
Inhibiting CK2 among Promising Therapeutic Strategies for Gliomas and Several Other Neoplasms


Abstract
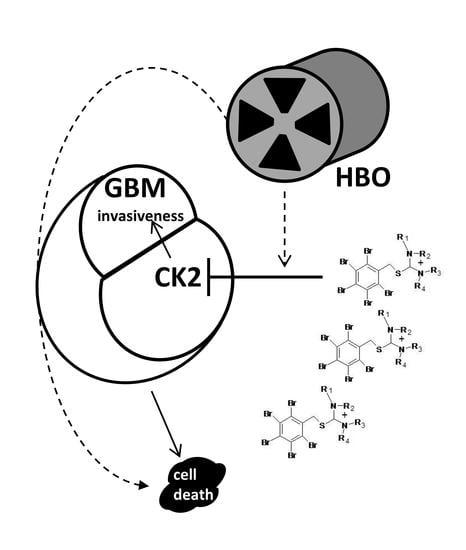
1. Introduction
2. Neoplasms and the Significant Role of CK2 in Tumour Biology
2.1. Glioblastoma
2.2. Medulloblastoma
2.3. Acute Lymphoblastic Leukaemia
2.4. Chronic Lymphocytic Leukaemia
2.5. Acute Myeloid Leukaemia
2.6. Acute Promyelocytic Leukaemia
2.7. Adrenocortical Cancer
2.8. Colorectal Cancer
2.9. Breast Cancer
2.10. Cholangiocarcinoma
2.11. Human Cervical Cancer
3. CK2 Structure and Function
4. DRB/DRB Derivatives CX-4945 and ZKK
4.1. DRB
4.2. TBI
4.3. DMAT
4.4. TBB
4.5. TDB
4.6. CX-4945
4.7. ZKKs
5. Other Inhibitors of Kinases
5.1. EGFR Inhibitors
5.2. PI3K/Akt/mTOR Inhibitors
5.3. Therapies Combined with CK2 Inhibitors
6. Effects of Silencing CK2 on Glioma Development
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
List of Abbreviations
| ABCB1 | P-glycoprotein, also known as ABCB1 |
| AKT | protein kinase B |
| AP23573 | ridaforolimus |
| ATP | adenosine 5′-triphosphate |
| AZD8055 | mTOR inhibitor |
| Bax | Bcl-2-associated protein X |
| BBB | blood–brain -barrier |
| BCL-XL | B-cell lymphoma-extra large |
| Bcl-2 | B-cell CLL/lymphoma 2 |
| BEN | S-benzylisothiourea hydrochloride |
| BRAFV600 | mutation of the B-Raf gene in which valine is substituted by glutamic acid at amino acid 600 |
| BKM120 | buparlisib |
| CCL-779 | temsirolimus |
| CDC34 | cell division cycle 34 |
| CDKN2A/B | cyclin-dependent kinase inhibitor 2A/B |
| CEM | human T-lymphoblastoid cells |
| CK2 | casein kinase 2 |
| CK2α | casein kinase 2 alpha |
| CK2β | casein kinase 2 beta |
| CLK2 | CDC-like kinase 2 |
| c-Myc | myelocytomatosis viral oncogene homolog |
| c-Myb | V-myb avian myeloblastosis viral oncogene homolog |
| CNS | central nervous system |
| CSNK2A1 | gene encoding casein kinase 2 alpha 1 |
| CSNK2A2 | gene encoding casein kinase 2 alpha 2 |
| CSNK2B | gene encoding casein kinase 2 beta |
| CXCL12/SDF-1 | stromal derived factor-1 |
| CX-4945 | (silmitasertib) 5-((3-Chlorophenyl)amino)benzo[c][2,6]naphthyridine-8-carboxylic acid) |
| NVP-BEZ235 | dactolisib |
| DMAT | 2-dimethylamino-4,5,6,7-1H-tetrabromobenzimidazole |
| DNA | deoxyribonucleic acid |
| DRB | 5,6-dichloro-1-beta-D-ribofuranosylbenzimidazole |
| DYRK1a | dual-specificity tyrosine phosphorylated and-regulated kinase 1a |
| EGFRvIII | epidermal growth factor receptor variant III |
| EGFR | epidermal growth factor receptor |
| ERK | extracellular signal-regulated kinase |
| FDA | Food and Drug Administration |
| FasL | Fas ligand |
| GSC | glioblastoma stem cells |
| GTP | guanosine-5’-triphosphate |
| HBO | hyperbaric oxygen |
| HeLa | human cervical cancer cells |
| HIF-1α | hypoxia-inducible factor 1α |
| HIF-2α | hypoxia- inducible factor 2α |
| HIPK2 | homeodomain-interacting protein kinase 2 |
| HL-60 | cell line human promyelocytic leukaemia |
| H295R | human adrenocortical cancer cell line |
| IDH1/IDH2 | isocitrate dehydrogenase 1/2 |
| IGF-1R | insulin-like growth factor-1 receptor |
| IR | insulin receptor |
| IR-A | insulin receptor isoform A |
| IR-B | insulin receptor isoform B |
| JAK | Janus kinase |
| K-562 | cell line human chronic erythromyeloblastoid leukaemia |
| KG-1 | cell line human acute myelogenous leukaemia |
| LN229 | glioma cell line |
| MAPK | mitogen-activated protein kinases |
| MCF-7 | breast cancer cells |
| MDR1 | multidrug resistance 1 |
| mTOR | mammalian target of rapamycin |
| mTORC1 | mammalian target of rapamycin complex 1 |
| mTORC2 | mammalian target of rapamycin complex 2 |
| NANOG | NANOG homeobox |
| NEK2a | never in mitosis (NIMA)-related kinase 2a |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| OCT4 | octamer-binding transcription factor 4 |
| OLIG2 | oligodendrocyte transcription factor 2 |
| AZD9291 | osimertinib |
| MK-3475 | pembrolizumab, monoclonal antibodies |
| PI3K | phosphatidyl-inositol 3-kinase |
| PIM | proviral insertion site in Moloney murine leukaemia virus |
| PIM2 | kinase PIM2 |
| PIM3 | kinase PIM3 |
| PIP2 | phosptidyl-inositol-4,5-diphosphate |
| PIP3 | phosphatidyl-inositol-3,4,5-triphosphate |
| PKCα | protein kinase C alpha |
| PKD | protein kinase D |
| PKD1 | protein kinase D1 |
| PTEN | phosphatase and tensin homolog deleted on chromosome 10 |
| pTERT | telomerase reverse transcriptase promoter |
| p53 | tumour protein |
| p70S6K1 | ribosomal S6 kinase p70 |
| RAD001 | everolimus |
| RNA | ribonucleic acid |
| rRNA | ribosomal RNA |
| SDF-1 | chemokine and the CXCR4 receptor |
| SEGA | subependymal giant cell astrocytoma |
| SGC-CK2-1 | N-(5-(3-Cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-ylamino)-2-methylphenyl) propionamide |
| SHH | Sonic hedgehog |
| shRNAs | short hairpin RNAs |
| siRNA | small interfering RNA |
| STAT3 | signal transducer and activator of transcription 3 |
| S6Ks | S6 kinases |
Appendix A

{kind=link}
{kind=link}
{kind=link}
| Compound | Activity and Cytotoxicity on Different Cell Models | Possible Off-Target Interactions | Reference |
| 5,6-dichloro-1-β-D-ribofuranosyl-1H-benzimidazole (DRB) | IC50 (15 μM) -glioma cells | -inhibition of TNFα-mediated NF-κB activation and sensitizing of cells to TNFα-induced apoptosis | Cozza et al., 2013 Dixit et al., 2012 |
| 4,5,6,7-tetrabromo-1H-benzimidazole (TBI/TBBz/TBBi) | IC50 (0.50 μM) -C6 (rat glioma cells) -T98G (glioblastoma cells) -SEGA (subependymal giant cell astrocytoma) | -modulation and transduction of many signalling pathways, including mTOR kinase-related pathways -inhibition of PIM, PKD1, HIPK2, DYRK1a kinase | Duncan et al., 2008 Kaminska et al., 2009 Pucko et al., 2019 Pagano et al., 2008 |
| 4,5,6,7-tetrabromo-1H-benzimidazole-2-N, N-dimethylamine (DMAT) | IC50 (0.14 μM) -T98G (glioblastoma cells) -LN229 glioblastoma cells -MCF-7 (human breast cancer cells) -KG-1 (human acute leukaemia myeloid cells) -H295R (human adrenocortical cancer cell line) -colorectal cancer | -activation of caspases 3, 7, and 8; increased expression of FasL and Fas; weakened membrane potential and mitochondrial function -induction of cell apoptosis | Duncan et al., 2008 Pucko et al., 2019 Kaminska et al., 2009 Koronkiewicz et al., 2013 Lawnicka et al., 2010 Tapia et al., 2006 |
| 4,5,6,7-tetrabromo-1H-benzotriazole (TBB/TBBt) | IC50 (0.50 μM) -T98G (glioblastoma cells) -CLL (chronic lymphocytic leukaemia cells) | -induction of cell apopotosis -inhibition of PIM family kinases including PIM1 and PIM3 -reduction in PTEN and phosphorylation kinase Akt | Duncan et al., 2008 Kaminska et al., 2009; Pucko et al., 2019 Andrzejewska et al., 2003 Pagano et al., 2008 Shehata et al., 2010 |
| 1-β-D-2′-deoxyribofuranosyl-4,5,6,7-tetrabromo-1H-benzimidazole TDB/K164 | IC50 (32 nM) -CEM (human T-lymphoblastoid cells) -HeLa (human cervical cancer cells) | -inhibition of PIM1, CLK2, DYRK1A kinases -induced apoptosis | Girardi et al., 2015 G. Cozza at al., 2014 |
| 5- (3-chlorophenylamino) benzo [c] [2,6] naphthyridine-8-carboxylic acid CX-4945 | IC50 (0.3 nM) -human hematological malignancies -solid tumours -cholangiocarcinoma -medulloblastoma cell lines -acute lymphoblastic leukaemia glioblastoma | -may act synergistically with several anticancer drugs such as gemcitabine, cisplatin, and bortezomib against cholangiocarcinoma and acute lymphoblastic leukaemia -induction of apoptosis -synergistic action with gefitinib exerted a strong antiproliferative effect on glioblastoma | Zhou et al., 2017 D’Amore et al., 2020 Buontempo et al., 2016 Nitta et al., 2019 Zakharia et al., 2019 Rowse et al., 2017 |
| N-(5-(3-Cyano-7-(cyclopropylamino)pyrazolo[1,5-a]pyrimidin-5-ylamino)-2-methylphenyl)propionamide SGC-CK2-1 | IC50 (36 nM) U-87MG (glioblastoma cells) | showed no antiproliferative activity | Salvi et al., 2021 |
| Isothiourea derivative (pentabromobenzylisothioureas) ZKK | IC50 (7–50 μM) -LN229 (glioblastoma cells) -C6 (rat glioma cells) -T98G (glioblastoma cells) -HL-60 (human promyelocytic leukaemia) -K-562l (human chronic erythromyeloblastic leukaemia) | -induction of apoptosis -combination of HBO with the ZKK3 increased cytotoxicity and made T98G cells more sensitive to antitumour effect -ZKK3 inhibited PIM, IGF-1R, IR, PKD1 kinases | Kaminska et al., 2009 Pucko et al., 2018 Koronkiewicz et al., 2013 Zembrzuska et al., 2019 Koronkiewicz et al., 2013 |
References
- Avendaño, C.; Menéndez, J.C. Chapter 9-Drugs That Inhibit Signalling Pathways for Tumor Cell Growth and Proliferation. In Medicinal Chemistry of Anticancer Drugs; Avendaño, C., Menéndez, J.C., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; pp. 251–305. [Google Scholar]
- Cohen, P. The origins of protein phosphorylation. Nat. Cell Biol. 2002, 4, E127–E130. [Google Scholar] [CrossRef] [PubMed]
- Prudent, R.; Cochet, C. New Protein Kinase CK2 Inhibitors: Jumping out of the Catalytic Box. Chem. Biol. 2009, 16, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, P. The Story of Imatinib in GIST-A Journey through the Development of a Targeted Therapy. Oncol. Res. Treat. 2018, 41, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.; Lightner, D.D.; Barnholtz-Sloan, J.; Villano, J.L. Epidemiologic and Molecular Prognostic Review of Glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, F.J.; Orr, B.A.; Ligon, K.L.; Eberhart, C.G. Neoplastic cells are a rare component in human glioblastoma microvasculature. Oncotarget 2012, 3, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO Classification of Tumours of the Central Nervous System. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Malzkorn, B.; Reifenberger, G. Integrated diagnostics of diffuse astrocytic and oligodendroglial tumors. Der Pathol. 2019, 40, 9–17. [Google Scholar] [CrossRef]
- Herrlinger, U.; Tzaridis, T.; Mack, F.; Steinbach, J.P.; Schlegel, U.; Sabel, M.; Hau, P.; Kortmann, R.-D.; Krex, D.; Grauer, O.; et al. Lomustine-temozolomide combination therapy versus standard temozolomide therapy in patients with newly diagnosed glioblastoma with methylated MGMT promoter (CeTeG/NOA–09): A randomised, open-label, phase 3 trial. Lancet 2019, 393, 678–688. [Google Scholar] [CrossRef]
- Agarwal, M.; Nitta, R.T.; Li, G. Casein Kinase 2: A Novel Player in Glioblastoma Therapy and Cancer Stem Cells. J. Mol. Genet. Med. 2013, 8, 1000094. [Google Scholar]
- Oliver, L.; Lalier, L.; Salaud, C.; Heymann, D.; Cartron, P.F.; Vallette, F. Drug Resistance in Glioblastoma: Are Persisters the Key to Therapy? Cancer Drug Resist. 2020, 3, 287–301. [Google Scholar] [CrossRef]
- Pardridge, W.M. Csf, Blood-Brain Barrier, and Brain Drug Delivery. Expert Opin. Drug Deliv. 2016, 13, 963–975. [Google Scholar] [CrossRef] [PubMed]
- Purow, B.; Schiff, D. Advances in the Genetics of Glioblastoma: Are We Reaching Critical Mass? Nat. Rev. Neurol. 2009, 5, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Shergalis, A.; Bankhead, A.; Luesakul, U.; Muangsin, N.; Neamati, N. Current Challenges and Opportunities in Treating Glioblastoma. Pharmacol. Rev. 2018, 70, 412–445. [Google Scholar] [CrossRef]
- Sharifzad, F.; Ghavami, S.; Verdi, J.; Mardpour, S.; Sisakht, M.M.; Azizi, Z.; Taghikhani, A.; Łos, M.J.; Fakharian, E.; Ebrahimi, M.; et al. Glioblastoma cancer stem cell biology: Potential theranostic targets. Drug Resist. Updat. 2019, 42, 35–45. [Google Scholar] [CrossRef]
- Dixit, D.; Sharma, V.; Ghosh, S.; Mehta, V.S.; Sen, E. Inhibition of Casein kinase-2 induces p53-dependent cell cycle arrest and sensitizes glioblastoma cells to tumor necrosis factor (TNFα)-induced apoptosis through SIRT1 inhibition. Cell Death Dis. 2012, 3, e271. [Google Scholar] [CrossRef]
- Nitta, R.T.; Bolin, S.; Luo, E.; Solow-Codero, D.E.; Samghabadi, P.; Purzner, T.; Aujla, P.S.; Nwagbo, G.; Cho, Y.-J.; Li, G. Casein kinase 2 inhibition sensitizes medulloblastoma to temozolomide. Oncogene 2019, 38, 6867–6879. [Google Scholar] [CrossRef]
- Onciu, M. Acute Lymphoblastic Leukemia. Hematol. Oncol. Clin. N. Am. 2009, 23, 655–674. [Google Scholar] [CrossRef]
- Gowda, C.; Song, C.; Kapadia, M.; Payne, J.; Hu, T.; Ding, Y.; Dovat, S. Regulation of cellular proliferation in acute lymphoblastic leukemia by Casein Kinase II (CK2) and Ikaros. Adv. Biol. Regul. 2016, 63, 71–80. [Google Scholar] [CrossRef]
- Chon, H.J.; Bae, K.J.; Lee, Y.; Kim, J. The Casein Kinase 2 Inhibitor, Cx-4945, as an Anti-Cancer Drug in Treatment of Human Hematological Malignancies. Front. Pharmacol. 2015, 6, 70. [Google Scholar] [CrossRef]
- Chiorazzi, N.; Rai, K.R.; Ferrarini, M. Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2005, 352, 804–815. [Google Scholar] [CrossRef] [PubMed]
- Martins, L.R.; Lúcio, P.; Silva, M.C.; Gameiro, P.; Silva, M.G.; Barata, J.T. On CK2 regulation of chronic lymphocytic leukemia cell viability. Mol. Cell. Biochem. 2011, 356, 51–55. [Google Scholar] [CrossRef] [PubMed]
- De Kouchkovsky, I.; Abdul-Hay, M. Acute Myeloid Leukemia: A Comprehensive Review and 2016 Update. Blood Cancer J. 2016, 6, e441. [Google Scholar] [CrossRef]
- Rosales, M.; Pérez, G.V.; Ramón, A.C.; Cruz, Y.; Rodriguez-Ulloa, A.; Besada, V.; Ramos, Y.; Vazquez-Blomquist, D.; Caballero, E.; Aguilar, D.; et al. Targeting of Protein Kinase Ck2 in Acute Myeloid Leukemia Cells Using the Clinical-Grade Synthetic-Peptide Cigb-300. Biomedicines 2021, 9, 766. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, J.J.; Chale, R.S.; Abad, A.C.; Schally, A.V. Acute promyelocytic leukemia (APL): A review of the literature. Oncotarget 2020, 11, 992–1003. [Google Scholar] [CrossRef]
- Gurrieri, C.; Piazza, F.A.; Ruzzene, M.; Tubi, L.Q.; Tosoni, K.; Gnoato, M.; Cabrelle, A.; Bonanni, L.; Trentin, L.; Zambello, R.; et al. Role of Protein Kinase CK2 in the Retinoic Acid-Induced Differentiation of Acute Promyelocytic Leukemia Cells. Blood 2007, 110, 879. [Google Scholar] [CrossRef]
- Nicolson, N.G.; Korah, R.; Carling, T. Adrenocortical cancer cell line mutational profile reveals aggressive genetic background. J. Mol. Endocrinol. 2019, 62, 179–186. [Google Scholar] [CrossRef]
- Tanaka, T.; Ohe, H.; Nomiyama, T.; Yanase, T. OR02-2 CX-4945 as a Potential Drug for Adrenorcortical Carcinoma That induces Multiple Exon-Skipping and Circular RNA of NR5A1. J. Endocr. Soc. 2019, 3 (Suppl. 1), OR02-2. [Google Scholar] [CrossRef]
- Haraldsdottir, S.; Einarsdottir, H.M.; Smaradottir, A.; Gunnlaugsson, A.; Halfdanarson, T.R. Colorectal Cancer-Review. Laeknabladid 2014, 100, 75–82. [Google Scholar]
- Pistorius, K.; Seitz, G.; Remberger, K.; Issinger, O.G. Differential Ckii Activities in Human Colorectal Mucosa, Adenomas and Carcinomas. Oncol. Res. Treat 1991, 14, 256–260. [Google Scholar] [CrossRef]
- Siddiqui-Jain, A.; Drygin, D.; Streiner, N.; Chua, P.; Pierre, F.; O’Brien, S.E.; Bliesath, J.; Omori, M.; Huser, N.; Ho, C.; et al. CX-4945, an Orally Bioavailable Selective Inhibitor of Protein Kinase CK2, Inhibits Prosurvival and Angiogenic Signaling and Exhibits Antitumor Efficacy. Cancer Res. 2010, 70, 10288–10298. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.-S.; Zhao, Z.; Yang, Z.-N.; Xu, F.; Lu, H.-J.; Zhu, Z.-Y.; Shi, W.; Jiang, J.; Yao, P.-P.; Zhu, H.-P. Risk Factors and Preventions of Breast Cancer. Int. J. Biol. Sci. 2017, 13, 1387–1397. [Google Scholar] [CrossRef] [PubMed]
- Ruzzene, M.; Pinna, L.A. Addiction to Protein Kinase Ck2: A Common Denominator of Diverse Cancer Cells? Biochim. Biophys. Acta 2010, 1804, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Razumilava, N.; Gores, G.J. Cholangiocarcinoma. Lancet 2014, 383, 2168–2179. [Google Scholar] [CrossRef]
- Zhou, F.; Xu, J.; Ding, G.; Cao, L. Overexpressions of CK2β and XIAP are Associated with Poor Prognosis of Patients with Cholangiocarcinoma. Pathol. Oncol. Res. 2013, 20, 73–79. [Google Scholar] [CrossRef]
- Nahand, J.S.; Moghoofei, M.; Salmaninejad, A.; Bahmanpour, Z.; Karimzadeh, M.; Nasiri, M.; Mirzaei, H.R.; Pourhanifeh, M.H.; Bokharaei-Salim, F.; Mirzaei, H.; et al. Pathogenic role of exosomes and microRNAs in HPV-mediated inflammation and cervical cancer: A review. Int. J. Cancer 2019, 146, 305–320. [Google Scholar] [CrossRef]
- Chua, M.M.J.; Lee, M.; Dominguez, I. Cancer-type dependent expression of CK2 transcripts. PLoS ONE 2017, 12, e0188854. [Google Scholar] [CrossRef]
- APinna, L. A historical view of protein kinase CK2. Cell. Mol. Biol. Res. 1994, 40, 383–390. [Google Scholar]
- Litchfield, D.W. Protein kinase CK2: Structure, regulation and role in cellular decisions of life and death. Biochem. J. 2003, 369, 1–15. [Google Scholar] [CrossRef]
- Montenarh, M.; Götz, C. Protein kinase CK2 and ion channels (Review). Biomed. Rep. 2020, 13, 55. [Google Scholar] [CrossRef]
- Borgo, C.; D’Amore, C.; Sarno, S.; Salvi, M.; Ruzzene, M. Protein kinase CK2: A potential therapeutic target for diverse human diseases. Signal Transduct. Target. Ther. 2021, 6, 183. [Google Scholar] [CrossRef] [PubMed]
- Dubois, N.; Willems, M.; Nguyen-Khac, M.-T.; Kroonen, J.; Goffart, N.; Deprez, M.; Bours, V.; Robe, P.A. Constitutive activation of casein kinase 2 in glioblastomas: Absence of class restriction and broad therapeutic potential. Int. J. Oncol. 2016, 48, 2445–2452. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ferrer-Font, L.; Villamañan, L.; Arias-Ramos, N.; Vilardell, J.; Plana, M.; Ruzzene, M.; Pinna, L.A.; Itarte, E.; Arús, C.; Candiota, A.P. Targeting Protein Kinase CK2: Evaluating CX-4945 Potential for GL261 Glioblastoma Therapy in Immunocompetent Mice. Pharmaceuticals 2017, 10, 24. [Google Scholar] [CrossRef] [PubMed]
- Rowse, A.L.; Gibson, S.A.; Meares, G.P.; Rajbhandari, R.; Nozell, S.E.; Dees, K.J.; Hjelmeland, A.B.; McFarland, B.C.; Benveniste, E.N. Protein kinase CK2 is important for the function of glioblastoma brain tumor initiating cells. J. Neuro-Oncol. 2017, 132, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; McFarland, B.C.; Drygin, D.; Yu, H.; Bellis, S.L.; Kim, H.; Bredel, M.; Benveniste, E.N. Targeting Protein Kinase CK2 Suppresses Prosurvival Signaling Pathways and Growth of Glioblastoma. Clin. Cancer Res. 2013, 19, 6484–6494. [Google Scholar] [CrossRef] [PubMed]
- Purzner, T.; Purzner, J.; Buckstaff, T.; Cozza, G.; Gholamin, S.; Rusert, J.M.; Hartl, T.A.; Sanders, J.; Conley, N.; Ge, X.; et al. Developmental phosphoproteomics identifies the kinase CK2 as a driver of Hedgehog signaling and a therapeutic target in medulloblastoma. Sci. Signal. 2018, 11, eaau5147. [Google Scholar] [CrossRef]
- Kroonen, J.; Artesi, M.; Capraro, V.; Nguyen-Khac, M.-T.; Willems, M.; Chakravarti, A.; Bours, V.; Robe, P.A. Casein kinase 2 inhibition modulates the DNA damage response but fails to radiosensitize malignant glioma cells. Int. J. Oncol. 2012, 41, 776–782. [Google Scholar] [CrossRef]
- Duncan, J.S.; Litchfield, D.W. Too much of a good thing: The role of protein kinase CK2 in tumorigenesis and prospects for therapeutic inhibition of CK2. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2008, 1784, 33–47. [Google Scholar] [CrossRef]
- Duncan, J.S.; Gyenis, L.; Lenehan, J.; Bretner, M.; Graves, L.M.; Haystead, T.A.; Litchfield, D.W. An Unbiased Evaluation of Ck2 Inhibitors by Chemoproteomics: Characterization of Inhibitor Effects on Ck2 and Identification of Novel Inhibitor Targets. Mol. Cell Proteom. 2008, 7, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Iegre, J.; Atkinson, E.L.; Brear, P.D.; Cooper, B.M.; Hyvönen, M.; Spring, D.R. Chemical probes targeting the kinase CK2: A journey outside the catalytic box. Org. Biomol. Chem. 2021, 19, 4380–4396. [Google Scholar] [CrossRef]
- Faust, M.; Montenarh, M. Subcellular localization of protein kinase CK2. Cell Tissue Res. 2000, 301, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Tawfic, S.; Yu, S.; Wang, H.; Faust, R.; Davis, A.; Ahmed, K. Protein kinase CK2 signal in neoplasia. Histol. Histopathol. 2001, 16, 573–582. [Google Scholar] [PubMed]
- Cozza, G. The Development of CK2 Inhibitors: From Traditional Pharmacology to in Silico Rational Drug Design. Pharmaceuticals 2017, 10, 26. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.A.; Andrzejewska, M.; Ruzzene, M.; Sarno, S.; Cesaro, L.; Bain, J.; Elliott, M.; Meggio, F.; Kazimierczuk, Z.; Pinna, L.A. Optimization of Protein Kinase Ck2 Inhibitors Derived from 4,5,6,7-Tetrabromobenzimidazole. J. Med. Chem. 2004, 47, 6239–6247. [Google Scholar] [CrossRef]
- Meggio, F.; Pagano, M.A.; Moro, S.; Zagotto, G.; Ruzzene, M.; Sarno, S.; Cozza, G.; Bain, J.; Elliott, M.; Deana, A.D.; et al. Pinna. Inhibition of Protein Kinase Ck2 by Condensed Polyphenolic Derivatives. An in Vitro and in Vivo Study. Biochemistry 2004, 43, 12931–12936. [Google Scholar] [CrossRef]
- Sarno, S.; de Moliner, E.; Ruzzene, M.; Pagano, M.A.; Battistutta, R.; Bain, J.; Fabbro, D.; Schoepfer, J.; Elliott, M.; Furet, P.; et al. Biochemical and Three-Dimensional-Structural Study of the Specific Inhibition of Protein Kinase Ck2 by [5-Oxo-5,6-Dihydroindolo-(1,2-a)Quinazolin-7-Yl]Acetic Acid (Iqa). Biochem. J. 2003, 374, 639–646. [Google Scholar] [CrossRef]
- Kubiński, K.; Masłyk, M.; Orzeszko, A. Benzimidazole inhibitors of protein kinase CK2 potently inhibit the activity of atypical protein kinase Rio1. Mol. Cell. Biochem. 2016, 426, 195–203. [Google Scholar] [CrossRef]
- Schaefer, S.; Svenstrup, T.H.; Fischer, M.; Guerra, B. D11-Mediated Inhibition of Protein Kinase CK2 Impairs HIF-1α-Mediated Signaling in Human Glioblastoma Cells. Pharmaceuticals 2017, 10, 5. [Google Scholar] [CrossRef]
- Malric, L.; Monferran, S.; Gilhodes, J.; Boyrie, S.; Dahan, P.; Skuli, N.; Sesen, J.; Filleron, T.; Kowalski-Chauvel, A.; Moyal, E.C.-J.; et al. Interest of integrins targeting in glioblastoma according to tumor heterogeneity and cancer stem cell paradigm: An update. Oncotarget 2017, 8, 86947–86968. [Google Scholar] [CrossRef]
- Di Maira, G.; Salvi, M.; Arrigoni, G.; Marin, O.; Sarno, S.; Brustolon, F.; Pinna, L.A.; Ruzzene, M. Protein kinase CK2 phosphorylates and upregulates Akt/PKB. Cell Death Differ. 2005, 12, 668–677. [Google Scholar] [CrossRef]
- Ponce, D.P.; Yefi, R.; Cabello, P.; Maturana, J.L.; Niechi, I.; Silva, E.; Galindo, M.; Antonelli, M.; Marcelain, K.; Armisen, R.; et al. CK2 functionally interacts with AKT/PKB to promote the β-catenin-dependent expression of survivin and enhance cell survival. Mol. Cell. Biochem. 2011, 356, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Ge, Z.; Ding, Y.; Tan, B.-H.; Desai, D.; Gowda, K.; Amin, S.G.; Gowda, R.; Robertson, G.P.; Yue, F.; et al. IKAROS and CK2 regulate expression of BCL-XL and chemosensitivity in high-risk B-cell acute lymphoblastic leukemia. Blood 2020, 136, 1520–1534. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Awah, C.U.; Sonabend, A.M. Topoisomerase II Poisons for Glioblastoma; Existing Challenges and Opportunities to Personalize Therapy. Front. Neurol. 2018, 9, 459. [Google Scholar] [CrossRef] [PubMed]
- Nager, M.; Bhardwaj, D.; Cantí, C.; Medina, L.; Nogués, P.; Herreros, J. Β-Catenin Signalling in Glioblastoma Multiforme and Glioma-Initiating Cells. Chemother Res. Pract. 2012, 2012, 192362. [Google Scholar] [CrossRef]
- Nitta, R.T.; Gholamin, S.; Feroze, A.H.; Agarwal, M.; Cheshier, S.H.; Mitra, S.S.; Li, G. Casein kinase 2α regulates glioblastoma brain tumor-initiating cell growth through the β-catenin pathway. Oncogene 2014, 34, 3688–3699. [Google Scholar] [CrossRef] [PubMed]
- Alves, A.L.V.; Gomes, I.N.F.; Carloni, A.C.; Rosa, M.N.; da Silva, L.S.; Evangelista, A.F.; Reis, R.M.; Silva, V.A.O. Role of glioblastoma stem cells in cancer therapeutic resistance: A perspective on antineoplastic agents from natural sources and chemical derivatives. Stem Cell Res. Ther. 2021, 12, 206. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhang, M.; Yan, G.; Ma, Q.; Yan, Z.; Wang, L.; Yang, K.; Guo, D. Nanog Promotes Stem-Like Traits of Glioblastoma Cells. Front. Biosci. 2021, 26, 552–565. [Google Scholar]
- Zhou, J.; Tien, A.-C.; Alberta, J.A.; Ficarro, S.B.; Griveau, A.; Sun, Y.; Deshpande, J.S.; Card, J.D.; Morgan-Smith, M.; Michowski, W.; et al. A Sequentially Priming Phosphorylation Cascade Activates the Gliomagenic Transcription Factor Olig2. Cell Rep. 2017, 18, 3167–3177. [Google Scholar] [CrossRef]
- Zandomeni, R.; Zandomeni, M.C.; Shugar, D.; Weinmann, R. Casein kinase type II is involved in the inhibition by 5,6-dichloro-1-beta-D-ribofuranosylbenzimidazole of specific RNA polymerase II transcription. J. Biol. Chem. 1986, 261, 3414–3419. [Google Scholar] [CrossRef]
- Cozza, G.; Sarno, S.; Ruzzene, M.; Girardi, C.; Orzeszko, A.; Kazimierczuk, Z.; Zagotto, G.; Bonaiuto, E.; Di Paolo, M.L.; Pinna, L.A. Exploiting the repertoire of CK2 inhibitors to target DYRK and PIM kinases. Biochim. Et Biophys. Acta (BBA) Proteins Proteom. 2013, 1834, 1402–1409. [Google Scholar] [CrossRef]
- Pagano, M.A.; Bain, J.; Kazimierczuk, Z.; Sarno, S.; Ruzzene, M.; Di Maira, G.; Elliott, M.; Orzeszko, A.; Cozza, G.; Meggio, F.; et al. The selectivity of inhibitors of protein kinase CK2: An update. Biochem. J. 2008, 415, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Pucko, E.; Ostrowski, R.; Matyja, E. Novel small molecule protein kinase CK2 inhibitors exert potent antitumor effects on T98G and SEGA cells in vitro. Folia Neuropathol. 2019, 57, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Kaminska, B.; Ellert-Miklaszewska, A.; Oberbek, A.; Wisniewski, P.; Kaza, B.; Makowska, M.; Bretner, M.; Kazimierczuk, Z. Efficacy and mechanism of anti-tumor action of new potential CK2 inhibitors toward glioblastoma cells. Int. J. Oncol. 2009, 35, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.A.; Meggio, F.; Ruzzene, M.; Andrzejewska, M.; Kazimierczuk, Z.; Pinna, L.A. 2-Dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole: A novel powerful and selective inhibitor of protein kinase CK2. Biochem. Biophys. Res. Commun. 2004, 321, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Pertz, V.; Zhang, B.; Kaur, P.; Shimada, H.; Groffen, J.; Kazimierczuk, Z.; Pinna, L.A.; Heisterkamp, N. Treatment of P190 Bcr/Abl lymphoblastic leukemia cells with inhibitors of the serine/threonine kinase CK2. Leukemia 2006, 21, 178–180. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Reichert, A.; Cunnick, J.; Senadheera, D.; Hemmeryckx, B.; Heisterkamp, N.; Groffen, J. Protein Kinase Ckiialpha Interacts with the Bcr Moiety of Bcr/Abl and Mediates Proliferation of Bcr/Abl-Expressing Cells. Oncogene 2003, 22, 8255–8262. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tapia, J.C.; Torres, V.A.; Rodriguez, D.; Leyton, L.; Quest, A.F.G. Casein kinase 2 (CK2) increases survivin expression via enhanced beta-catenin-T cell factor/lymphoid enhancer binding factor-dependent transcription. Proc. Natl. Acad. Sci. USA 2006, 103, 15079–15084. [Google Scholar] [CrossRef]
- Koronkiewicz, M.; Chilmonczyk, Z.; Kazimierczuk, Z. Synergistic anti-leukemic effects of CK2 inhibitors and pentabromobenzylisothioureas in vitro. Anticancer Res. 2013, 33, 4891–4899. [Google Scholar]
- Lawnicka, H.; Kowalewicz-Kulbat, M.; Sicinska, P.; Kazimierczuk, Z.; Grieb, P.; Stepien, H. Anti-neoplastic effect of protein kinase CK2 inhibitor, 2-dimethylamino-4,5,6,7-tetrabromobenzimidazole (DMAT), on growth and hormonal activity of human adrenocortical carcinoma cell line (H295R) in vitro. Cell Tissue Res. 2010, 340, 371–379. [Google Scholar] [CrossRef]
- Szyszka, R.; Grankowski, N.; Felczak, K.; Shugar, D. Halogenated Benzimidazoles and Benzotriazoles as Selective Inhibitors of Protein Kinases CK-I and CK-II from Saccharomyces Cerevisiae and Other Sources. Biochem. Biophys. Res. Commun. 1995, 208, 418–424. [Google Scholar] [CrossRef]
- Andrzejewska, M.; Pagano, M.A.; Meggio, F.; Brunati, A.M.; Kazimierczuk, Z. Polyhalogenobenzimidazoles: Synthesis and Their inhibitory activity against casein kinases. Bioorg. Med. Chem. 2003, 11, 3997–4002. [Google Scholar] [CrossRef]
- Shehata, M.; Schnabl, S.; Demirtas, D.; Hilgarth, M.; Hubmann, R.; Ponath, E.; Badrnya, S.; Lehner, C.; Hoelbl, A.; Duechler, M.; et al. Reconstitution of PTEN activity by CK2 inhibitors and interference with the PI3-K/Akt cascade counteract the antiapoptotic effect of human stromal cells in chronic lymphocytic leukemia. Blood 2010, 116, 2513–2521. [Google Scholar] [CrossRef] [PubMed]
- Girardi, C.; Ottaviani, D.; Pinna, L.A.; Ruzzene, M. Different Persistence of the Cellular Effects Promoted by Protein Kinase CK2 Inhibitors CX-4945 and TDB. BioMed Res. Int. 2015, 2015, 185736. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Girardi, C.; Ranchio, A.; Lolli, G.; Sarno, S.; Orzeszko, A.; Kazimierczuk, Z.; Battistutta, R.; Ruzzene, M.; Pinna, L.A. Cell-permeable dual inhibitors of protein kinases CK2 and PIM-1: Structural features and pharmacological potential. Cell. Mol. Life Sci. 2014, 71, 3173–3185. [Google Scholar] [CrossRef]
- Borgo, C.; Ruzzene, M. Role of protein kinase CK2 in antitumor drug resistance. J. Exp. Clin. Cancer Res. 2019, 38, 287. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, N.; Tang, S.; Qi, X.; Zhao, L.; Zhong, R.; Peng, Y. Exploring the Pivotal Role of the CK2 Hinge Region Sub-Pocket in Binding with Tricyclic Quinolone Analogues by Computational Analysis. Molecules 2017, 22, 840. [Google Scholar] [CrossRef]
- Ku, M.J.; Park, J.W.; Ryu, B.J.; Son, Y.-J.; Kim, S.H.; Lee, S.Y. CK2 inhibitor CX4945 induces sequential inactivation of proteins in the signaling pathways related with cell migration and suppresses metastasis of A549 human lung cancer cells. Bioorg. Med. Chem. Lett. 2013, 23, 5609–5613. [Google Scholar] [CrossRef]
- D’Amore, C.; Borgo, C.; Sarno, S.; Salvi, M. Role of CK2 inhibitor CX-4945 in anti-cancer combination therapy–potential clinical relevance. Cell. Oncol. 2020, 43, 1003–1016. [Google Scholar] [CrossRef]
- Buontempo, F.; Orsini, E.; Lonetti, A.; Cappellini, A.; Chiarini, F.; Evangelisti, C.; Evangelisti, C.; Melchionda, F.; Pession, A.; Bertaina, A.; et al. Synergistic Cytotoxic Effects of Bortezomib and Ck2 Inhibitor Cx-4945 in Acute Lymphoblastic Leukemia: Turning Off the Prosurvival Er Chaperone Bip/Grp78 and Turning on the Pro-Apoptotic Nf-Κb. Oncotarget 2016, 7, 1323–1340. [Google Scholar] [CrossRef]
- Zakharia, K.; Miyabe, K.; Wang, Y.; Wu, D.; Moser, C.D.; Borad, M.J.; Roberts, L.R. Preclinical in Vitro and in Vivo Evidence of an Antitumor Effect of Cx-4945, a Casein Kinase Ii Inhibitor, in Cholangiocarcinoma. Transl. Oncol. 2019, 12, 143–153. [Google Scholar] [CrossRef]
- Eskilsson, E.; Røsland, G.V.; Solecki, G.; Wang, Q.; Harter, P.N.; Graziani, G.; Verhaak, R.G.; Winkler, F.; Bjerkvig, R.; Miletic, H. Egfr Heterogeneity and Implications for Therapeutic Intervention in Glioblastoma. Neuro Oncol. 2018, 20, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Furnari, F.; Cloughesy, T.F.; Cavenee, W.K.; Mischel, P.S. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat. Cancer 2015, 15, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Pierre, F.; Chua, P.C.; O’Brien, S.E.; Siddiqui-Jain, A.; Bourbon, P.; Haddach, M.; Michaux, J.; Nagasawa, J.; Schwaebe, M.K.; Stefan, E.; et al. Pre-clinical characterization of CX-4945, a potent and selective small molecule inhibitor of CK2 for the treatment of cancer. Mol. Cell. Biochem. 2011, 356, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, E.L.; Iegre, J.; Brear, P.D.; Zhabina, E.A.; Hyvönen, M.; Spring, D.R. Downfalls of Chemical Probes Acting at the Kinase Atp-Site: Ck2 as a Case Study. Molecules 2021, 26, 1977. [Google Scholar] [CrossRef] [PubMed]
- Salvi, M.; Borgo, C.; Pinna, L.A.; Ruzzene, M. Targeting Ck2 in Cancer: A Valuable Strategy or a Waste of Time? Cell Death Discov. 2021, 7, 325. [Google Scholar] [CrossRef]
- Pucko, E.; Matyja, E.; Koronkiewicz, M.; Ostrowski, R.P.; Kazimierczuk, Z. Potent Antitumour Effects of Novel Pentabromobenzylisothioureas Studied on Human Glial-derived Tumour Cell Lines. Anticancer Res. 2018, 38, 2691–2705. [Google Scholar] [CrossRef]
- Koronkiewicz, M.; Kazimierczuk, Z.; Szarpak, K.; Chilmonczyk, Z. Proapoptotic effects of new pentabromobenzylisothiouronium salts in a human prostate adenocarcinoma cell line. Acta Pol. Pharm. Drug Res. 2013, 69, 1325–1333. [Google Scholar]
- Ehtesham, M.; Winston, J.A.; Kabos, P.; Thompson, R.C. CXCR4 expression mediates glioma cell invasiveness. Oncogene 2006, 25, 2801–2806. [Google Scholar] [CrossRef]
- Ganju, R.K.; Brubaker, S.A.; Meyer, J.; Dutt, P.; Yang, Y.; Qin, S.; Newman, W.; Groopman, J.E. The α-Chemokine, Stromal Cell-derived Factor-1α, Binds to the Transmembrane G-protein-coupled CXCR-4 Receptor and Activates Multiple Signal Transduction Pathways. J. Biol. Chem. 1998, 273, 23169–23175. [Google Scholar] [CrossRef]
- Bian, X.-W.; Yang, S.-X.; Chen, J.-H.; Ping, Y.-F.; Zhou, X.-D.; Wang, Q.-L.; Jiang, X.-F.; Gong, W.; Xiao, H.-L.; Du, L.-L.; et al. Preferential Expression of Chemokine Receptor Cxcr4 By Highly Malignant Human Gliomas And Its Association With Poor Patient Survival. Neurosurgery 2007, 61, 570–579. [Google Scholar] [CrossRef]
- Zembrzuska, K.; Ostrowski, R.P.; Matyja, E. Hyperbaric oxygen increases glioma cell sensitivity to antitumor treatment with a novel isothiourea derivative in vitro. Oncol. Rep. 2019, 41, 2703–2716. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Gong, S.; Pan, J.; Wang, J.; Zou, D.; Xiong, S.; Zhao, L.; Yan, Q.; Deng, Y.; Wu, N.; et al. Hyperbaric oxygen promotes not only glioblastoma proliferation but also chemosensitization by inhibiting HIF1α/HIF2α-Sox2. Cell Death Discov. 2021, 7, 103. [Google Scholar] [CrossRef] [PubMed]
- Mottet, D.; Ruys, S.P.D.; Demazy, C.; Raes, M.; Michiels, C. Role for casein kinase 2 in the regulation of HIF-1 activity. Int. J. Cancer 2005, 117, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Boling, W.; Zhang, J.H. Hyperbaric oxygen therapy as adjunctive strategy in treatment of glioblastoma multiforme. Med. Gas Res. 2018, 8, 24–28. [Google Scholar] [CrossRef]
- Asati, V.; Mahapatra, D.K.; Bharti, S.K. PIM kinase inhibitors: Structural and pharmacological perspectives. Eur. J. Med. Chem. 2019, 172, 95–108. [Google Scholar] [CrossRef]
- Herzog, S.; Fink, M.A.; Weitmann, K.; Friedel, C.; Hadlich, S.; Langner, S.; Kindermann, K.; Holm, T.; Böhm, A.; Eskilsson, E.; et al. Pim1 kinase is upregulated in glioblastoma multiforme and mediates tumor cell survival. Neuro-Oncology 2014, 17, 223–242. [Google Scholar] [CrossRef]
- Quan, J.; Zhou, L.; Qu, J. Knockdown of Pim-3 suppresses the tumorigenicity of glioblastoma by regulating cell cycle and apoptosis. Cell. Mol. Biol. 2015, 61, 42–50. [Google Scholar]
- Zamykal, M.; Martens, T.; Matschke, J.; Günther, H.S.; Kathagen, A.; Schulte, A.; Peters, R.; Westphal, M.; Lamszus, K. Inhibition of intracerebral glioblastoma growth by targeting the insulin-like growth factor 1 receptor involves different context-dependent mechanisms. Neuro-Oncology 2014, 17, 1076–1085. [Google Scholar] [CrossRef]
- Vigneri, R.; Goldfine, I.D.; Frittitta, L. Insulin, Insulin Receptors, and Cancer. J. Endocrinol. Investig. 2016, 39, 1365–1376. [Google Scholar] [CrossRef]
- Ulanet, D.B.; Ludwig, D.L.; Kahn, C.R.; Hanahan, D. Insulin receptor functionally enhances multistage tumor progression and conveys intrinsic resistance to IGF-1R targeted therapy. Proc. Natl. Acad. Sci. USA 2010, 107, 10791–10798. [Google Scholar] [CrossRef]
- Brognard, J.; Hunter, T. Protein kinase signaling networks in cancer. Curr. Opin. Genet. Dev. 2011, 21, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef] [PubMed]
- Saadeh, F.S.; Mahfouz, R.; Assi, H.I. EGFR as a clinical marker in glioblastomas and other gliomas. Int. J. Biol. Markers 2017, 33, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Antonyak, M.A.; Singh, G.; Cerione, R.A. A Mechanism for the Upregulation of EGF Receptor Levels in Glioblastomas. Cell Rep. 2013, 3, 2008–2020. [Google Scholar] [CrossRef]
- Bode, U.; Massimino, M.; Bach, F.; Zimmermann, M.; Khuhlaeva, E.; Westphal, M.; Fleischhack, G. Nimotuzumab treatment of malignant gliomas. Expert Opin. Biol. Ther. 2012, 12, 1649–1659. [Google Scholar] [CrossRef]
- Liang, W.; Wu, X.; Fang, W.; Zhao, Y.; Yang, Y.; Wenhua, L.; Xue, C.; Zhang, J.; Zhang, J.; Ma, Y.; et al. Network Meta-Analysis of Erlotinib, Gefitinib, Afatinib and Icotinib in Patients with Advanced Non-Small-Cell Lung Cancer Harboring EGFR Mutations. PLoS ONE 2014, 9, e85245. [Google Scholar] [CrossRef]
- Kinsella, P.; Howley, R.; Doolan, P.; Clarke, C.; Madden, S.F.; Clynes, M.; Farrell, M.; Amberger-Murphy, V. Characterization and Response of Newly Developed High-Grade Glioma Cultures to the Tyrosine Kinase Inhibitors, Erlotinib, Gefitinib and Imatinib. Exp. Cell Res. 2012, 318, 641–652. [Google Scholar] [CrossRef]
- Liu, X.; Chen, X.; Shi, L.; Shan, Q.; Cao, Q.; Yue, C.; Li, H.; Li, S.; Wang, J.; Gao, S.; et al. The third-generation EGFR inhibitor AZD9291 overcomes primary resistance by continuously blocking ERK signaling in glioblastoma. J. Exp. Clin. Cancer Res. 2019, 38, 219. [Google Scholar] [CrossRef]
- Koul, D.; Fu, J.; Shen, R.; LaFortune, T.A.; Wang, S.; Tiao, N.; Kim, Y.-W.; Liu, J.-L.; Ramnarian, D.; Yuan, Y.; et al. Antitumor Activity of NVP-BKM120—A Selective Pan Class I PI3 Kinase Inhibitor Showed Differential Forms of Cell Death Based on p53 Status of Glioma Cells. Clin. Cancer Res. 2011, 18, 184–195. [Google Scholar] [CrossRef]
- Zhao, H.F.; Wang, J.; Shao, W.; Wu, C.P.; Chen, Z.P.; To, S.S.T.; Li, W.P. Recent Advances in the Use of Pi3k Inhibitors for Glioblastoma Multiforme: Current Preclinical and Clinical Development. Mol. Cancer 2017, 16, 100. [Google Scholar] [CrossRef]
- Gao, Q.; Lei, T.; Ye, F. Therapeutic targeting of EGFR-activated metabolic pathways in glioblastoma. Expert Opin. Investig. Drugs 2013, 22, 1023–1040. [Google Scholar] [CrossRef] [PubMed]
- Chresta, C.M.; Davies, B.R.; Hickson, I.; Harding, T.; Cosulich, S.; Critchlow, S.E.; Vincent, J.P.; Ellston, R.; Jones, D.; Sini, P.; et al. AZD8055 Is a Potent, Selective, and Orally Bioavailable ATP-Competitive Mammalian Target of Rapamycin Kinase Inhibitor with In vitro and In vivo Antitumor Activity. Cancer Res. 2009, 70, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Colella, B.; Colardo, M.; Iannone, G.; Contadini, C.; Saiz-Ladera, C.; Fuoco, C.; Barilà, D.; Velasco, G.; Segatto, M.; Di Bartolomeo, S. mTOR Inhibition Leads to Src-Mediated EGFR Internalisation and Degradation in Glioma Cells. Cancers 2020, 12, 2266. [Google Scholar] [CrossRef] [PubMed]
- Catalano, M.; D’Alessandro, G.; Lepore, F.; Corazzari, M.; Caldarola, S.; Valacca, C.; Faienza, F.; Esposito, V.; Limatola, C.; Cecconi, F.; et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol. Oncol. 2015, 9, 1612–1625. [Google Scholar] [CrossRef]
- Del Alcazar, C.R.G.; Hardebeck, M.C.; Mukherjee, B.; Tomimatsu, N.; Gao, X.; Yan, J.; Xie, X.J.; Bachoo, R.; Li, L.; Burma, S.; et al. Inhibition of DNA Double-Strand Break Repair by the Dual Pi3k/Mtor Inhibitor Nvp-Bez235 as a Strategy for Radiosensitization of Glioblastoma. Clin. Cancer Res. 2014, 20, 1235–1248. [Google Scholar] [CrossRef] [PubMed]
- Colardo, M.; Segatto, M.; Di Bartolomeo, S. Targeting Rtk-Pi3k-Mtor Axis in Gliomas: An Update. Int. J. Mol. Sci. 2021, 22, 4899. [Google Scholar] [CrossRef]
- Heffron, T.P.; Ndubaku, C.O.; Salphati, L.; Alicke, B.; Cheong, J.; Drobnick, J.; Edgar, K.; Gould, S.E.; Lee, L.B.; Lesnick, J.D.; et al. Discovery of Clinical Development Candidate Gdc-0084, a Brain Penetrant Inhibitor of Pi3k and Mtor. ACS Med. Chem. Lett. 2016, 7, 351–356. [Google Scholar] [CrossRef]
- Bliesath, J.; Huser, N.; Omori, M.; Bunag, D.; Proffitt, C.; Streiner, N.; Ho, C.; Siddiqui-Jain, A.; O’Brien, S.E.; Lim, J.K.; et al. Combined Inhibition of Egfr and Ck2 Augments the Attenuation of Pi3k-Akt-Mtor Signaling and the Killing of Cancer Cells. Cancer Lett. 2012, 322, 113–118. [Google Scholar] [CrossRef]
- Gober, M.K.; Flight, R.M.; Lambert, J.; Moseley, H.; Stromberg, A.; Black, E.P. Deregulation of a Network of mRNA and miRNA Genes Reveals That CK2 and MEK Inhibitors May Synergize to Induce Apoptosis KRAS-Active NSCLC. Cancer Inform. 2019, 18, 1176935119843507. [Google Scholar] [CrossRef]
- Lustri, A.M.; Di Matteo, S.; Fraveto, A.; Costantini, D.; Cantafora, A.; Napoletano, C.; Bragazzi, M.C.; Giuliante, F.; De Rose, A.M.; Berloco, P.B.; et al. TGF-β signaling is an effective target to impair survival and induce apoptosis of human cholangiocarcinoma cells: A study on human primary cell cultures. PLoS ONE 2017, 12, e0183932. [Google Scholar] [CrossRef]
- Whelan, R.; Hargaden, G.C.; Knox, A.J. Modulating the Blood & Ndash; Brain Barrier: A Comprehensive Review. Pharmaceutics 2021, 13, 1980. [Google Scholar] [PubMed]
- Liu, X.; Chen, J.; Li, W.; Hang, C.; Dai, Y. Inhibition of Casein Kinase II by CX-4945, But Not Yes-associated protein (YAP) by Verteporfin, Enhances the Antitumor Efficacy of Temozolomide in Glioblastoma. Transl. Oncol. 2019, 13, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Borgo, C.; Vilardell, J.; Bosello-Travain, V.; Pinna, L.A.; Venerando, A.; Salvi, M. Dependence of HSP27 cellular level on protein kinase CK2 discloses novel therapeutic strategies. Biochim. Biophys. Acta (BBA) Gen. Subj. 2018, 1862, 2902–2910. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.H.; Song, J.S.; Kim, S.H.; Kim, J. Pharmacokinetic characterization of CK2 inhibitor CX-4945. Arch. Pharmacal. Res. 2013, 36, 840–845. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pucko, E.B.; Ostrowski, R.P. Inhibiting CK2 among Promising Therapeutic Strategies for Gliomas and Several Other Neoplasms. Pharmaceutics 2022, 14, 331. https://doi.org/10.3390/pharmaceutics14020331
Pucko EB, Ostrowski RP. Inhibiting CK2 among Promising Therapeutic Strategies for Gliomas and Several Other Neoplasms. Pharmaceutics. 2022; 14(2):331. https://doi.org/10.3390/pharmaceutics14020331
Chicago/Turabian StylePucko, Emanuela B., and Robert P. Ostrowski. 2022. "Inhibiting CK2 among Promising Therapeutic Strategies for Gliomas and Several Other Neoplasms" Pharmaceutics 14, no. 2: 331. https://doi.org/10.3390/pharmaceutics14020331
APA StylePucko, E. B., & Ostrowski, R. P. (2022). Inhibiting CK2 among Promising Therapeutic Strategies for Gliomas and Several Other Neoplasms. Pharmaceutics, 14(2), 331. https://doi.org/10.3390/pharmaceutics14020331