Both Kinase and Non-Kinase Regions. PLoS ONE 2010, 5, e12460. [Google Scholar] [CrossRef] [PubMed]
- Borgo, C.; D’Amore, C.; Sarno, S.; Salvi, M.; Ruzzene, M. Protein kinase CK2: A potential therapeutic target for diverse human diseases. Signal Transduct. Target. Ther. 2021, 6, 183. [Google Scholar] [CrossRef] [PubMed]
- Ijomone, O.M.; Iroegbu, J.D.; Aschner, M.; Bornhorst, J. Impact of environmental toxicants on p38-and ERK-MAPK signaling pathways in the central nervous system. Neurotoxicology 2021, 86, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Patterson, H.; Nibbs, R.; McInnes, I.; Siebert, S. Protein kinase inhibitors in the treatment of inflammatory and autoimmune diseases. Clin. Exp. Immunol. 2014, 176, 1–10. [Google Scholar] [CrossRef]
- Abdellatif, K.R.A.; Bakr, R.B. Pyrimidine and fused pyrimidine derivatives as promising protein kinase inhibitors for cancer treatment. Med. Chem. Res. 2021, 30, 31–49. [Google Scholar] [CrossRef]
- Hanks, S.K.; Hunter, T. The eukaryotic protein kinase superfamily: Kinase (catalytic) domain structure and classification1. FASEB J. 1995, 9, 576–596. [Google Scholar] [CrossRef]
- Hunter, T. [1] Protein kinase classification. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1991; Volume 200, pp. 3–37. [Google Scholar]
- Modi, V.; Dunbrack, R.L., Jr. Kincore: A web resource for structural classification of protein kinases and their inhibitors. Nucleic Acids Res. 2021, 50, D654–D664. [Google Scholar] [CrossRef]
- Talele, T.T.; McLaughlin, M.L. Molecular docking/dynamics studies of Aurora A kinase inhibitors. J. Mol. Graph. Model. 2008, 26, 1213–1222. [Google Scholar] [CrossRef]
- Wang, Z.; Huang, W.; Zhou, K.; Ren, X.; Ding, K. Targeting the Non-Catalytic Functions: A New Paradigm for Kinase Drug Discovery? J. Med. Chem. 2022, 65, 1735–1748. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Angus, S.P.; Zawistowski, J.S.; Johnson, G.L. Epigenetic Mechanisms Regulating Adaptive Responses to Targeted Kinase Inhibitors in Cancer. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 209–229. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef] [PubMed]
- Kung, J.E.; Jura, N. Structural Basis for the Non-catalytic Functions of Protein Kinases. Structure 2016, 24, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Knapp, S. New opportunities for kinase drug repurposing and target discovery. Br. J. Cancer 2018, 118, 936–937. [Google Scholar] [CrossRef] [PubMed]
- Imran, M.; Asdaq, S.M.B.; Khan, S.A.; Unnikrishnan Meenakshi, D.; Alamri, A.S.; Alsanie, W.F.; Alhomrani, M.; Mohzari, Y.; Alrashed, A.; AlMotairi, M.; et al. Innovations and Patent Trends in the Development of USFDA Approved Protein Kinase Inhibitors in the Last Two Decades. Pharmaceuticals 2021, 14, 710. [Google Scholar] [CrossRef]
- Bidwell, G.L., 3rd; Raucher, D. Therapeutic peptides for cancer therapy. Part I-peptide inhibitors of signal transduction cascades. Expert Opin. Drug Deliv. 2009, 6, 1033–1047. [Google Scholar] [CrossRef]
- Liu, C.; Ke, P.; Zhang, J.; Zhang, X.; Chen, X. Protein kinase inhibitor peptide as a tool to specifically inhibit protein kinase A. Front. Physiol. 2020, 11, 1532. [Google Scholar] [CrossRef]
- Srikar, R.; Suresh, D.; Zambre, A.; Taylor, K.; Chapman, S.; Leevy, M.; Upendran, A.; Kannan, R. Targeted nanoconjugate co-delivering siRNA and tyrosine kinase inhibitor to KRAS mutant NSCLC dissociates GAB1-SHP2 post oncogene knockdown. Sci. Rep. 2016, 6, 30245. [Google Scholar] [CrossRef]
- Jain, A.; Mahira, S.; Majoral, J.P.; Bryszewska, M.; Khan, W.; Ionov, M. Dendrimer mediated targeting of siRNA against polo-like kinase for the treatment of triple negative breast cancer. J. Biomed. Mater. Res. A 2019, 107, 1933–1944. [Google Scholar] [CrossRef]
- Moradpour, Z.; Barghi, L. Novel Approaches for Efficient Delivery of Tyrosine Kinase Inhibitors. J. Pharm. Pharm. Sci. 2019, 22, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Smidova, V.; Michalek, P.; Goliasova, Z.; Eckschlager, T.; Hodek, P.; Adam, V.; Heger, Z. Nanomedicine of tyrosine kinase inhibitors. Theranostics 2021, 11, 1546–1567. [Google Scholar] [CrossRef] [PubMed]
- Reagan-Shaw, S.; Ahmad, N. Silencing of polo-like kinase (Plk) 1 via siRNA causes induction of apoptosis and impairment of mitosis machinery in human prostate cancer cells: Implications for the treatment of prostate cancer. FASEB J. 2005, 19, 611–613. [Google Scholar] [CrossRef]
- Tsutsumi, K.; Kasaoka, T.; Park, H.M.; Nishiyama, H.; Nakajima, M.; Honda, T. Tumor growth inhibition by synthetic and expressed siRNA targeting focal adhesion kinase. Int. J. Oncol. 2008, 33, 215–224. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gagnon, V.; Mathieu, I.; Sexton, E.; Leblanc, K.; Asselin, E. AKT involvement in cisplatin chemoresistance of human uterine cancer cells. Gynecol. Oncol. 2004, 94, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Karasarides, M.; Chiloeches, A.; Hayward, R.; Niculescu-Duvaz, D.; Scanlon, I.; Friedlos, F.; Ogilvie, L.; Hedley, D.; Martin, J.; Marshall, C.J. B-RAF is a therapeutic target in melanoma. Oncogene 2004, 23, 6292–6298. [Google Scholar] [CrossRef]
- Hojjat-Farsangi, M.; Ghaemimanesh, F.; Daneshmanesh, A.H.; Bayat, A.A.; Mahmoudian, J.; Jeddi-Tehrani, M.; Rabbani, H.; Mellstedt, H. Inhibition of the receptor tyrosine kinase ROR1 by anti-ROR1 monoclonal antibodies and siRNA induced apoptosis of melanoma cells. PLoS ONE 2013, 8, e61167. [Google Scholar] [CrossRef]
- Tao, Y.; Zhang, P.; Frascogna, V.; Lecluse, Y.; Auperin, A.; Bourhis, J.; Deutsch, E. Enhancement of radiation response by inhibition of Aurora-A kinase using siRNA or a selective Aurora kinase inhibitor PHA680632 in p53-deficient cancer cells. Br. J. Cancer 2007, 97, 1664–1672. [Google Scholar] [CrossRef]
- Asik, E.; Akpinar, Y.; Caner, A.; Kahraman, N.; Guray, T.; Volkan, M.; Albarracin, C.; Pataer, A.; Arun, B.; Ozpolat, B. EF2-kinase targeted cobalt-ferrite siRNA-nanotherapy suppresses BRCA1-mutated breast cancer. Nanomedicine 2019, 14, 2315–2338. [Google Scholar] [CrossRef]
- Goldberg, M.S.; Sharp, P.A. Pyruvate kinase M2-specific siRNA induces apoptosis and tumor regression. J. Exp. Med. 2012, 209, 217–224. [Google Scholar] [CrossRef]
- He, S.B.; Yuan, Y.; Wang, L.; Yu, M.J.; Zhu, Y.B.; Zhu, X.G. Effects of cyclin-dependent kinase 8 specific siRNA on the proliferation and apoptosis of colon cancer cells. J. Exp. Clin. Cancer Res. 2011, 30, 109. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Zhong, L.; Weng, Y.; Peng, L.; Huang, Y.; Zhao, Y.; Liang, X.J. Therapeutic siRNA: State of the art. Signal Transduct. Target. 2020, 5, 101. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzada, T.; Reid, G.; McKenzie, D.R. Fundamentals of siRNA and miRNA therapeutics and a review of targeted nanoparticle delivery systems in breast cancer. Biophys. Rev. 2018, 10, 69–86. [Google Scholar] [CrossRef]
- Jonker, D.J.; O’Callaghan, C.J.; Karapetis, C.S.; Zalcberg, J.R.; Tu, D.; Au, H.-J.; Berry, S.R.; Krahn, M.; Price, T.; Simes, R.J. Cetuximab for the treatment of colorectal cancer. N. Engl. J. Med. 2007, 357, 2040–2048. [Google Scholar] [CrossRef]
- von Minckwitz, G.; Procter, M.; de Azambuja, E.; Zardavas, D.; Benyunes, M.; Viale, G.; Suter, T.; Arahmani, A.; Rouchet, N.; Clark, E.; et al. Adjuvant Pertuzumab and Trastuzumab in Early HER2-Positive Breast Cancer. N. Engl. J. Med. 2017, 377, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Hudis, C.A. Trastuzumab—mechanism of action and use in clinical practice. N. Engl. J. Med. 2007, 357, 39–51. [Google Scholar] [CrossRef]
- Gharwan, H.; Groninger, H. Kinase inhibitors and monoclonal antibodies in oncology: Clinical implications. Nat. Rev. Clin. Oncol. 2016, 13, 209–227. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Cai, M.; Shao, L.; Zhang, J. Targeting Protein Kinases Degradation by PROTACs. Front. Chem. 2021, 9, 679120. [Google Scholar] [CrossRef]
- Kurimchak, A.M.; Herrera-Montávez, C.; Montserrat, S.; Araiza, D.; Hu, J.; Jin, J.; Duncan, J.S. MDR1 Drug Efflux Pump Promotes Intrinsic and Acquired Resistance to PROTACs in Cancer Cells. bioRxiv 2021. [Google Scholar] [CrossRef]
- Sakurai, R.; Villarreal, P.; Husain, S.; Liu, J.; Sakurai, T.; Tou, E.; Torday, J.S.; Rehan, V.K. Curcumin protects the developing lung against long-term hyperoxic injury. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2013, 305, L301–L311. [Google Scholar] [CrossRef]
- Masuda, M.; Wakasaki, T.; Toh, S.; Shimizu, M.; Adachi, S. Chemoprevention of Head and Neck Cancer by Green Tea Extract: EGCG-The Role of EGFR Signaling and “Lipid Raft”. J. Oncol. 2011, 2011, 540148. [Google Scholar] [CrossRef] [PubMed]
- Byun, S.; Lee, K.W.; Jung, S.K.; Lee, E.J.; Hwang, M.K.; Lim, S.H.; Bode, A.M.; Lee, H.J.; Dong, Z. Luteolin inhibits protein kinase C(epsilon) and c-Src activities and UVB-induced skin cancer. Cancer Res. 2010, 70, 2415–2423. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wang, J.J.; Chen, X.L.; Du, S.M.; Li, D.S.; Pei, Z.J.; Lan, H.; Wu, L.B. The JAK2/STAT3 and mitochondrial pathways are essential for quercetin nanoliposome-induced C6 glioma cell death. Cell Death Dis. 2013, 4, e746. [Google Scholar] [CrossRef]
- Boly, R.; Gras, T.; Lamkami, T.; Guissou, P.; Serteyn, D.; Kiss, R.; Dubois, J. Quercetin inhibits a large panel of kinases implicated in cancer cell biology. Int. J. Oncol. 2011, 38, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Aljarbou, A.N.; Aldebasi, Y.H.; Faisal, S.M.; Khan, M.A. Resveratrol suppresses the proliferation of breast cancer cells by inhibiting fatty acid synthase signaling pathway. Cancer Epidemiol. 2014, 38, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; International Natural Product Sciences Taskforce; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Sankarapandian, V.; Venmathi Maran, B.A.; Rajendran, R.L.; Jogalekar, M.P.; Gurunagarajan, S.; Krishnamoorthy, R.; Gangadaran, P.; Ahn, B.C. An Update on the Effectiveness of Probiotics in the Prevention and Treatment of Cancer. Life 2022, 12, 59. [Google Scholar] [CrossRef]
- Asoudeh-Fard, A.; Barzegari, A.; Dehnad, A.; Bastani, S.; Golchin, A.; Omidi, Y. Lactobacillus plantarum induces apoptosis in oral cancer KB cells through upregulation of PTEN and downregulation of MAPK signalling pathways. BioImpacts BI 2017, 7, 193. [Google Scholar] [CrossRef]
- Mandel, A.; Larsson, P.; Sarwar, M.; Semenas, J.; Syed Khaja, A.S.; Persson, J.L. The interplay between AR, EGF receptor and MMP-9 signaling pathways in invasive prostate cancer. Mol. Med. 2018, 24, 1–13. [Google Scholar] [CrossRef]
- Kurose, H.; Ueda, K.; Kondo, R.; Ogasawara, S.; Kusano, H.; Sanada, S.; Naito, Y.; Nakiri, M.; Nishihara, K.; Kakuma, T. Elevated expression of EPHA2 is associated with poor prognosis after radical prostatectomy in prostate Cancer. Anticancer Res. 2019, 39, 6249–6257. [Google Scholar] [CrossRef]
- Taddei, M.L.; Parri, M.; Angelucci, A.; Onnis, B.; Bianchini, F.; Giannoni, E.; Raugei, G.; Calorini, L.; Rucci, N.; Teti, A.; et al. Kinase-dependent and -independent roles of EphA2 in the regulation of prostate cancer invasion and metastasis. Am. J. Pathol. 2009, 174, 1492–1503. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.J.; Ma, Q.; Zhu, J.; Li, J.; Xue, B.X.; Gao, J.; Sun, C.Y.; Zang, Y.C.; Zhou, Y.B.; Yang, D.R. Combined inhibition of JAK1, 2/Stat3-PD-L1 signaling pathway suppresses the immune escape of castration-resistant prostate cancer to NK cells in hypoxia. Mol. Med. Rep. 2018, 17, 8111–8120. [Google Scholar] [PubMed]
- Luo, Y.; Yang, X.; Basourakos, S.P.; Zuo, X.; Wei, D.; Zhao, J.; Li, M.; Li, Q.; Feng, T.; Guo, P. Enzalutamide-Resistant Progression of Castration-Resistant Prostate Cancer Is Driven via the JAK2/STAT1-Dependent Pathway. Front. Mol. Biosci. 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Wang, W.; Zhou, W.; Coleman, I.; Cai, Q.; Dong, B.; Ittmann, M.M.; Creighton, C.J.; Bian, Y.; Meng, Y.; et al. MAPK4 promotes prostate cancer by concerted activation of androgen receptor and AKT. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Alwanian, W.M.; Tyner, A.L. Protein tyrosine kinase 6 signaling in prostate cancer. Am. J. Clin. Exp. Urol. 2020, 8, 1–8. [Google Scholar]
- Cronin, R.; Brooke, G.N.; Prischi, F. The role of the p90 ribosomal S6 kinase family in prostate cancer progression and therapy resistance. Oncogene 2021, 40, 3775–3785. [Google Scholar] [CrossRef]
- Yamamura, A.; Nayeem, M.J.; Muramatsu, H.; Nakamura, K.; Sato, M. MAZ51 Blocks the Tumor Growth of Prostate Cancer by Inhibiting Vascular Endothelial Growth Factor Receptor 3. Front. Pharm. 2021, 12, 667474. [Google Scholar] [CrossRef]
- Suzuki, A.; Lu, J.; Kusakai, G.-i.; Kishimoto, A.; Ogura, T.; Esumi, H. ARK5 is a tumor invasion-associated factor downstream of Akt signaling. Mol. Cell. Biol. 2004, 24, 3526–3535. [Google Scholar] [CrossRef]
- Laderoute, K.R.; Amin, K.; Calaoagan, J.M.; Knapp, M.; Le, T.; Orduna, J.; Foretz, M.; Viollet, B. 5′-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol. Cell. Biol. 2006, 26, 5336–5347. [Google Scholar] [CrossRef]
- Bonini, M.G.; Gantner, B.N. The multifaceted activities of AMPK in tumor progression—Why the “one size fits all” definition does not fit at all? IUBMB Life 2013, 65, 889–896. [Google Scholar] [CrossRef]
- Park, H.U.; Suy, S.; Danner, M.; Dailey, V.; Zhang, Y.; Li, H.; Hyduke, D.R.; Collins, B.T.; Gagnon, G.; Kallakury, B. AMP-activated protein kinase promotes human prostate cancer cell growth and survival. Mol. Cancer Ther. 2009, 8, 733–741. [Google Scholar] [CrossRef]
- Tennakoon, J.B.; Shi, Y.; Han, J.J.; Tsouko, E.; White, M.A.; Burns, A.R.; Zhang, A.; Xia, X.; Ilkayeva, O.R.; Xin, L. Androgens regulate prostate cancer cell growth via an AMPK-PGC-1α-mediated metabolic switch. Oncogene 2014, 33, 5251–5261. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, Y.; Zichu Yang, I.A.; Nixon, C.; Salt, I.P.; Leung, H.Y. AMP-activated protein kinase (AMPK) as a potential therapeutic target independent of PI3K/Akt signaling in prostate cancer. Oncoscience 2014, 1, 446. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.S.; Frigo, D.E. A spatiotemporal hypothesis for the regulation, role, and targeting of AMPK in prostate cancer. Nat. Rev. Urol. 2017, 14, 164–180. [Google Scholar] [CrossRef] [PubMed]
- Popovics, P.; Frigo, D.E.; Schally, A.V.; Rick, F.G. Targeting the 5′-AMP-activated protein kinase and related metabolic pathways for the treatment of prostate cancer. Expert Opin. Targets 2015, 19, 617–632. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.G.; Nam, Y.; Shin, K.J.; Yoon, S.; Park, W.S.; Joung, J.Y.; Seo, J.K.; Jang, J.; Lee, S.; Nam, D.; et al. Androgen-induced expression of DRP1 regulates mitochondrial metabolic reprogramming in prostate cancer. Cancer Lett. 2020, 471, 72–87. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Song, Y.; Sun, Y.; Li, X.; Chen, L.; Yang, L.; Xing, Y. AMPK/GSK3beta/beta-catenin cascade-triggered overexpression of CEMIP promotes migration an.nd invasion in anoikis-resistant prostate cancer cells by enhancing metabolic reprogramming. FASEB J. 2018, 32, 3924–3935. [Google Scholar] [CrossRef] [PubMed]
- Kerr, M.C.; Teasdale, R.D. Defining macropinocytosis. Traffic 2009, 10, 364–371. [Google Scholar] [CrossRef]
- Dharmawardhane, S.; Schurmann, A.; Sells, M.A.; Chernoff, J.; Schmid, S.L.; Bokoch, G.M. Regulation of macropinocytosis by p21-activated kinase-1. Mol. Biol. Cell 2000, 11, 3341–3352. [Google Scholar] [CrossRef]
- Ridley, A.J.; Paterson, H.F.; Johnston, C.L.; Diekmann, D.; Hall, A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell 1992, 70, 401–410. [Google Scholar] [CrossRef]
- Commisso, C.; Davidson, S.M.; Soydaner-Azeloglu, R.G.; Parker, S.J.; Kamphorst, J.J.; Hackett, S.; Grabocka, E.; Nofal, M.; Drebin, J.A.; Thompson, C.B. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013, 497, 633–637. [Google Scholar] [CrossRef]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Phin, S.; Moore, M.W.; Cotter, P.D. Genomic Rearrangements of PTEN in Prostate Cancer. Front. Oncol. 2013, 3, 240. [Google Scholar] [CrossRef] [PubMed]
- M Dillon, L.; W Miller, T. Therapeutic targeting of cancers with loss of PTEN function. Curr. Drug Targets 2014, 15, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.M.; Abate-Shen, C. Molecular genetics of prostate cancer: New prospects for old challenges. Genes Dev. 2010, 24, 1967–2000. [Google Scholar] [CrossRef]
- Araki, N.; Johnson, M.T.; Swanson, J.A. A role for phosphoinositide 3-kinase in the completion of macropinocytosis and phagocytosis by macrophages. J. Cell Biol. 1996, 135, 1249–1260. [Google Scholar] [CrossRef]
- Kim, S.M.; Nguyen, T.T.; Ravi, A.; Kubiniok, P.; Finicle, B.T.; Jayashankar, V.; Malacrida, L.; Hou, J.; Robertson, J.; Gao, D. PTEN deficiency and AMPK activation promote nutrient scavenging and anabolism in prostate cancer cells. Cancer Discov. 2018, 8, 866–883. [Google Scholar] [CrossRef]
- Sapio, L.; Di Maiolo, F.; Illiano, M.; Esposito, A.; Chiosi, E.; Spina, A.; Naviglio, S. Targeting protein kinase A in cancer therapy: An update. EXCLI J. 2014, 13, 843–855. [Google Scholar]
- Caretta, A.; Mucignat-Caretta, C. Protein kinase a in cancer. Cancers 2011, 3, 913–926. [Google Scholar] [CrossRef]
- Eder, I.E.; Egger, M.; Neuwirt, H.; Seifarth, C.; Maddalo, D.; Desiniotis, A.; Schäfer, G.; Puhr, M.; Bektic, J.; Cato, A.C. Enhanced inhibition of prostate tumor growth by dual targeting the androgen receptor and the regulatory subunit type Iα of protein kinase A in vivo. Int. J. Mol. Sci. 2013, 14, 11942–11962. [Google Scholar] [CrossRef]
- Sarwar, M.; Sandberg, S.; Abrahamsson, P.A.; Persson, J.L. Protein kinase A (PKA) pathway is functionally linked to androgen receptor (AR) in the progression of prostate cancer. Urol. Oncol. 2014, 32, 25.e1–25.e12. [Google Scholar] [CrossRef] [PubMed]
- Dagar, M.; Singh, J.P.; Dagar, G.; Tyagi, R.K.; Bagchi, G. Phosphorylation of HSP90 by protein kinase A is essential for the nuclear translocation of androgen receptor. Urol. Oncol. Semin. Orig. Investig. 2019, 294, 8699–8710. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Sun, J.; Xie, S.; Chi, C.; Zhu, Y.; Pan, J.; Dong, B.; Huang, Y.; Xia, W.; Sha, J. PRKAR2B-HIF-1α loop promotes aerobic glycolysis and tumour growth in prostate cancer. Cell Prolif. 2020, 53, e12918. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Giridhar, K.V.; Zhang, L.; Xu, S.; Wang, Q.J. A protein kinase C/protein kinase D pathway protects LNCaP prostate cancer cells from phorbol ester-induced apoptosis by promoting ERK1/2 and NF-{kappa}B activities. Carcinogenesis 2011, 32, 1198–1206. [Google Scholar] [CrossRef]
- Aljameeli, A.; Thakkar, A.; Shah, G. Calcitonin receptor increases invasion of prostate cancer cells by recruiting zonula occludens-1 and promoting PKA-mediated TJ disassembly. Cell. Signal. 2017, 36, 1–13. [Google Scholar] [CrossRef]
- Menon, J.; Doebele, R.C.; Gomes, S.; Bevilacqua, E.; Reindl, K.M.; Rosner, M.R. A novel interplay between Rap1 and PKA regulates induction of angiogenesis in prostate cancer. PLoS ONE 2012, 7, e49893. [Google Scholar] [CrossRef]
- Boutin, B.; Tajeddine, N.; Monaco, G.; Molgo, J.; Vertommen, D.; Rider, M.; Parys, J.B.; Bultynck, G.; Gailly, P. Endoplasmic reticulum Ca2+ content decrease by PKA-dependent hyperphosphorylation of type 1 IP3 receptor contributes to prostate cancer cell resistance to androgen deprivation. Cell Calcium 2015, 57, 312–320. [Google Scholar] [CrossRef]
- Shan, K.; Feng, N.; Cui, J.; Wang, S.; Qu, H.; Fu, G.; Li, J.; Chen, H.; Wang, X.; Wang, R.; et al. Resolvin D1 and D2 inhibit tumour growth and inflammation via modulating macrophage polarization. J. Cell Mol. Med. 2020, 24, 8045–8056. [Google Scholar] [CrossRef]
- Lindsley, C.W. The Akt/PKB family of protein kinases: A review of small molecule inhibitors and progress towards target validation: A 2009 update. Curr. Top. Med. Chem. 2010, 10, 458–477. [Google Scholar] [CrossRef]
- Yoeli-Lerner, M.; Toker, A. Akt/PKB signaling in cancer: A function in cell motility and invasion. Cell Cycle 2006, 5, 603–605. [Google Scholar] [CrossRef]
- Shorning, B.Y.; Dass, M.S.; Smalley, M.J.; Pearson, H.B. The PI3K-AKT-mTOR Pathway and Prostate Cancer: At the Crossroads of AR, MAPK, and WNT Signaling. Int. J. Mol. Sci. 2020, 21, 4507. [Google Scholar] [CrossRef]
- Ghosh, P.M.; Malik, S.; Bedolla, R.; Kreisberg, J.I. Akt in prostate cancer: Possible role in androgen-independence. Curr. Drug Metab. 2003, 4, 487–496. [Google Scholar] [CrossRef]
- Li, B.; Sun, A.; Youn, H.; Hong, Y.; Terranova, P.F.; Thrasher, J.B.; Xu, P.; Spencer, D. Conditional Akt activation promotes androgen-independent progression of prostate cancer. Carcinogenesis 2007, 28, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Hu, M.C.; Makino, K.; Spohn, B.; Bartholomeusz, G.; Yan, D.-H.; Hung, M.-C. HER-2/neu promotes androgen-independent survival and growth of prostate cancer cells through the Akt pathway. Cancer Res. 2000, 60, 6841–6845. [Google Scholar] [PubMed]
- Pungsrinont, T.; Kallenbach, J.; Baniahmad, A. Role of PI3K-AKT-mTOR Pathway as a Pro-Survival Signaling and Resistance-Mediating Mechanism to Therapy of Prostate Cancer. Int. J. Mol. Sci. 2021, 22, 1088. [Google Scholar] [CrossRef]
- Shukla, S.; Maclennan, G.T.; Hartman, D.J.; Fu, P.; Resnick, M.I.; Gupta, S. Activation of PI3K-Akt signaling pathway promotes prostate cancer cell invasion. Int. J. Cancer 2007, 121, 1424–1432. [Google Scholar] [CrossRef] [PubMed]
- Van de Sande, T.; Roskams, T.; Lerut, E.; Joniau, S.; Van Poppel, H.; Verhoeven, G.; Swinnen, J.V. High-level expression of fatty acid synthase in human prostate cancer tissues is linked to activation and nuclear localization of Akt/PKB. J. Pathol. 2005, 206, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Shukla, M.; Maclennan, G.T.; Fu, P.; Gupta, S. Deregulation of FOXO3A during prostate cancer progression. Int. J. Oncol. 2009, 34, 1613–1620. [Google Scholar] [CrossRef][Green Version]
- Das, T.; Suman, S.; Alatassi, H.; Ankem, M.; Damodaran, C. Inhibition of AKT promotes FOXO3a-dependent apoptosis in prostate cancer. Cell Death Dis. 2016, 7, e2111. [Google Scholar] [CrossRef]
- Shukla, S.; Bhaskaran, N.; Maclennan, G.T.; Gupta, S. Deregulation of FoxO3a accelerates prostate cancer progression in TRAMP mice. Prostate 2013, 73, 1507–1517. [Google Scholar] [CrossRef]
- Black, J.D. PKC and control of the cell cycle. In Protein Kinase C in Cancer Signaling and Therapy; Springer: Berlin/Heidelberg, Germany, 2010; pp. 155–188. [Google Scholar]
- Reyland, M.E.; Bradford, A.P. PKC and the Control of Apoptosis. In Protein Kinase C in Cancer Signaling and Therapy; Springer: Berlin/Heidelberg, Germany, 2010; pp. 189–222. [Google Scholar]
- Reina-Campos, M.; Diaz-Meco, M.T.; Moscat, J. The Dual Roles of the Atypical Protein Kinase Cs in Cancer. Cancer Cell 2019, 36, 218–235. [Google Scholar] [CrossRef] [PubMed]
- Apostolatos, A.H.; Ratnayake, W.S.; Win-Piazza, H.; Apostolatos, C.A.; Smalley, T.; Kang, L.; Salup, R.; Hill, R.; Acevedo-Duncan, M. Inhibition of atypical protein kinase C-ι effectively reduces the malignancy of prostate cancer cells by downregulating the NF-κB signaling cascade. Int. J. Oncol. 2018, 53, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Baek, H.-S.; Park, N.; Kwon, Y.-J.; Ye, D.-J.; Shin, S.; Chun, Y.-J. Annexin A5 suppresses cyclooxygenase-2 expression by downregulating the protein kinase C-ζ–nuclear factor-κB signaling pathway in prostate cancer cells. Oncotarget 2017, 8, 74263. [Google Scholar] [CrossRef]
- Reyland, M.E. Protein kinase C isoforms: Multi-functional regulators of cell life and death. Front. BioSci. (Landmark Ed.) 2009, 14, 2386–2399. [Google Scholar] [CrossRef] [PubMed]
- Ponguta, L.A.; Gregory, C.W.; French, F.S.; Wilson, E.M. Site-specific androgen receptor serine phosphorylation linked to epidermal growth factor-dependent growth of castration-recurrent prostate cancer. J. Biol. Chem. 2008, 283, 20989–21001. [Google Scholar] [CrossRef]
- Patek, S.; Willder, J.; Heng, J.; Taylor, B.; Horgan, P.; Leung, H.; Underwood, M.; Edwards, J. Androgen receptor phosphorylation status at serine 578 predicts poor outcome in prostate cancer patients. Oncotarget 2017, 8, 4875–4887. [Google Scholar] [CrossRef] [PubMed]
- Aziz, M.H.; Manoharan, H.T.; Church, D.R.; Dreckschmidt, N.E.; Zhong, W.; Oberley, T.D.; Wilding, G.; Verma, A.K. Protein kinase Cε interacts with signal transducers and activators of transcription 3 (Stat3), phosphorylates Stat3Ser727, and regulates its constitutive activation in prostate cancer. Cancer Res. 2007, 67, 8828–8838. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Blando, J.M.; Perez, C.J.; Abba, M.C.; Benavides, F.; Kazanietz, M.G. Protein kinase C epsilon cooperates with PTEN loss for prostate tumorigenesis through the CXCL13-CXCR5 pathway. Cell Rep. 2017, 19, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Zeng, F.; Li, S.; Li, G.; Lai, X.; Wang, Q.J.; Deng, F. Crosstalk of protein kinase C epsilon with Smad2/3 promotes tumor cell proliferation in prostate cancer cells by enhancing aerobic glycolysis. Cell Mol. Life Sci. 2018, 75, 4583–4598. [Google Scholar] [CrossRef]
- Ivaska, J.; Vuoriluoto, K.; Huovinen, T.; Izawa, I.; Inagaki, M.; Parker, P.J. PKCε-mediated phosphorylation of vimentin controls integrin recycling and motility. EMBO J. 2005, 24, 3834–3845. [Google Scholar] [CrossRef]
- McJilton, M.A.; Van Sikes, C.; Wescott, G.G.; Wu, D.; Foreman, T.L.; Gregory, C.W.; Weidner, D.A.; Harris Ford, O.; Morgan Lasater, A.; Mohler, J.L.; et al. Protein kinase Cepsilon interacts with Bax and promotes survival of human prostate cancer cells. Oncogene 2003, 22, 7958–7968. [Google Scholar] [CrossRef]
- Usman, S.; Waseem, N.H.; Nguyen, T.K.N.; Mohsin, S.; Jamal, A.; Teh, M.T.; Waseem, A. Vimentin Is at the Heart of Epithelial Mesenchymal Transition (EMT) Mediated Metastasis. Cancers 2021, 13, 4985. [Google Scholar] [CrossRef]
- Ratnayake, W.S.; Apostolatos, C.A.; Breedy, S.; Dennison, C.L.; Hill, R.; Acevedo-Duncan, M. Atypical PKCs activate Vimentin to facilitate prostate cancer cell motility and invasion. Cell Adhes. Migr. 2021, 15, 37–57. [Google Scholar] [CrossRef]
- Roy, A.; Ye, J.; Deng, F.; Wang, Q.J. Protein kinase D signaling in cancer: A friend or foe? Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 283–294. [Google Scholar] [CrossRef]
- Zhang, X.; Connelly, J.; Chao, Y.; Wang, Q.J. Multifaceted Functions of Protein Kinase D in Pathological Processes and Human Diseases. Biomolecules 2021, 11, 483. [Google Scholar] [CrossRef]
- Durand, N.; Bastea, L.I.; Döppler, H.; Eiseler, T.; Storz, P. Src-mediated tyrosine phosphorylation of protein kinase D2 at focal adhesions regulates cell adhesion. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Khachaturyan, G. The Role of Protein Kinase D Signaling and the Thermal Microenvironment on Single Cell Migration. Ph.D. Thesis, Ruperto-Carola University, Heidelberg, Germany, 2020. [Google Scholar]
- Roy, A.; Wang, Q.J. Protein Kinase D: A Potential Therapeutic Target in Prostate Cancer. Mol. Cell Pharm. 2017, 9, 1–4. [Google Scholar]
- Li, L.; Hua, L.; Fan, H.; He, Y.; Xu, W.; Zhang, L.; Yang, J.; Deng, F.; Zeng, F. Interplay of PKD3 with SREBP1 Promotes Cell Growth via Upregulating Lipogenesis in Prostate Cancer Cells. J. Cancer 2019, 10, 6395–6404. [Google Scholar] [CrossRef]
- Zou, Z.; Zeng, F.; Xu, W.; Wang, C.; Ke, Z.; Wang, Q.J.; Deng, F. PKD2 and PKD3 promote prostate cancer cell invasion by modulating NF-kappaB- and HDAC1-mediated expression and activation of uPA. J. Cell Sci. 2012, 125, 4800–4811. [Google Scholar] [CrossRef]
- Durand, N.; Borges, S.; Storz, P. Protein kinase D enzymes as regulators of EMT and cancer cell invasion. J. Clin. Med. 2016, 5, 20. [Google Scholar] [CrossRef]
- Hamshaw, I.; Ajdarirad, M.; Mueller, A. The role of PKC and PKD in CXCL12 directed prostate cancer migration. Biochem. Biophys. Res. Commun. 2019, 519, 86–92. [Google Scholar] [CrossRef]
- Anderson, C.; Lees-Miller, S. The nuclear serine/threonine protein kinase DNA-PK. Crit. Rev. Eukaryot. Gene Expr. 1992, 2, 283–314. [Google Scholar]
- Yang, H.; Yao, F.; Marti, T.M.; Schmid, R.A.; Peng, R.W. Beyond DNA Repair: DNA-PKcs in Tumor Metastasis, Metabolism and Immunity. Cancers 2020, 12, 3389. [Google Scholar] [CrossRef]
- Törmänen Persson, H.; Aksaas, A.K.; Kvissel, A.K.; Punga, T.; Engström, Å.; Skålhegg, B.S.; Akusjärvi, G. Two cellular protein kinases, DNA-PK and PKA, phosphorylate the adenoviral L4-33K protein and have opposite effects on L1 alternative RNA splicing. PLoS ONE 2012, 7, e31871. [Google Scholar] [CrossRef]
- Medova, M.; Medo, M.; Hovhannisyan, L.; Munoz-Maldonado, C.; Aebersold, D.M.; Zimmer, Y. DNA-PK in human malignant disorders: Mechanisms and implications for pharmacological interventions. Pharmacol. Ther. 2020, 215, 107617. [Google Scholar] [CrossRef]
- Kothari, V.; Goodwin, J.F.; Zhao, S.G.; Drake, J.M.; Yin, Y.; Chang, S.L.; Evans, J.R.; Wilder-Romans, K.; Gabbara, K.; Dylgjeri, E. DNA-dependent protein kinase drives prostate cancer progression through transcriptional regulation of the Wnt signaling pathway. Clin. Cancer Res. 2019, 25, 5608–5622. [Google Scholar] [CrossRef]
- Dylgjeri, E.; McNair, C.; Goodwin, J.F.; Raymon, H.K.; McCue, P.A.; Shafi, A.A.; Leiby, B.E.; De Leeuw, R.; Kothari, V.; McCann, J.J. Pleiotropic impact of DNA-PK in cancer and implications for therapeutic strategies. Clin. Cancer Res. 2019, 25, 5623–5637. [Google Scholar] [CrossRef]
- Giguère, V. DNA-PK, nuclear mTOR, and the androgen pathway in prostate cancer. Trends Cancer 2020, 6, 337–347. [Google Scholar] [CrossRef]
- Patel, R.S.; Rupani, R.; Impreso, S.; Lui, A.; Patel, N.A. Role of alternatively spliced, pro-survival Protein Kinase C delta VIII (PKCδVIII) in ovarian cancer. FASEB BioAdv. 2021, 1–19. [Google Scholar] [CrossRef]
- Jain, P.; Karthikeyan, C.; Moorthy, N.S.H.; Waiker, D.; Jain, A.K.; Trivedi, P. Human CDC2-like kinase 1 (CLK1): A novel target for Alzheimer’s disease. Curr. Drug Targets 2014, 15, 539–550. [Google Scholar] [CrossRef]
- Lindberg, M.F.; Meijer, L. Dual-Specificity, Tyrosine Phosphorylation-Regulated Kinases (DYRKs) and cdc2-Like Kinases (CLKs) in Human Disease, an Overview. Int. J. Mol. Sci. 2021, 22, 6047. [Google Scholar] [CrossRef] [PubMed]
- Bullock, A.N.; Das, S.; Debreczeni, J.É.; Rellos, P.; Fedorov, O.; Niesen, F.H.; Guo, K.; Papagrigoriou, E.; Amos, A.L.; Cho, S.; et al. Kinase Domain Insertions Define Distinct Roles of CLK Kinases in SR Protein Phosphorylation. Structure 2009, 17, 352–362. [Google Scholar] [CrossRef]
- Zhou, Z.; Fu, X.-D. Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma 2013, 122, 191–207. [Google Scholar] [CrossRef] [PubMed]
- Martín Moyano, P.; Němec, V.; Paruch, K. Cdc-Like Kinases (CLKs): Biology, Chemical Probes, and Therapeutic Potential. Int. J. Mol. Sci. 2020, 21, 7549. [Google Scholar] [CrossRef]
- Kulkarni, P.; Uversky, V.N. Cancer/Testis Antigens: “Smart” Biomarkers for Diagnosis and Prognosis of Prostate and Other Cancers. Int. J. Mol. Sci. 2017, 18, 740. [Google Scholar] [CrossRef]
- Kulkarni, P.; Jolly, M.K.; Jia, D.; Mooney, S.M.; Bhargava, A.; Kagohara, L.T.; Chen, Y.; Hao, P.; He, Y.; Veltri, R.W.; et al. Phosphorylation-induced conformational dynamics in an intrinsically disordered protein and potential role in phenotypic heterogeneity. Proc. Natl. Acad. Sci. USA 2017, 114, E2644–E2653. [Google Scholar] [CrossRef]
- Lin, X.; Roy, S.; Jolly, M.K.; Bocci, F.; Schafer, N.P.; Tsai, M.-Y.; Chen, Y.; He, Y.; Grishaev, A.; Weninger, K.; et al. PAGE4 and Conformational Switching: Insights from Molecular Dynamics Simulations and Implications for Prostate Cancer. J. Mol. Biol. 2018, 430, 2422–2438. [Google Scholar] [CrossRef]
- Giannakouros, T.; Nikolakaki, E.; Mylonis, I.; Georgatsou, E. Serine-arginine protein kinases: A small protein kinase family with a large cellular presence. FEBS J. 2011, 278, 570–586. [Google Scholar] [CrossRef]
- Nikas, I.P.; Themistocleous, S.C.; Paschou, S.A.; Tsamis, K.I.; Ryu, H.S. Serine-Arginine Protein Kinase 1 (SRPK1) as a Prognostic Factor and Potential Therapeutic Target in Cancer: Current Evidence and Future Perspectives. Cells 2020, 9, 19. [Google Scholar] [CrossRef]
- Patel, M.; Sachidanandan, M.; Adnan, M. Serine arginine protein kinase 1 (SRPK1): A moonlighting protein with theranostic ability in cancer prevention. Mol. Biol. Rep. 2019, 46, 1487–1497. [Google Scholar] [CrossRef]
- Ngo, J.C.K.; Gullingsrud, J.; Giang, K.; Yeh, M.J.; Fu, X.-D.; Adams, J.A.; McCammon, J.A.; Ghosh, G. SR Protein Kinase 1 Is Resilient to Inactivation. Structure 2007, 15, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Mavrou, A.; Brakspear, K.; Hamdollah-Zadeh, M.; Damodaran, G.; Babaei-Jadidi, R.; Oxley, J.; Gillatt, D.A.; Ladomery, M.R.; Harper, S.J.; Bates, D.O.; et al. Serine–arginine protein kinase 1 (SRPK1) inhibition as a potential novel targeted therapeutic strategy in prostate cancer. Oncogene 2015, 34, 4311–4319. [Google Scholar] [CrossRef] [PubMed]
- Bullock, N.; Potts, J.; Simpkin, A.J.; Koupparis, A.; Harper, S.J.; Oxley, J.; Oltean, S. Serine-arginine protein kinase 1 (SRPK1), a determinant of angiogenesis, is upregulated in prostate cancer and correlates with disease stage and invasion. J. Clin. Pathol. 2016, 69, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Melegh, Z.; Oltean, S. Targeting Angiogenesis in Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 2676. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Guo, Y.; Zhang, X.; Liu, H.; Yin, M.; Chen, X.; Peng, C. Pyruvate kinase M2 (PKM2) in cancer and cancer therapeutics. Cancer Lett. 2021, 503, 240–248. [Google Scholar] [CrossRef]
- Guo, W.; Zhang, Z.; Li, G.; Lai, X.; Gu, R.; Xu, W.; Chen, H.; Xing, Z.; Chen, L.; Qian, J. Pyruvate kinase M2 promotes prostate cancer metastasis through regulating ERK1/2-COX-2 signaling. Front. Oncol. 2020, 10, 2022. [Google Scholar] [CrossRef]
- Giannoni, E.; Taddei, M.L.; Morandi, A.; Comito, G.; Calvani, M.; Bianchini, F.; Richichi, B.; Raugei, G.; Wong, N.; Tang, D.; et al. Targeting stromal-induced pyruvate kinase M2 nuclear translocation impairs oxphos and prostate cancer metastatic spread. Oncotarget 2015, 6, 24061–24074. [Google Scholar] [CrossRef]
- Dai, J.; Escara-Wilke, J.; Keller, J.M.; Jung, Y.; Taichman, R.S.; Pienta, K.J.; Keller, E.T. Primary prostate cancer educates bone stroma through exosomal pyruvate kinase M2 to promote bone metastasis. J. Exp. Med. 2019, 216, 2883–2899. [Google Scholar] [CrossRef]
- Zhu, F.; Zykova, T.A.; Kang, B.S.; Wang, Z.; Ebeling, M.C.; Abe, Y.; Ma, W.Y.; Bode, A.M.; Dong, Z. Bidirectional Signals Transduced by TOPK-ERK Interaction Increase Tumorigenesis of HCT116 Colorectal Cancer Cells. Gastroenterology 2007, 133, 219–231. [Google Scholar] [CrossRef]
- Han, Z.; Li, L.; Huang, Y.; Zhao, H.; Luo, Y. PBK/TOPK: A Therapeutic Target Worthy of Attention. Cells 2021, 10, 371. [Google Scholar] [CrossRef]
- Herbert, K.J.; Ashton, T.M.; Prevo, R.; Pirovano, G.; Higgins, G.S. T-LAK cell-originated protein kinase (TOPK): An emerging target for cancer-specific therapeutics. Cell Death Dis. 2018, 9, 1089. [Google Scholar] [CrossRef] [PubMed]
- Alhawas, L.; Amin, K.S.; Salla, B.; Banerjee, P.P. T-LAK cell-originated protein kinase (TOPK) enhances androgen receptor splice variant (ARv7) and drives androgen-independent growth in prostate cancer. Carcinogenesis 2021, 42, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.M.; Graham, N.A.; Stoyanova, T.; Sedghi, A.; Goldstein, A.S.; Cai, H.; Smith, D.A.; Zhang, H.; Komisopoulou, E.; Huang, J.; et al. Oncogene-specific activation of tyrosine kinase networks during prostate cancer progression. Proc. Natl. Acad. Sci. USA 2012, 109, 1643–1648. [Google Scholar] [CrossRef]
- Chauhan, A.; Khan, T. Focal adhesion kinase—An emerging viable target in cancer and development of focal adhesion kinase inhibitors. Chem. Biol. Drug Des. 2021, 97, 774–794. [Google Scholar] [CrossRef]
- Marcellus, K.A.; Crawford Parks, T.E.; Almasi, S.; Jasmin, B.J. Distinct roles for the RNA-binding protein Staufen1 in prostate cancer. BMC Cancer 2021, 21, 120. [Google Scholar] [CrossRef] [PubMed]
- Slack, J.K.; Adams, R.B.; Rovin, J.D.; Bissonette, E.A.; Stoker, C.E.; Parsons, J.T. Alterations in the focal adhesion kinase/Src signal transduction pathway correlate with increased migratory capacity of prostate carcinoma cells. Oncogene 2001, 20, 1152–1163. [Google Scholar] [CrossRef]
- Ok Atılgan, A.; Özdemir, B.H.; Yılmaz Akçay, E.; Tepeoğlu, M.; Börcek, P.; Dirim, A. Association between focal adhesion kinase and matrix metalloproteinase-9 expression in prostate adenocarcinoma and their influence on the progression of prostatic adenocarcinoma. Ann. Diagn. Pathol. 2020, 45, 151480. [Google Scholar] [CrossRef]
- Kimura, S.H.; Tsuruga, H.; Yabuta, N.; Endo, Y.; Nojima, H. Structure, expression, and chromosomal localization of human GAK. Genomics 1997, 44, 179–187. [Google Scholar] [CrossRef]
- Kanaoka, Y.; Kimura, S.H.; Okazaki, I.; Ikeda, M.; Nojima, H. GAK: A cyclin G associated kinase contains a tensin/auxilin-like domain. FEBS Lett. 1997, 402, 73–80. [Google Scholar] [CrossRef]
- Sato, J.; Shimizu, H.; Kasama, T.; Yabuta, N.; Nojima, H. GAK, a regulator of clathrin-mediated membrane trafficking, localizes not only in the cytoplasm but also in the nucleus. Genes Cells 2009, 14, 627–641. [Google Scholar] [CrossRef]
- Zhang, C.X.; Engqvist-Goldstein, A.E.; Carreno, S.; Owen, D.J.; Smythe, E.; Drubin, D.G. Multiple roles for cyclin G-associated kinase in clathrin-mediated sorting events. Traffic 2005, 6, 1103–1113. [Google Scholar] [CrossRef]
- Eisenberg, E.; Greene, L.E. Multiple roles of auxilin and hsc70 in clathrin-mediated endocytosis. Traffic 2007, 8, 640–646. [Google Scholar] [CrossRef]
- Sakurai, M.A.; Ozaki, Y.; Okuzaki, D.; Naito, Y.; Sasakura, T.; Okamoto, A.; Tabara, H.; Inoue, T.; Hagiyama, M.; Ito, A.; et al. Gefitinib and luteolin cause growth arrest of human prostate cancer PC-3 cells via inhibition of cyclin G-associated kinase and induction of miR-630. PLoS ONE 2014, 9, e100124. [Google Scholar] [CrossRef]
- Ray, M.R.; Wafa, L.A.; Cheng, H.; Snoek, R.; Fazli, L.; Gleave, M.; Rennie, P.S. Cyclin G-associated kinase: A novel androgen receptor-interacting transcriptional coactivator that is overexpressed in hormone refractory prostate cancer. Int. J. Cancer 2006, 118, 1108–1119. [Google Scholar] [CrossRef]
- Asquith, C.R.M.; Bennett, J.M.; Su, L.; Laitinen, T.; Elkins, J.M.; Pickett, J.E.; Wells, C.I.; Li, Z.; Willson, T.M.; Zuercher, W.J. Towards the Development of an In vivo Chemical Probe for Cyclin G Associated Kinase (GAK). Molecules 2019, 24, 4016. [Google Scholar] [CrossRef]
- Asquith, C.R.M.; Berger, B.T.; Wan, J.; Bennett, J.M.; Capuzzi, S.J.; Crona, D.J.; Drewry, D.H.; East, M.P.; Elkins, J.M.; Fedorov, O.; et al. SGC-GAK-1: A Chemical Probe for Cyclin G Associated Kinase (GAK). J. Med. Chem 2019, 62, 2830–2836. [Google Scholar] [CrossRef]
- Macedo-Silva, C.; Benedetti, R.; Ciardiello, F.; Cappabianca, S.; Jerónimo, C.; Altucci, L. Epigenetic mechanisms underlying prostate cancer radioresistance. Clin. Epigenetics 2021, 13, 125. [Google Scholar] [CrossRef]
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A Landscape Takes Shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef]
- Upadhyay, N.; Tilekar, K.; Hess, J.D.; Pokrovsky, V.S.; Aguilera, R.J.; Ramaa, C.S. Benefits and pitfalls: Epigenetic modulators in prostate cancer intervention. Curr. Res. Chem. Biol. 2021, 1, 100006. [Google Scholar] [CrossRef]
- Wang, R.; Liu, X. Epigenetic regulation of prostate cancer. Genes Dis. 2020, 7, 606–613. [Google Scholar] [CrossRef]
- Cheng, L.; MacLennan, G.T.; Lopez-Beltran, A.; Montironi, R. Anatomic, morphologic and genetic heterogeneity of prostate cancer: Implications for clinical practice. Expert Rev. Anticancer Ther. 2012, 12, 1371–1374. [Google Scholar] [CrossRef][Green Version]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.-M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef]
- Spangle, J.M.; Roberts, T.M. Epigenetic regulation of RTK signaling. J. Mol. Med. 2017, 95, 791–798. [Google Scholar] [CrossRef]
- Lim, P.S.; Sutton, C.R.; Rao, S. Protein kinase C in the immune system: From signalling to chromatin regulation. Immunology 2015, 146, 508–522. [Google Scholar] [CrossRef]
- Jurkowska, R.Z.; Jurkowski, T.P.; Jeltsch, A. Structure and function of mammalian DNA methyltransferases. Chembiochem 2011, 12, 206–222. [Google Scholar] [CrossRef]
- Arzate-Mejía, R.G.; Valle-García, D.; Recillas-Targa, F. Signaling epigenetics: Novel insights on cell signaling and epigenetic regulation. IUBMB Life 2011, 63, 881–895. [Google Scholar] [CrossRef]
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef]
- Biswas, S.; Rao, C.M. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur. J. Pharmacol. 2018, 837, 8–24. [Google Scholar] [CrossRef]
- Pakneshan, P.; Xing, R.H.; Rabbani, S.A. Methylation status of uPA promoter as a molecular mechanism regulating prostate cancer invasion and growth in vitro and in vivo. FASEB J. 2003, 17, 1081–1088. [Google Scholar] [CrossRef]
- Chin, S.P.; Dickinson, J.L.; Holloway, A.F. Epigenetic regulation of prostate cancer. Clin. Epigenet. 2011, 2, 151–169. [Google Scholar] [CrossRef]
- Panigrahi, A.R.; Pinder, S.E.; Chan, S.Y.; Paish, E.C.; Robertson, J.F.; Ellis, I.O. The role of PTEN and its signalling pathways, including AKT, in breast cancer; an assessment of relationships with other prognostic factors and with outcome. J. Pathol. 2004, 204, 93–100. [Google Scholar] [CrossRef]
- Wozniak, D.J.; Kajdacsy-Balla, A.; Macias, V.; Ball-Kell, S.; Zenner, M.L.; Bie, W.; Tyner, A.L. PTEN is a protein phosphatase that targets active PTK6 and inhibits PTK6 oncogenic signaling in prostate cancer. Nat. Commun. 2017, 8, 1508. [Google Scholar] [CrossRef]
- Kang, Y.-H.; Lee, H.S.; Kim, W.H. Promoter Methylation and Silencing of PTEN in Gastric Carcinoma. Lab. Investig. 2002, 82, 285–291. [Google Scholar] [CrossRef]
- Yegnasubramanian, S.; Haffner, M.C.; Zhang, Y.; Gurel, B.; Cornish, T.C.; Wu, Z.; Irizarry, R.A.; Morgan, J.; Hicks, J.; DeWeese, T.L.; et al. DNA hypomethylation arises later in prostate cancer progression than CpG island hypermethylation and contributes to metastatic tumor heterogeneity. Cancer Res. 2008, 68, 8954–8967. [Google Scholar] [CrossRef]
- Baxter, E.; Windloch, K.; Gannon, F.; Lee, J.S. Epigenetic regulation in cancer progression. Cell BioSci. 2014, 4, 1–11. [Google Scholar] [CrossRef]
- Valk-Lingbeek, M.E.; Bruggeman, S.W.; van Lohuizen, M. Stem cells and cancer: The polycomb connection. Cell 2004, 118, 409–418. [Google Scholar] [CrossRef]
- Metzger, E.; Yin, N.; Wissmann, M.; Kunowska, N.; Fischer, K.; Friedrichs, N.; Patnaik, D.; Higgins, J.M.; Potier, N.; Scheidtmann, K.-H. Phosphorylation of histone H3 at threonine 11 establishes a novel chromatin mark for transcriptional regulation. Nat. Cell Biol. 2008, 10, 53–60. [Google Scholar] [CrossRef]
- Kim, J.Y.; Banerjee, T.; Vinckevicius, A.; Luo, Q.; Parker, J.B.; Baker, M.R.; Radhakrishnan, I.; Wei, J.J.; Barish, G.D.; Chakravarti, D. A role for WDR5 in integrating threonine 11 phosphorylation to lysine 4 methylation on histone H3 during androgen signaling and in prostate cancer. Mol. Cell 2014, 54, 613–625. [Google Scholar] [CrossRef]
- Mahajan, K.; Malla, P.; Lawrence, H.R.; Chen, Z.; Kumar-Sinha, C.; Malik, R.; Shukla, S.; Kim, J.; Coppola, D.; Lawrence, N.J.; et al. ACK1/TNK2 Regulates Histone H4 Tyr88-phosphorylation and AR Gene Expression in Castration-Resistant Prostate Cancer. Cancer Cell 2017, 31, 790–803.e798. [Google Scholar] [CrossRef]
- Kim, E.H.; Cao, D.; Mahajan, N.P.; Andriole, G.L.; Mahajan, K. ACK1–AR and AR–HOXB13 signaling axes: Epigenetic regulation of lethal prostate cancers. NAR Cancer 2020, 2, zcaa018. [Google Scholar] [CrossRef]
- Xu, B.; Tao, T.; Wang, Y.; Fang, F.; Huang, Y.; Chen, S.; Zhu, W.; Chen, M. hsa-miR-135a-1 inhibits prostate cancer cell growth and migration by targeting EGFR. Tumor Biol. 2016, 37, 14141–14151. [Google Scholar] [CrossRef]
- Zangoue, M.; Zangouei, A.S.; Mojarrad, M.; Moghbeli, M. MicroRNAs as the critical regulators of protein kinases in prostate and bladder cancers. Egypt. J. Med. Hum. Genet. 2021, 22, 72. [Google Scholar] [CrossRef]
- Hagman, Z.; Haflidadottir, B.S.; Ansari, M.; Persson, M.; Bjartell, A.; Edsjö, A.; Ceder, Y. The tumour suppressor miR-34c targets MET in prostate cancer cells. Br. J. Cancer 2013, 109, 1271–1278. [Google Scholar] [CrossRef]
- Nam, R.K.; Benatar, T.; Wallis, C.J.D.; Kobylecky, E.; Amemiya, Y.; Sherman, C.; Seth, A. MicroRNA-139 is a predictor of prostate cancer recurrence and inhibits growth and migration of prostate cancer cells through cell cycle arrest and targeting IGF1R and AXL. Prostate 2019, 79, 1422–1438. [Google Scholar] [CrossRef]
- Zhang, G.M.; Bao, C.Y.; Wan, F.N.; Cao, D.L.; Qin, X.J.; Zhang, H.L.; Zhu, Y.; Dai, B.; Shi, G.H.; Ye, D.W. MicroRNA-302a Suppresses Tumor Cell Proliferation by Inhibiting AKT in Prostate Cancer. PLoS ONE 2015, 10, e0124410. [Google Scholar] [CrossRef]
- Hernando Polo, S.; Moreno Muñoz, D.; Rosero Rodríguez, A.C.; Silva Ruiz, J.; Rosero Rodríguez, D.I.; Couñago, F. Changing the History of Prostate Cancer with New Targeted Therapies. Biomedicines 2021, 9, 392. [Google Scholar] [CrossRef]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.-M.; Nguyen, H.D.; Ho, C.K.; Kwan, T.T.; Morris, R.; Lauffer, S.; Nussenzweig, A. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef]
- Kim, H.; Xu, H.; George, E.; Hallberg, D.; Kumar, S.; Jagannathan, V.; Medvedev, S.; Kinose, Y.; Devins, K.; Verma, P. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Teyssonneau, D.; Margot, H.; Cabart, M.; Anonnay, M.; Sargos, P.; Vuong, N.-S.; Soubeyran, I.; Sevenet, N.; Roubaud, G. Prostate cancer and PARP inhibitors: Progress and challenges. J. Hematol. Oncol. 2021, 14, 51. [Google Scholar] [CrossRef]
- Neeb, A.; Herranz, N.; Arce-Gallego, S.; Miranda, S.; Buroni, L.; Yuan, W.; Athie, A.; Casals, T.; Carmichael, J.; Rodrigues, D.N.; et al. Advanced Prostate Cancer with ATM Loss: PARP and ATR Inhibitors. Eur. Urol. 2021, 79, 200–211. [Google Scholar] [CrossRef]
- Twardowski, P.W.; Beumer, J.H.; Chen, C.; Kraft, A.S.; Chatta, G.S.; Mitsuhashi, M.; Ye, W.; Christner, S.M.; Lilly, M.B. A phase II trial of dasatinib in patients with metastatic castration-resistant prostate cancer treated previously with chemotherapy. Anti-Cancer Drugs 2013, 24, 743. [Google Scholar] [CrossRef]
- Araujo, J.C.; Trudel, G.C.; Saad, F.; Armstrong, A.J.; Evan, Y.Y.; Bellmunt, J.; Wilding, G.; McCaffrey, J.; Serrano, S.V.; Matveev, V.B. Docetaxel and dasatinib or placebo in men with metastatic castration-resistant prostate cancer (READY): A randomised, double-blind phase 3 trial. Lancet Oncol. 2013, 14, 1307–1316. [Google Scholar] [CrossRef]
- Nickols, N.G.; Nazarian, R.; Zhao, S.G.; Tan, V.; Uzunangelov, V.; Xia, Z.; Baertsch, R.; Neeman, E.; Gao, A.C.; Thomas, G.V. MEK-ERK signaling is a therapeutic target in metastatic castration resistant prostate cancer. Prostate Cancer Prostatic Dis. 2019, 22, 531–538. [Google Scholar] [CrossRef]
- Soria, J.; Massard, C.; Magne, N.; Bader, T.; Mansfield, C.; Blay, J.; Bui, B.; Moussy, A.; Hermine, O.; Armand, J. Phase 1 dose-escalation study of oral tyrosine kinase inhibitor masitinib in advanced and/or metastatic solid cancers. Eur. J. Cancer 2009, 45, 2333–2341. [Google Scholar] [CrossRef]
- Michaelson, M.D.; Regan, M.; Oh, W.; Kaufman, D.; Olivier, K.; Michaelson, S.; Spicer, B.; Gurski, C.; Kantoff, P.; Smith, M. Phase II study of sunitinib in men with advanced prostate cancer. Ann. Oncol. 2009, 20, 913–920. [Google Scholar] [CrossRef]
- Michaelson, M.D.; Oudard, S.; Ou, Y.-C.; Sengeløv, L.; Saad, F.; Houede, N.; Ostler, P.; Stenzl, A.; Daugaard, G.; Jones, R. Randomized, placebo-controlled, phase III trial of sunitinib plus prednisone versus prednisone alone in progressive, metastatic, castration-resistant prostate cancer. J. Clin. Oncol. 2014, 32, 76–82. [Google Scholar] [CrossRef]
- Kelly, W.K.; Halabi, S.; Carducci, M.; George, D.; Mahoney, J.F.; Stadler, W.M.; Morris, M.; Kantoff, P.; Monk, J.P.; Kaplan, E. Randomized, double-blind, placebo-controlled phase III trial comparing docetaxel and prednisone with or without bevacizumab in men with metastatic castration-resistant prostate cancer: CALGB 90401. J. Clin. Oncol. 2012, 30, 1534. [Google Scholar] [CrossRef]
- Dahut, W.L.; Madan, R.A.; Karakunnel, J.J.; Adelberg, D.; Gulley, J.L.; Turkbey, I.B.; Chau, C.H.; Spencer, S.D.; Mulquin, M.; Wright, J. Phase II clinical trial of cediranib in patients with metastatic castration-resistant prostate cancer. Br. J. Urol. 2013, 111, 1269. [Google Scholar] [CrossRef]
- Basch, E.M.; Scholz, M.; de Bono, J.S.; Vogelzang, N.; de Souza, P.; Marx, G.; Vaishampayan, U.; George, S.; Schwarz, J.K.; Antonarakis, E.S.; et al. Cabozantinib Versus Mitoxantrone-prednisone in Symptomatic Metastatic Castration-resistant Prostate Cancer: A Randomized Phase 3 Trial with a Primary Pain Endpoint. Eur. Urol. 2019, 75, 929–937. [Google Scholar] [CrossRef]
- Gravis, G.; Bladou, F.; Salem, N.; Gonçalves, A.; Esterni, B.; Walz, J.; Bagattini, S.; Marcy, M.; Brunelle, S.; Viens, P. Results from a monocentric phase II trial of erlotinib in patients with metastatic prostate cancer. Ann. Oncol. 2008, 19, 1624–1628. [Google Scholar] [CrossRef]
- Saura, C.; Roda, D.; Roselló, S.; Oliveira, M.; Macarulla, T.; Pérez-Fidalgo, J.A.; Morales-Barrera, R.; Sanchis-García, J.M.; Musib, L.; Budha, N.; et al. A First-in-Human Phase I Study of the ATP-Competitive AKT Inhibitor Ipatasertib Demonstrates Robust and Safe Targeting of AKT in Patients with Solid Tumors. Cancer Discov. 2017, 7, 102–113. [Google Scholar] [CrossRef]
- De Bono, J.S.; De Giorgi, U.; Rodrigues, D.N.; Massard, C.; Bracarda, S.; Font, A.; Arranz Arija, J.A.; Shih, K.C.; Radavoi, G.D.; Xu, N.; et al. Randomized Phase II Study Evaluating Akt Blockade with Ipatasertib, in Combination with Abiraterone, in Patients with Metastatic Prostate Cancer with and without PTEN Loss. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 928–936. [Google Scholar] [CrossRef]
- De Bono, J.; Bracarda, S.; Sternberg, C.; Chi, K.; Olmos, D.; Sandhu, S.; Massard, C.; Matsubara, N.; Alekseev, B.; Gafanov, R. LBA4 IPATential150: Phase III study of ipatasertib (ipat) plus abiraterone (abi) vs placebo (pbo) plus abi in metastatic castration-resistant prostate cancer (mCRPC). Ann. Oncol. 2020, 31, S1153–S1154. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Target | Result | Type of Study | Reference |
|---|---|---|---|---|
| Dasatinib | SRC Tyr kinase family |
| Phase II trial | [226] |
| Phase III trial | [227] | ||
| Trametinib | MAPK |
| Phase II trial | [228] |
| Masitinib | FAK |
| Phase I trial | [229] |
| Sunitinib | RTK |
| Phase II trial | [230] |
| Phase II trial | [231] | ||
| Bevacizumab | VEGFR Tyr kinase |
| Phase III trial | [232] |
| Cediranib | RTK |
| Phase II trial | [233] |
| Cabozantinib | RTK |
| Phase III trial | [234] |
| Erlotinib | VEGFR Tyr kinase |
| Phase II trial | [235] |
| Ipatasertib | Akt |
| Phase I trial | [236] |
| Phase II trial | [237] | ||
| Phase III trial | [238] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bagheri, S.; Rahban, M.; Bostanian, F.; Esmaeilzadeh, F.; Bagherabadi, A.; Zolghadri, S.; Stanek, A. Targeting Protein Kinases and Epigenetic Control as Combinatorial Therapy Options for Advanced Prostate Cancer Treatment. Pharmaceutics 2022, 14, 515. https://doi.org/10.3390/pharmaceutics14030515
Bagheri S, Rahban M, Bostanian F, Esmaeilzadeh F, Bagherabadi A, Zolghadri S, Stanek A. Targeting Protein Kinases and Epigenetic Control as Combinatorial Therapy Options for Advanced Prostate Cancer Treatment. Pharmaceutics. 2022; 14(3):515. https://doi.org/10.3390/pharmaceutics14030515
Chicago/Turabian StyleBagheri, Soghra, Mahdie Rahban, Fatemeh Bostanian, Fatemeh Esmaeilzadeh, Arash Bagherabadi, Samaneh Zolghadri, and Agata Stanek. 2022. "Targeting Protein Kinases and Epigenetic Control as Combinatorial Therapy Options for Advanced Prostate Cancer Treatment" Pharmaceutics 14, no. 3: 515. https://doi.org/10.3390/pharmaceutics14030515
APA StyleBagheri, S., Rahban, M., Bostanian, F., Esmaeilzadeh, F., Bagherabadi, A., Zolghadri, S., & Stanek, A. (2022). Targeting Protein Kinases and Epigenetic Control as Combinatorial Therapy Options for Advanced Prostate Cancer Treatment. Pharmaceutics, 14(3), 515. https://doi.org/10.3390/pharmaceutics14030515