
In Vitro-In Silico Tools for Streamlined Development of Acalabrutinib Amorphous Solid Dispersion Tablets

 , ,
, ,  and
and
Abstract
:
1. Introduction
- Dispersion polymer screening
- Spray drying and characterization of ASD intermediates
- In vitro dissolution testing of ASD intermediates versus crystalline acalabrutinib
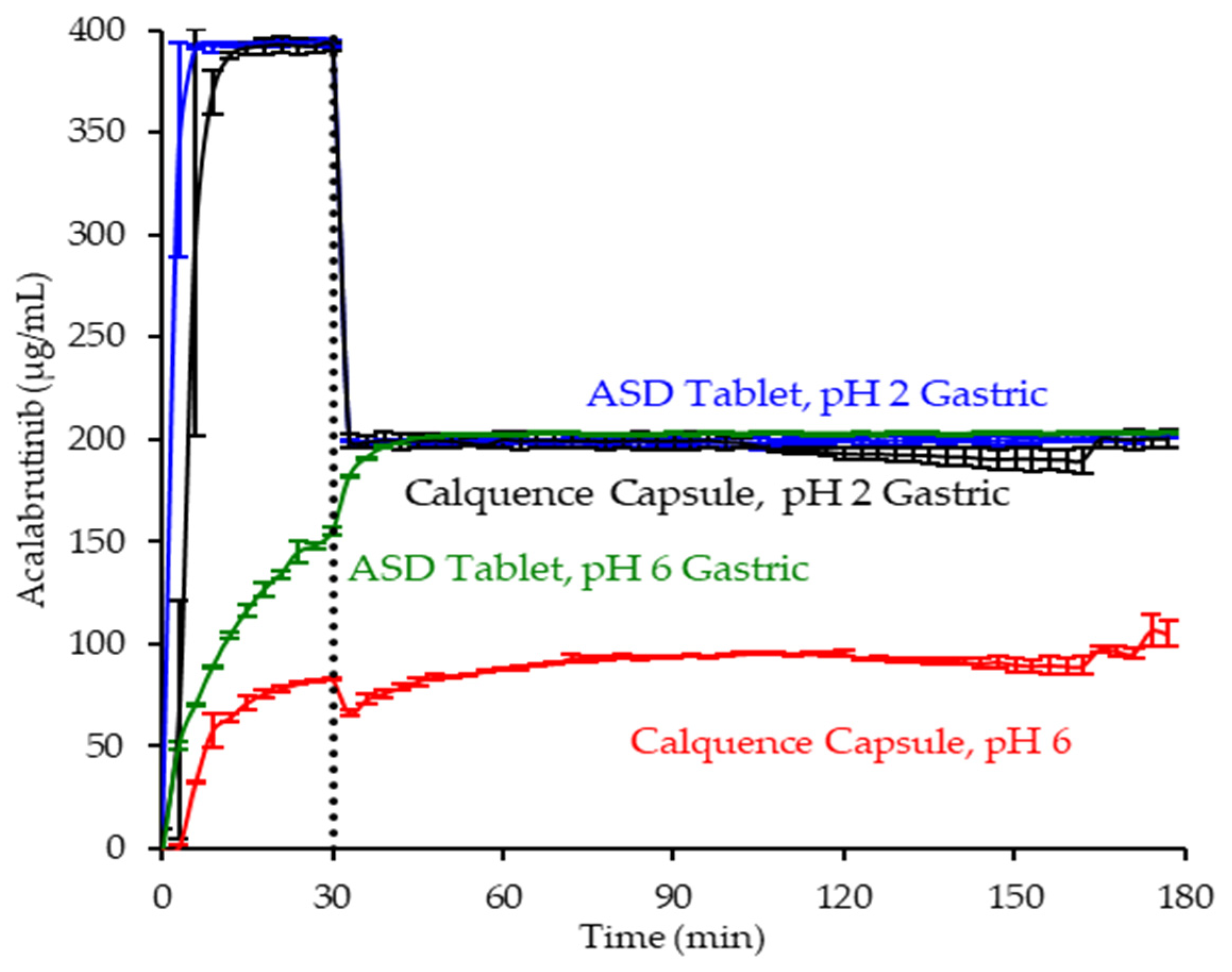
- In vitro dissolution testing of lead 50/50 acalabrutinib/HPMCAS-H ASD tablet versus commercially available Calquence
- Physicochemical property measurements of lead ASD intermediate
- In silico predictions of in vivo performance
2. Materials and Methods
2.1. Material Sourcing
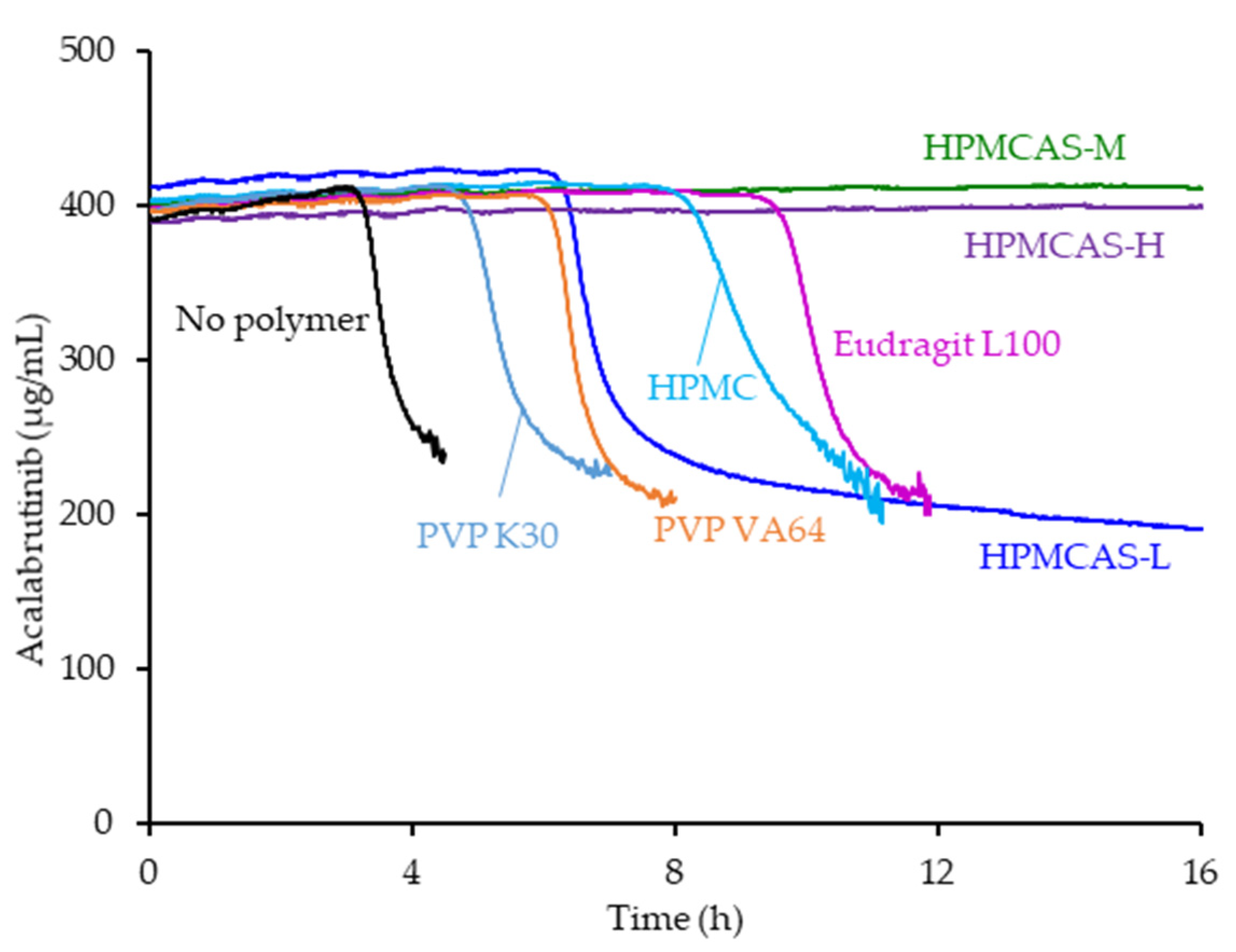
2.2. Dispersion Polymer Screening
2.3. ASD and ASD Tablet Manufacturing and Characterization
2.4. In Vitro Dissolution Testing of Intermediates
2.5. In Vitro Dissolution Testing of Dosage Forms
2.6. Physicochemical Property Measurements of Lead ASD Intermediate
2.6.1. Amorphous Acalabrutinib Slurry pH
2.6.2. Amorphous Acalabrutinib pH-Solubility
2.7. In Silico Predictions of In Vivo Performance
2.7.1. Physicochemical Properties and In Vitro Bioperformance Inputs
2.7.2. Physiology
2.7.3. PK

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Properties | |||||
|---|---|---|---|---|---|
| Parameter | Value | Source | |||
| Molecular weight (g/mol) | 465.52 | ADMET Predictor | |||
| pKa | 3.5, 5.8 (basic) | Pepin et al. a, FDA Biopharm review | |||
| log D (pH 7.4) | 1.14 | ADMET Predictor | |||
| Effective permeability (× 10−4 cm/s) | 5.4 | Pepin et al. for humans a Assumed dog permeability is similar to human | |||
| Solubility vs. pH (mg/mL) | ASD Tablet | Calquence Capsule | ASD Tablet (measured) Calquence capsule (Pepin et al. a) | ||
| pH | Solubility | pH | Solubility | ||
| 4 | 6.47 | 4 | 3.9 | ||
| 4.5 | 2.62 | 5 | 0.34 | ||
| 5 | 0.81 | 6 | 0.08 | ||
| 6 | 0.43 | 7 | 0.05 | ||
| Biorelevant solubility—6.7 mM SIF b, pH 6.5 (mg/mL) | 0.71 (ASD Tablet) 0.11 (Calquence capsule) | Measured | |||
| Dissolution model (z-factor, mL/mg/s) | ASD Tablet | Calquence capsule | Calculated from in vitro dissolution data for Calquence capsule and ASD tablet (Section 3.4) | ||
| pH | z-factor | pH | z-factor | ||
| 4.6 | 0.007 | 4 | 0.001 | ||
| 6.3 | 0.005 | 6 | 0.03 | ||
| 6.5 (SIF b) | 0.011 | 6.5 (SIF b) | 0.02 | ||
| Mean precipitation time (s) | 100,000 | Pepin et al. a | |||
| PK Parameters in Beagle Dog (Single Compartment) | |||||
| Parameter | Value | Source | |||
| Clearance (L/h/kg) | 0.75 | Calculated from Podoll et al. c | |||
| Volume of distribution (L/kg) | 1.27 | ||||
| Elimination half-life (h) | 1.18 | ||||
| First-pass extraction (%) | 25 | Pepin et al. a | |||
3. Results
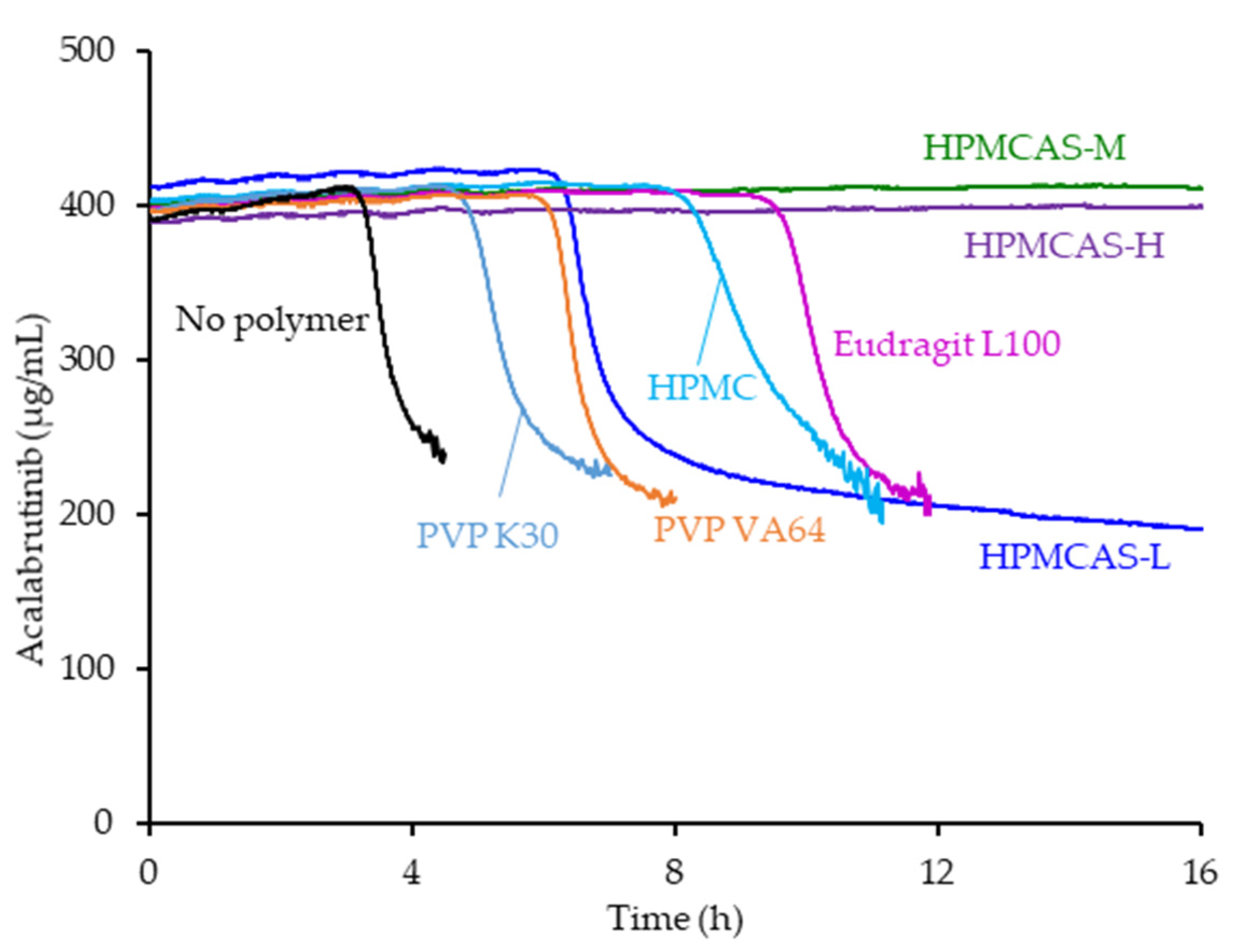
3.1. Dispersion Polymer Screening
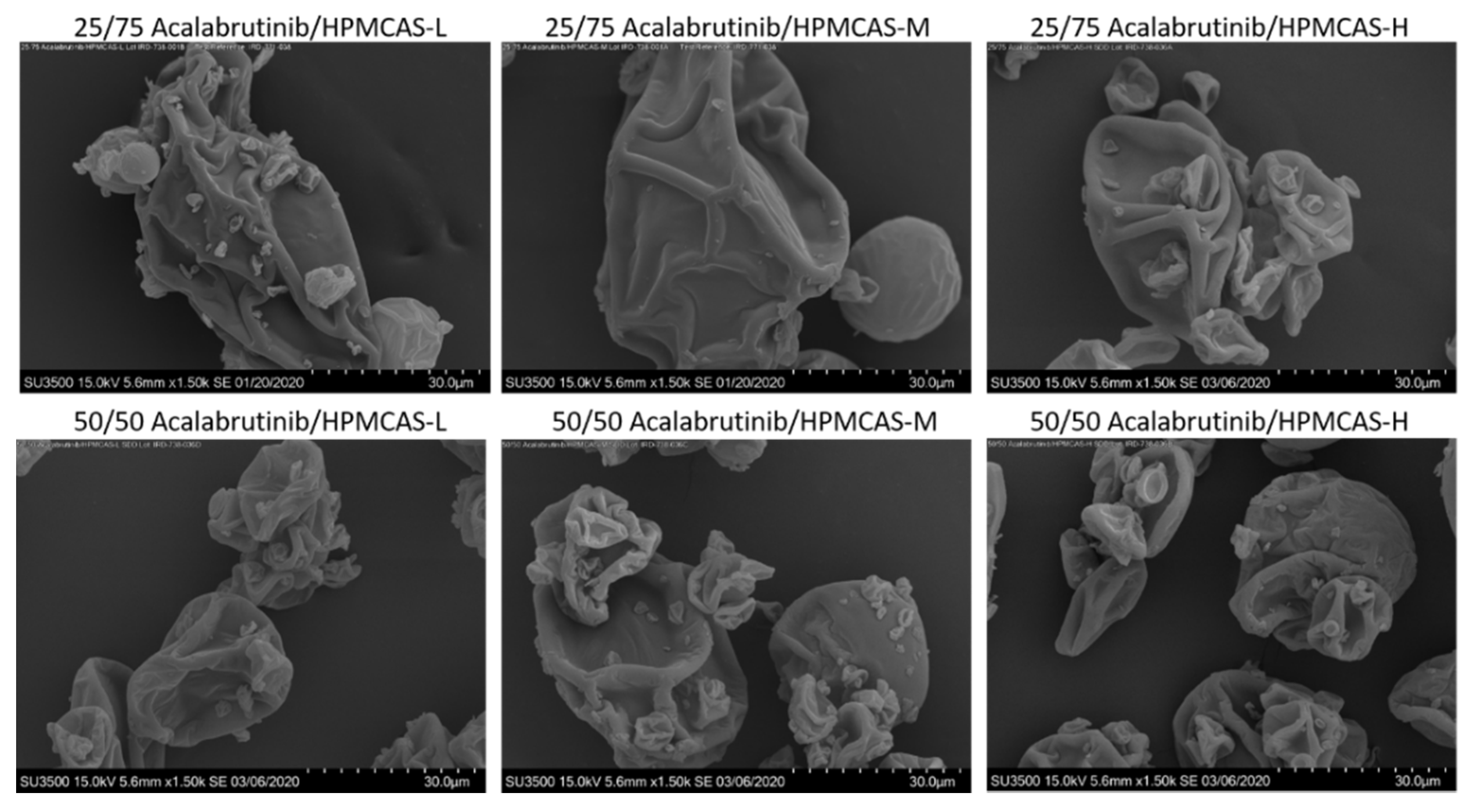
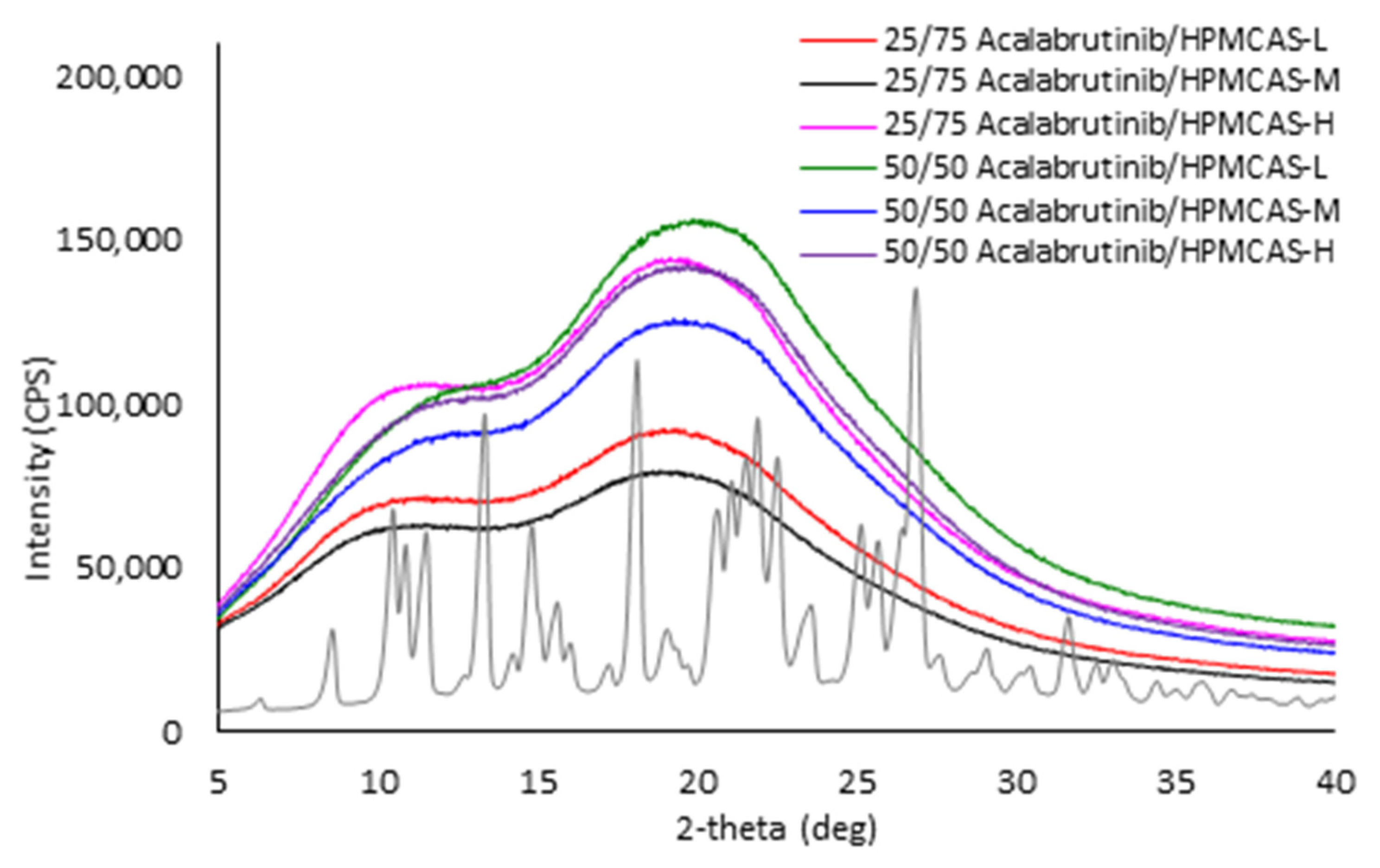
3.2. ASD Manufacturing and Characterization
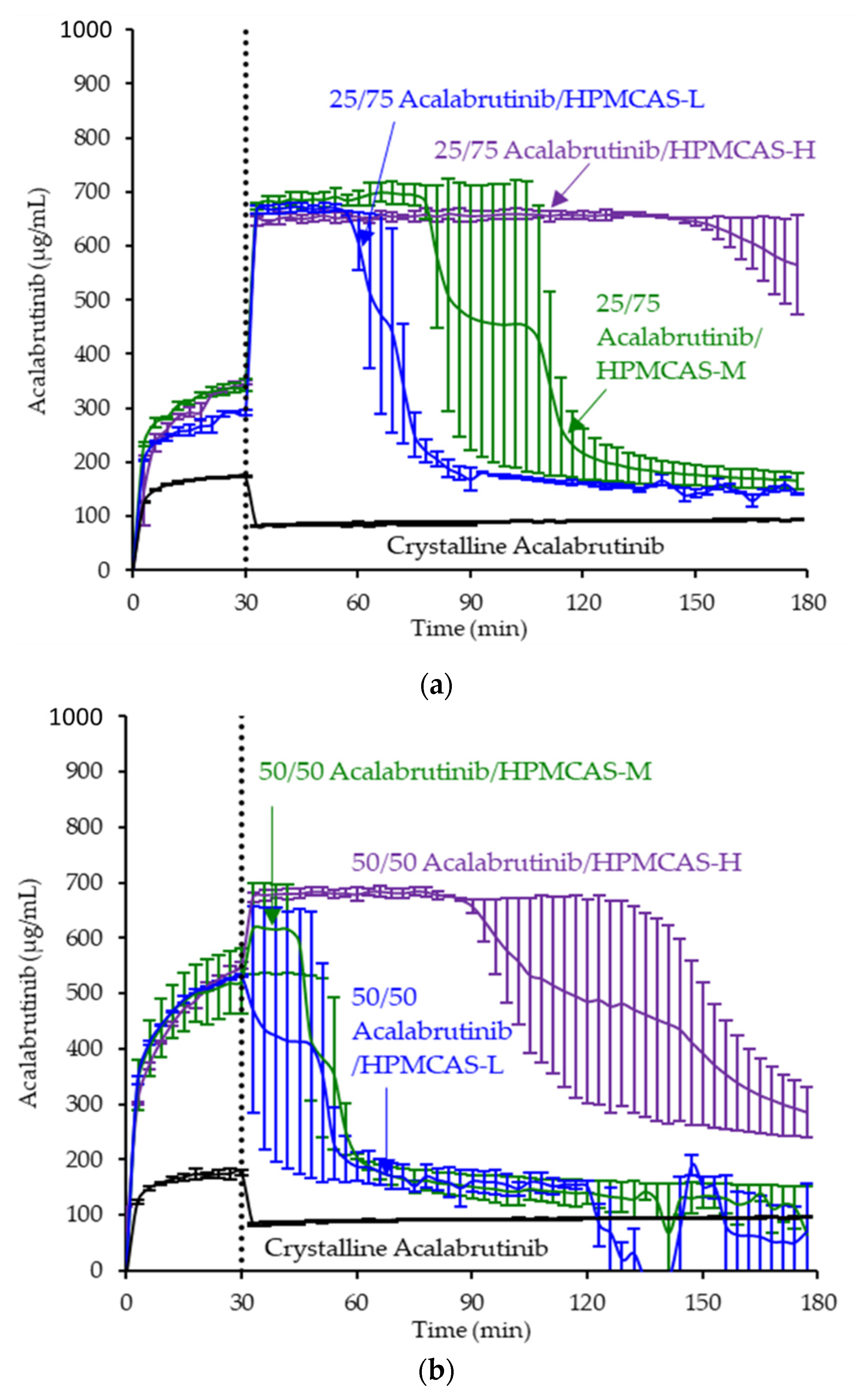
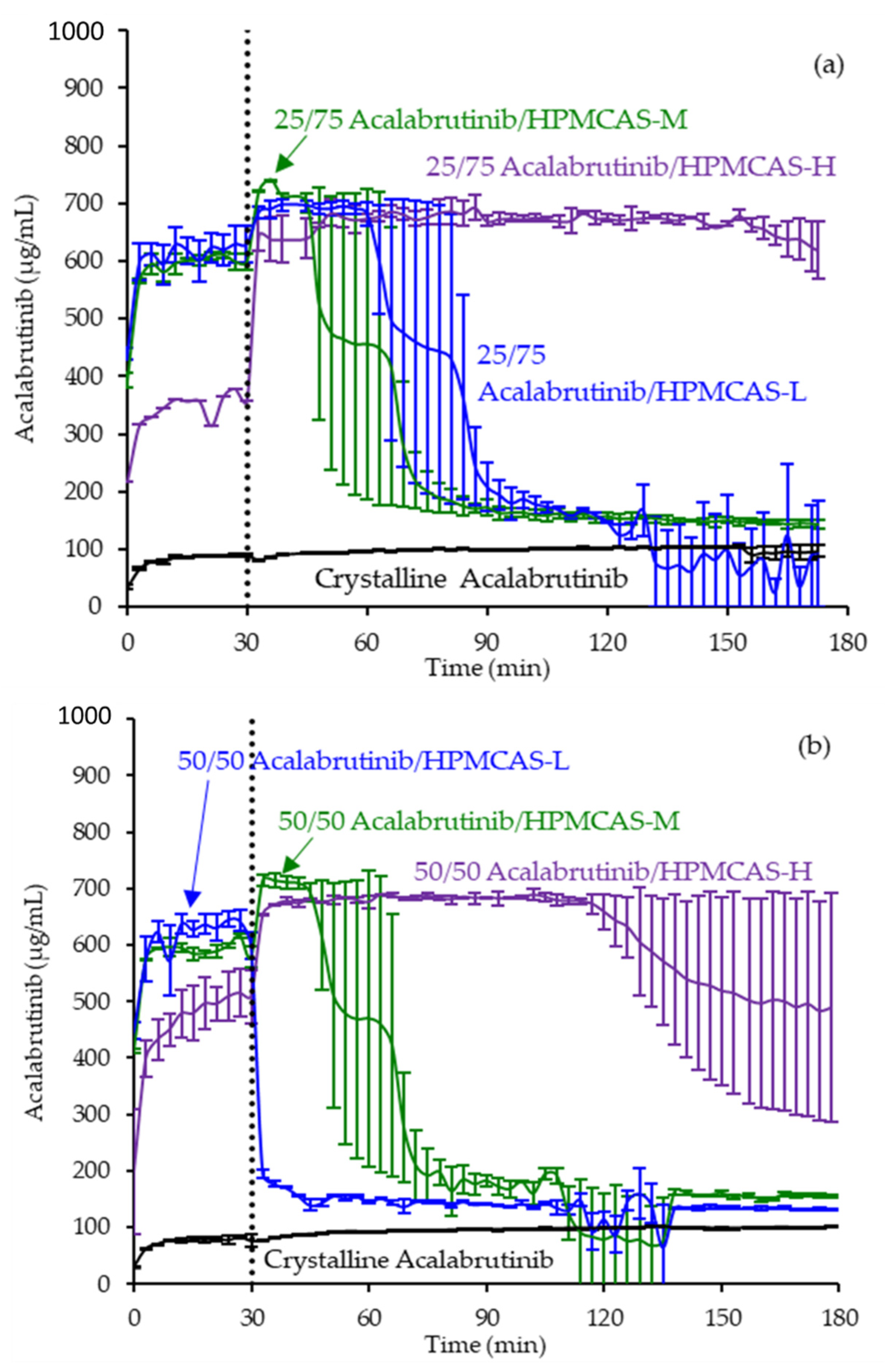
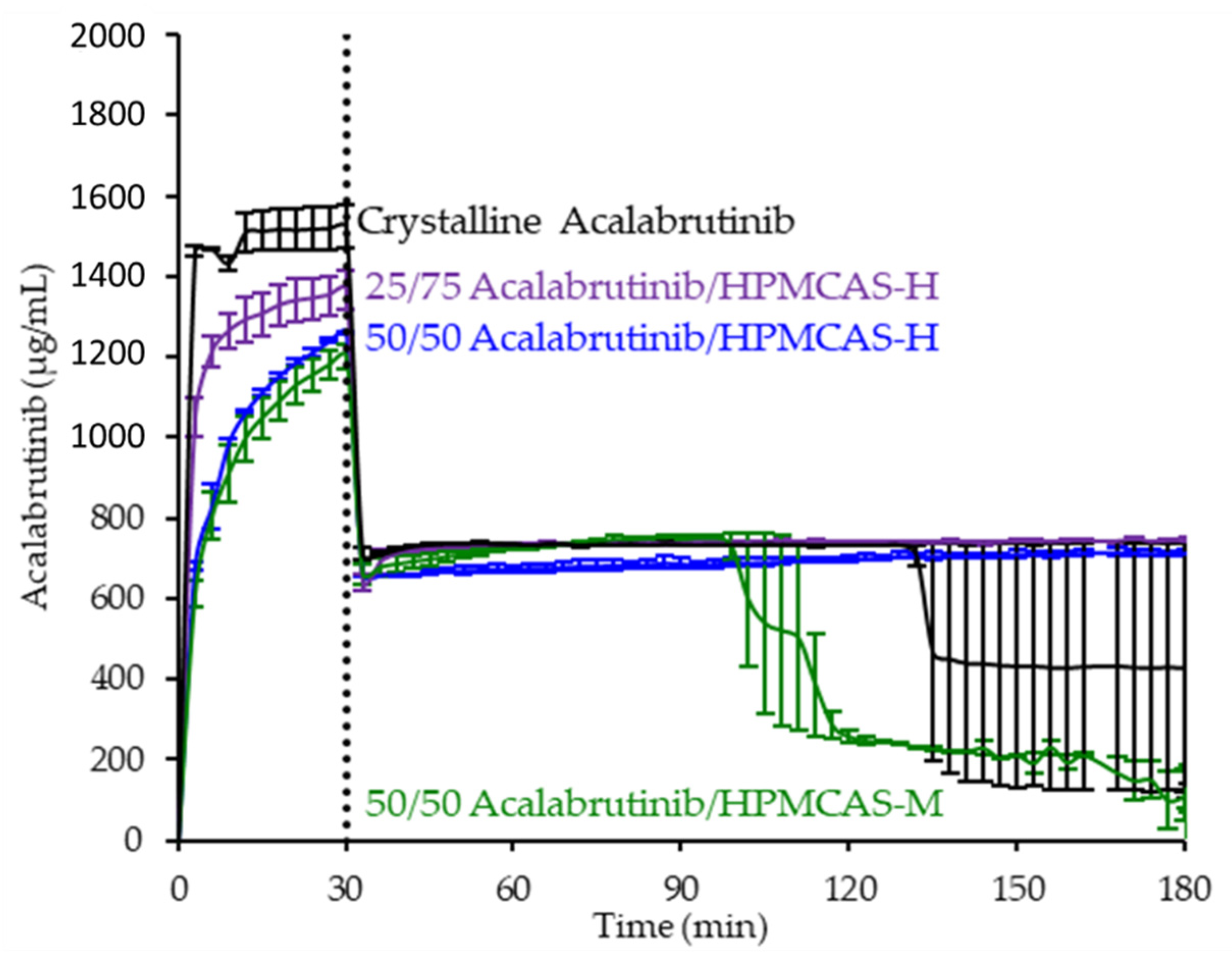
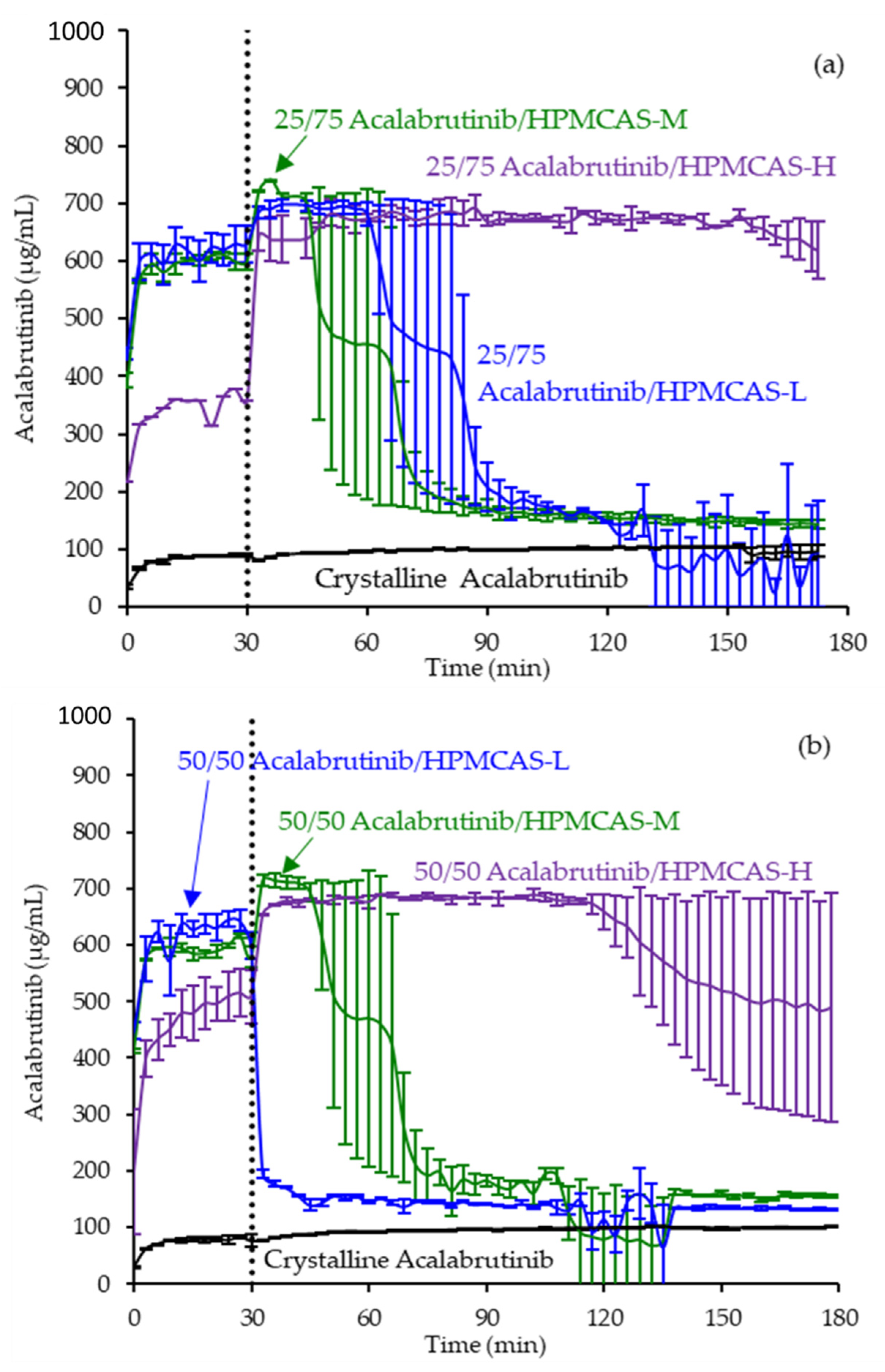
3.3. In Vitro Dissolution Testing of Intermediates
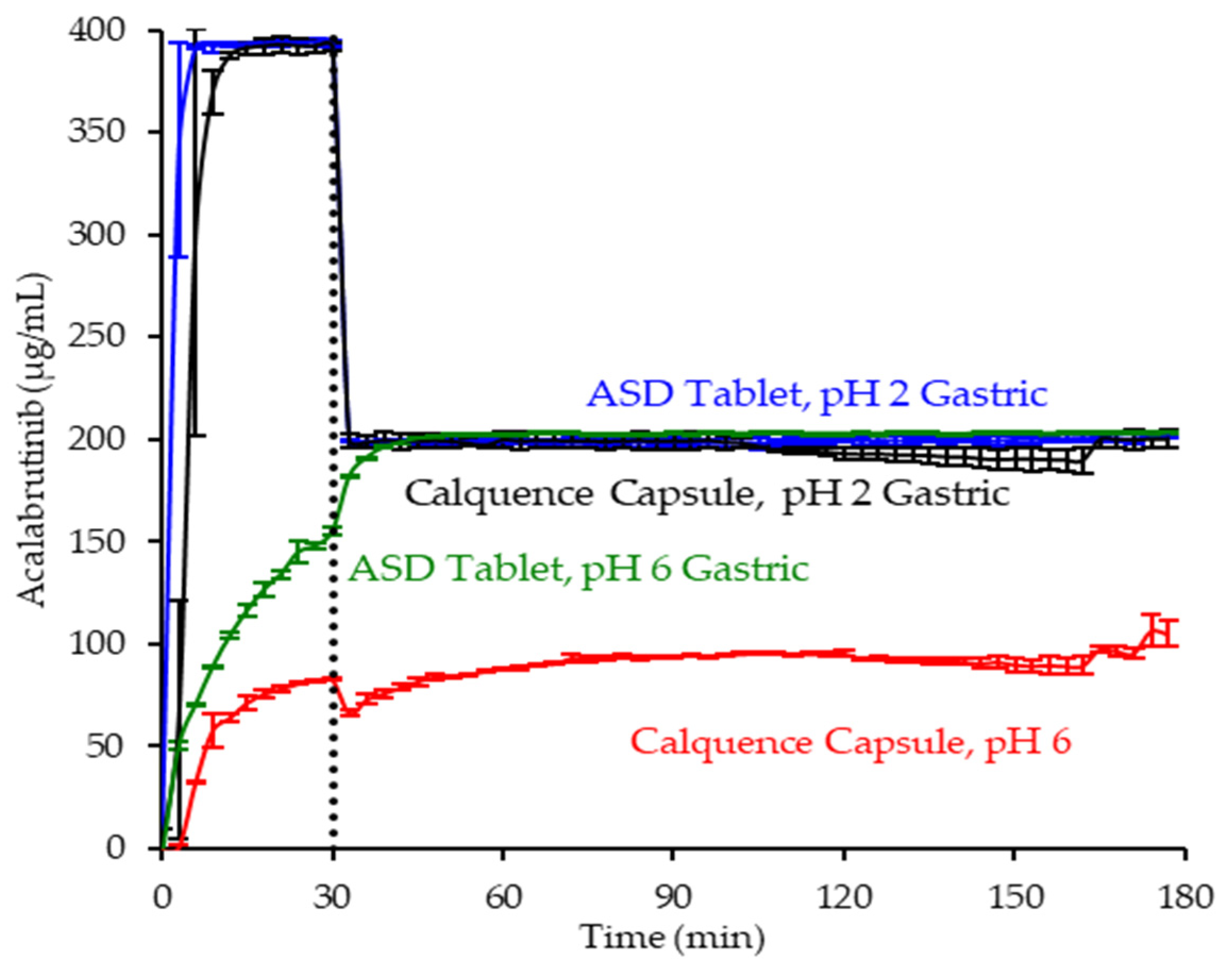
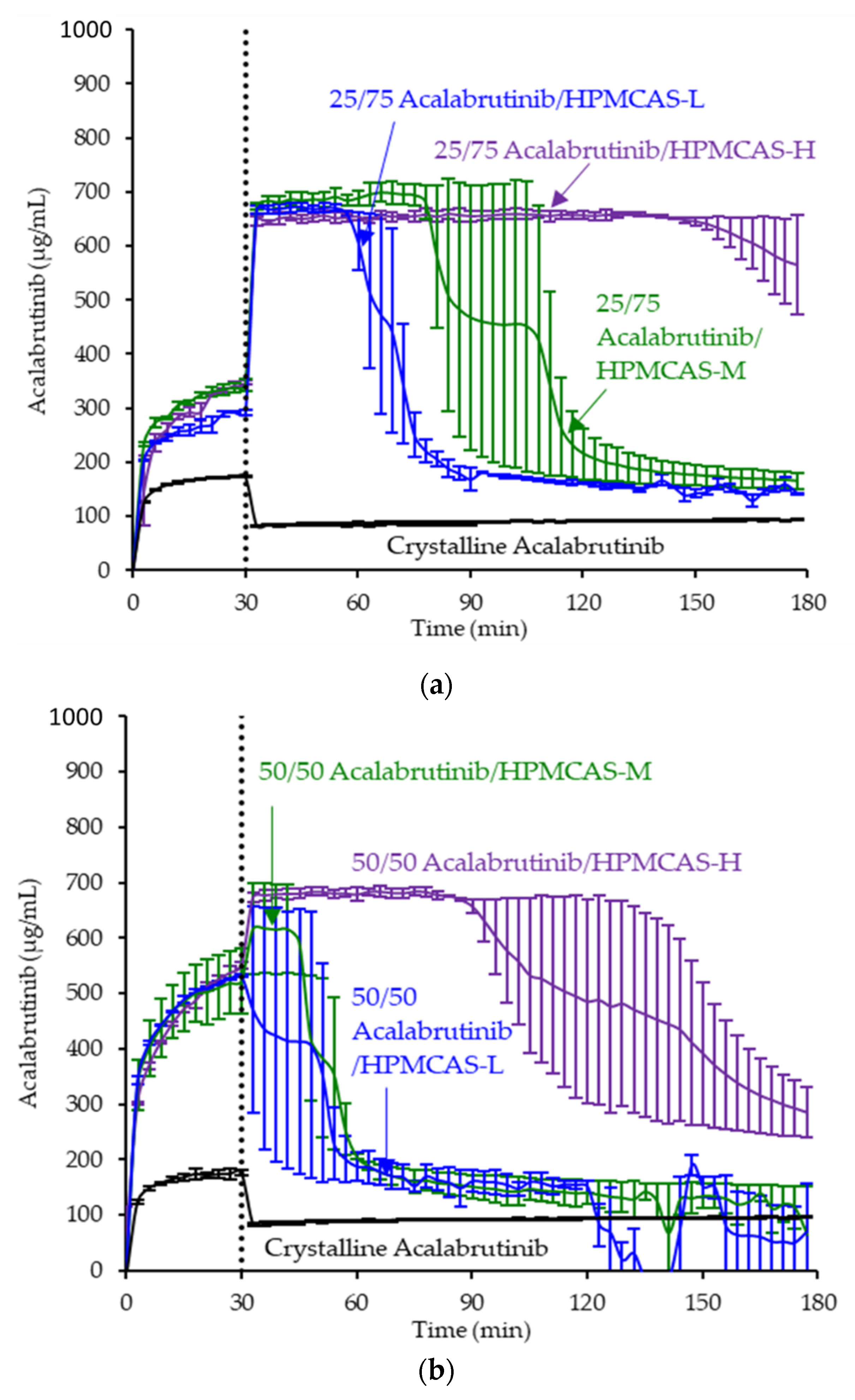
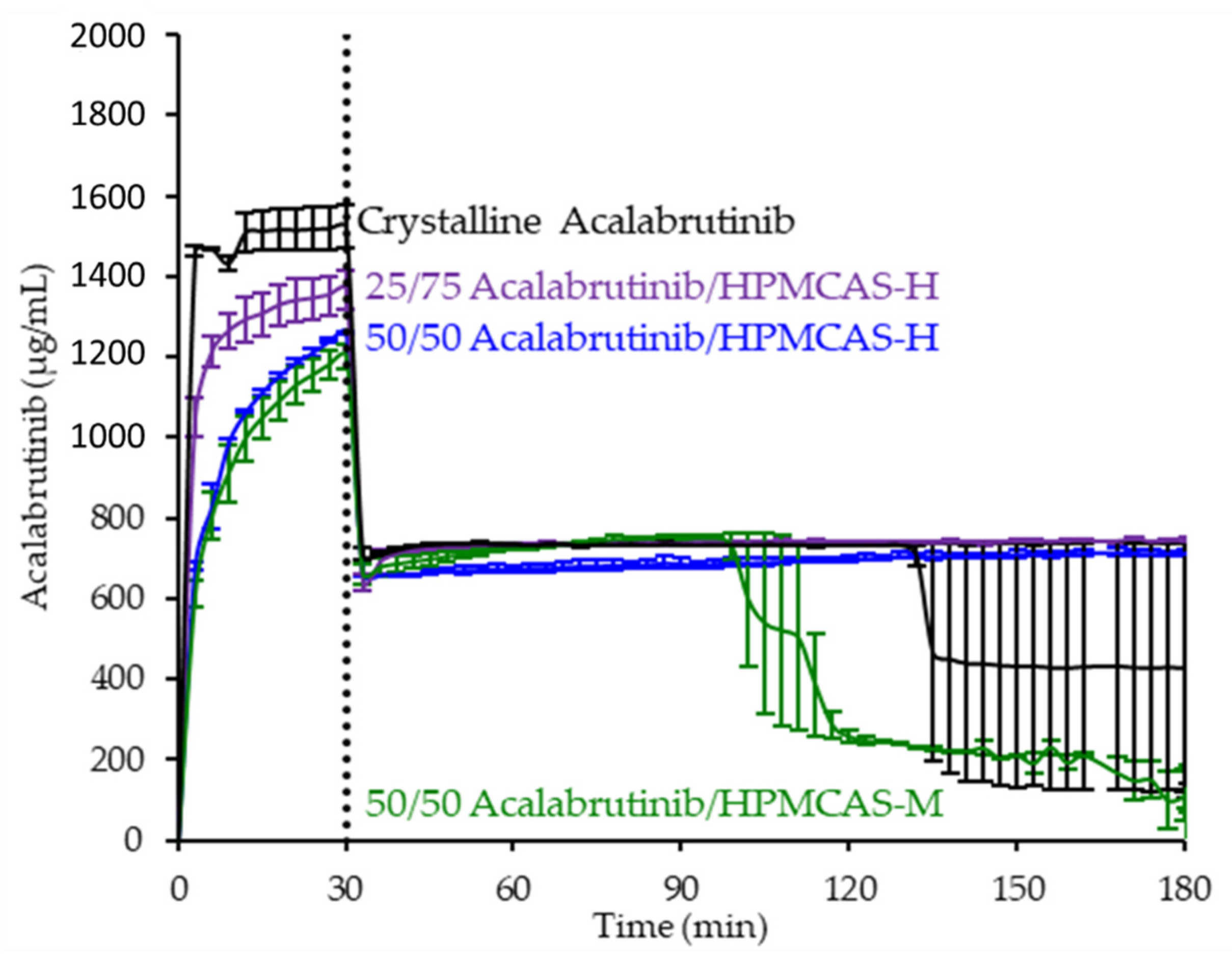
3.4. In Vitro Dissolution Testing of Dosage Forms
3.5. Physicochemical Property Measurements of Lead ASD Intermediate
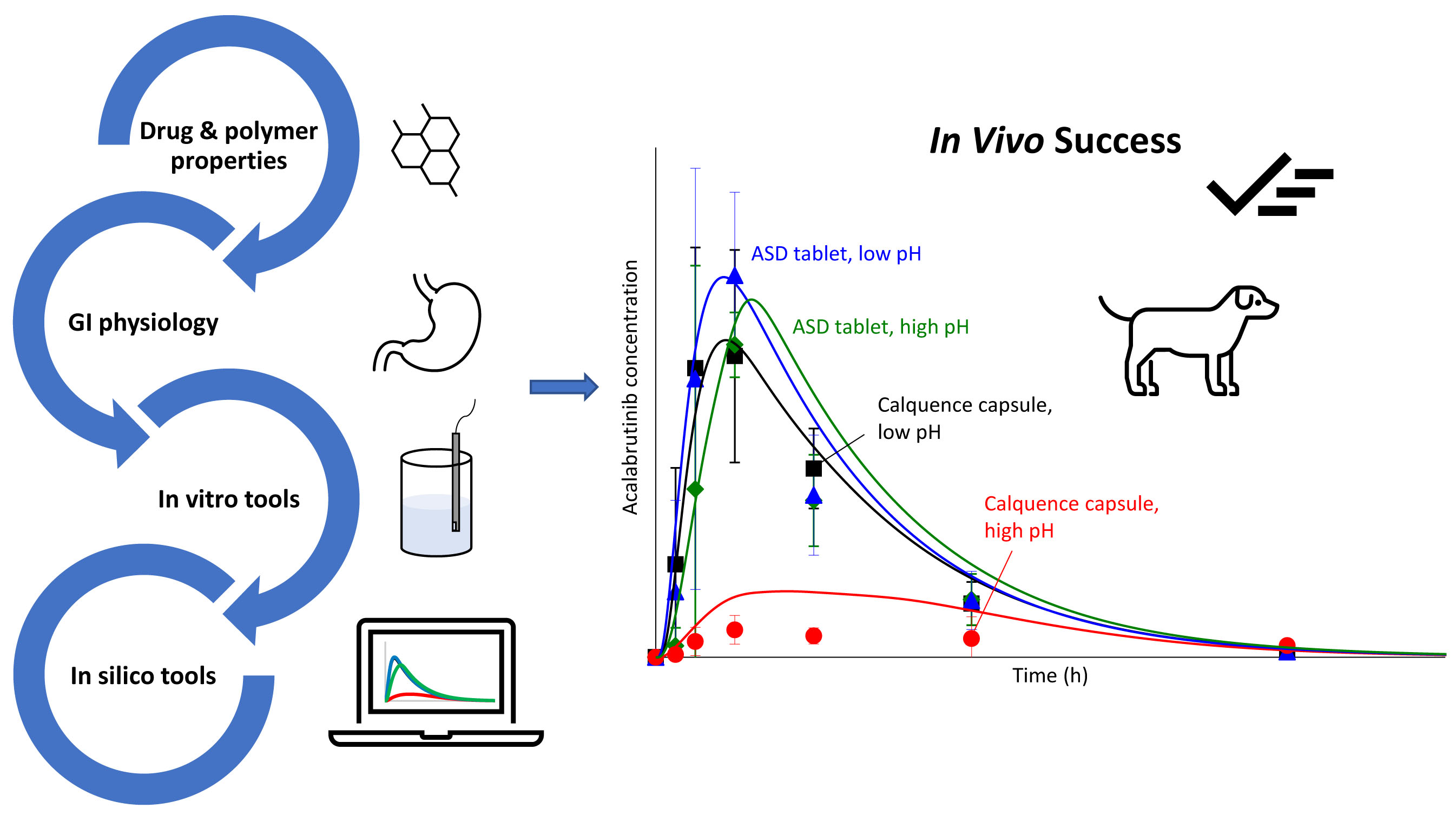
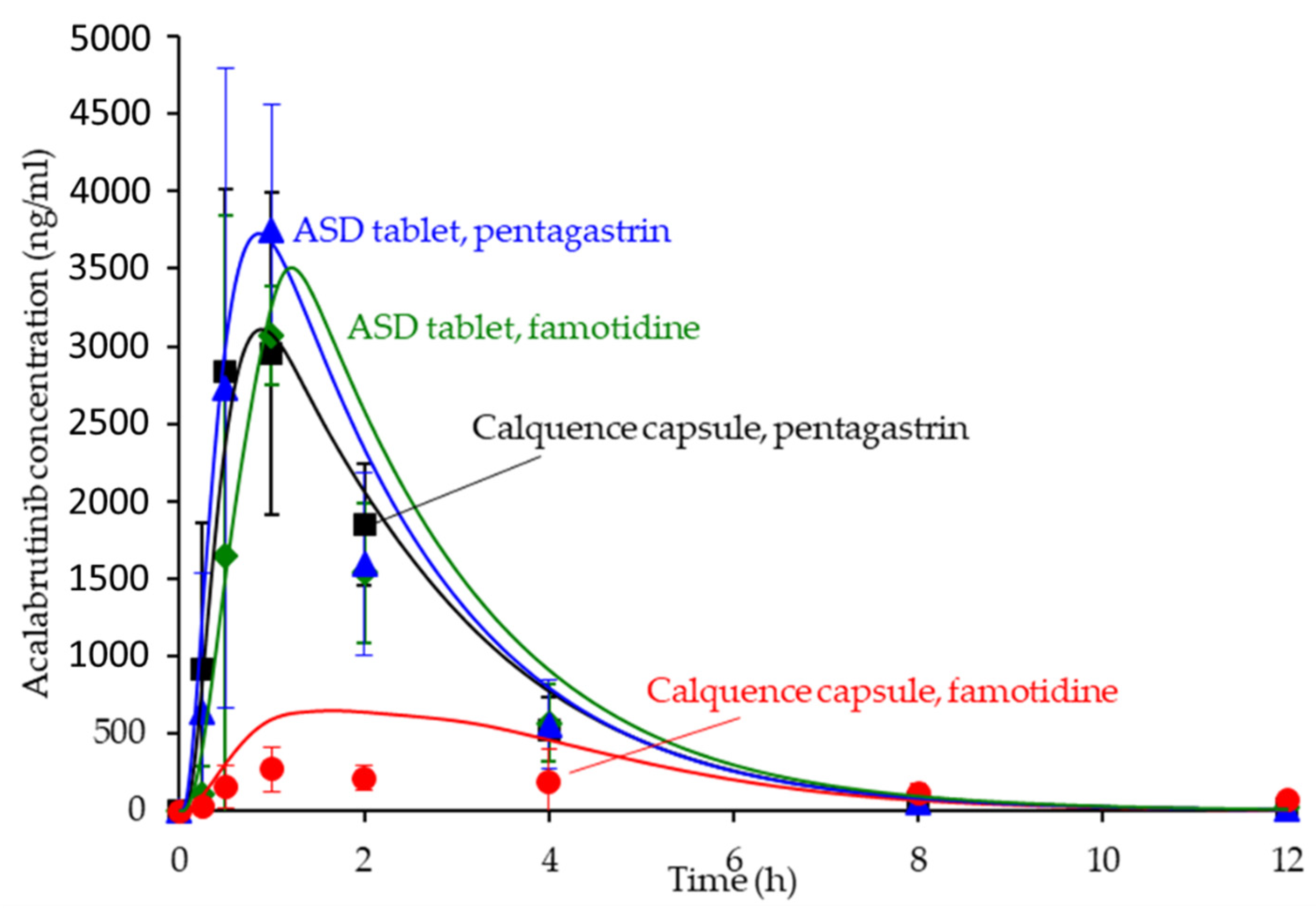
3.6. In Silico Predictions Versus In Vivo Performance
4. Discussion
4.1. Benefits of Streamlined, In Vitro–In Silico ASD Development Approach
4.2. Relative In Vitro Performance of HPMCAS ASDs
4.3. In Silico PK Simulation Tools in Early Development
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
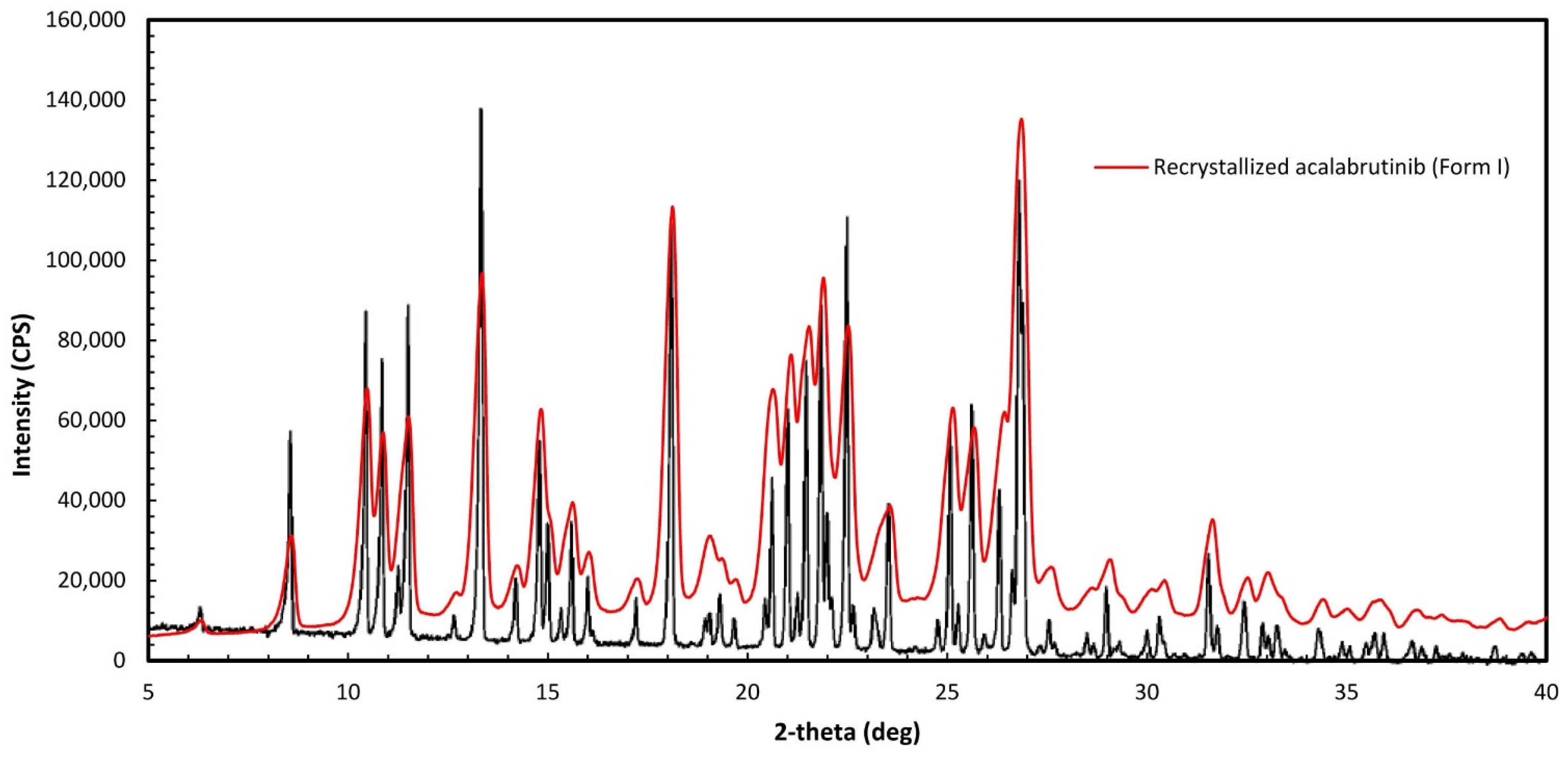
Appendix A.1. Acalabrutinib Form 1 Preparation
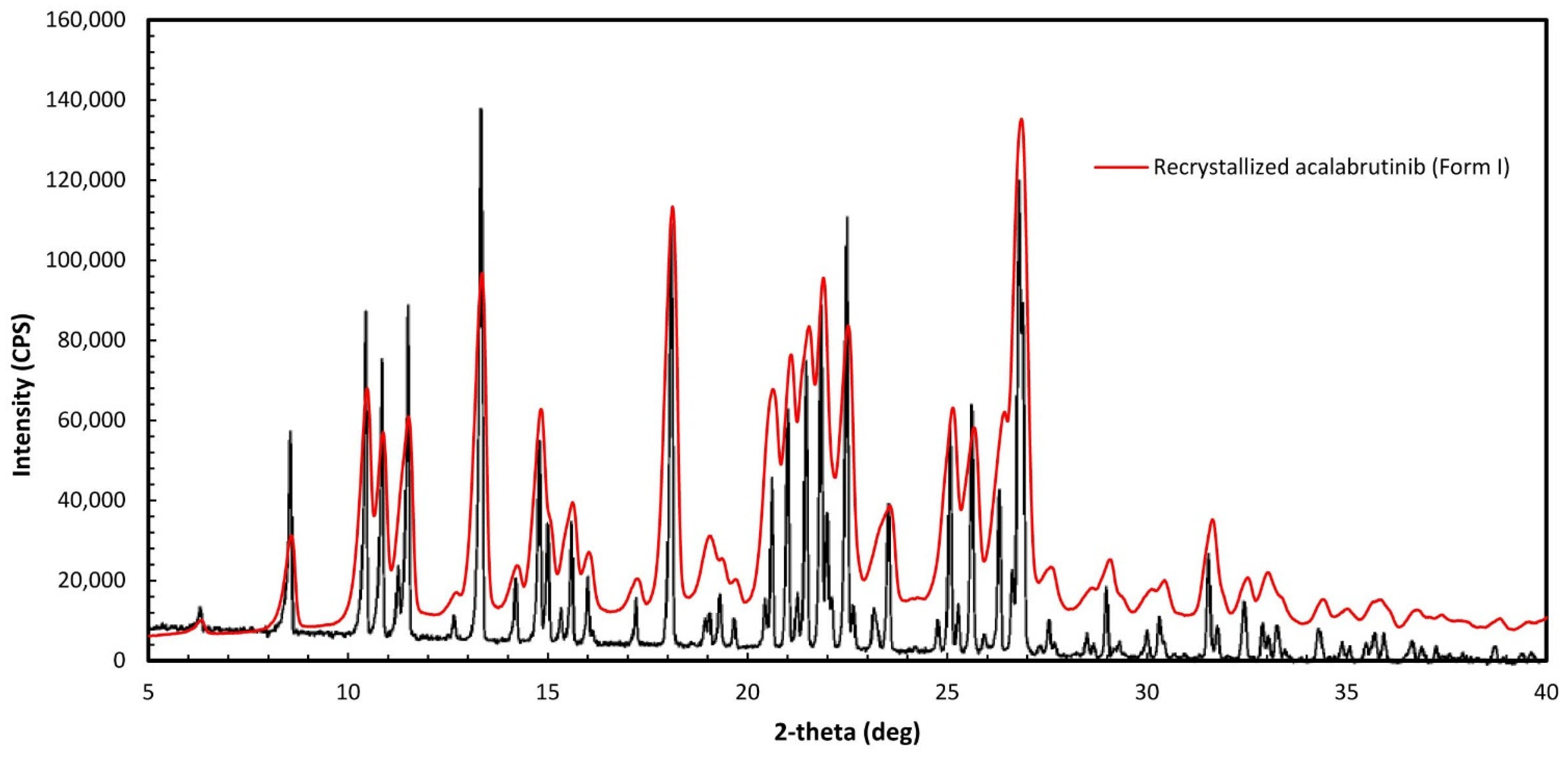
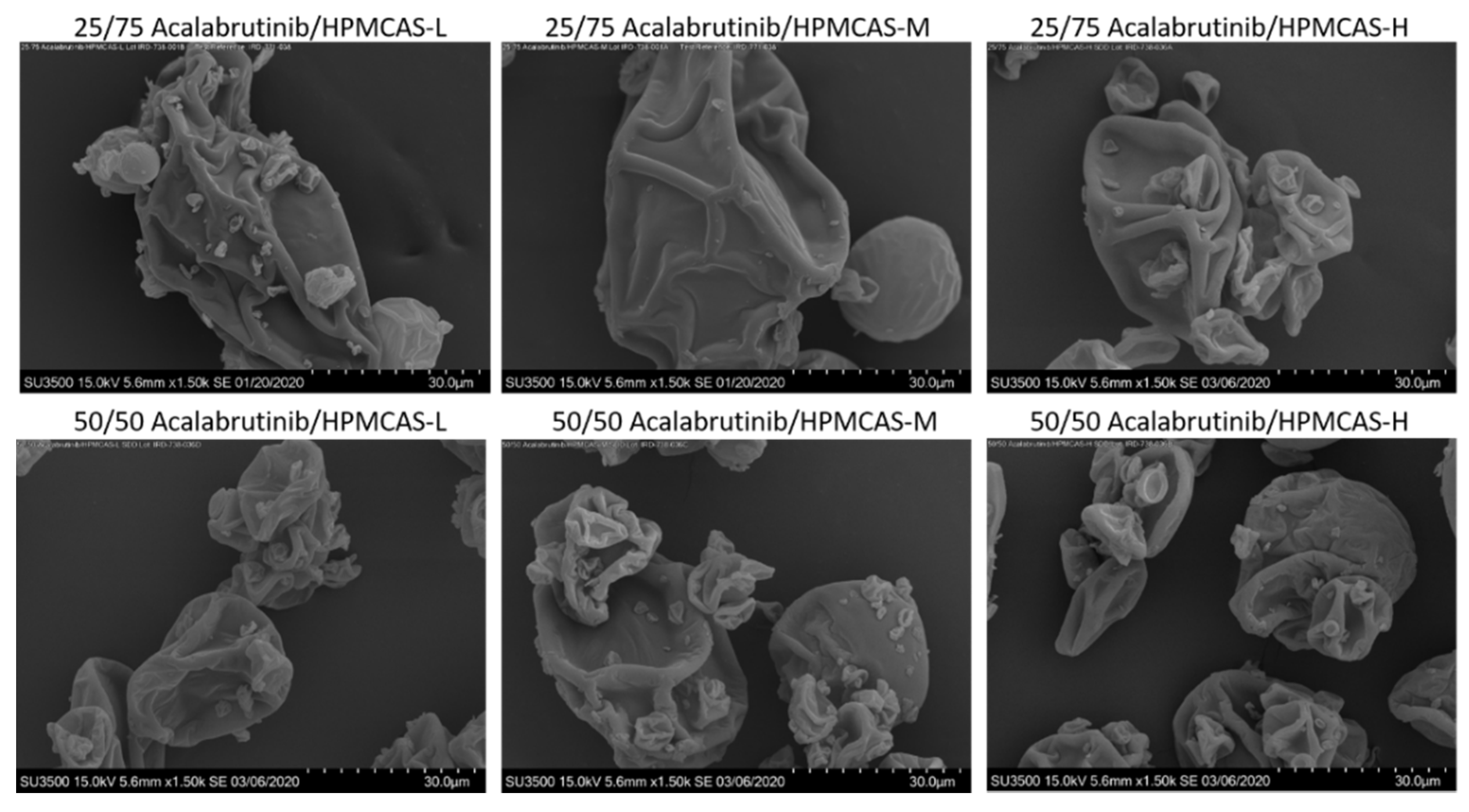
Appendix A.2. Amorphous Solid Dispersion (ASD) and Active Pharmaceutical Ingredient (API) Characterization Methods
Appendix A.2.1. Powder X-ray Diffraction (PXRD)
Appendix A.2.2. Modulated Differential Scanning Calorimetry (mDSC)
Appendix A.2.3. Scanning Electron Micrography (SEM)
Appendix A.3. In Vitro Dissolution Methods
Appendix A.3.1. In Vitro Dissolution Testing of Intermediates
| Ingredient | Mass Concentration (g/L) | Molar Concentration (mM) |
|---|---|---|
| Gastric medium (pH 5) | ||
| NaOAc | 0.83 | 10 |
| Adjust to pH 5.0 dropwise using 12.1 N HCl | ||
| Gastric medium (pH 6) | ||
| Na2HPO4∙7H2O | 5.36 | 20 |
| KH2PO4 | 6.36 | 47 |
| NaCl | 4.8 | 82 |
| Adjust to pH 6.0 dropwise using 12.1 N HCl | ||
| Gastric medium (pH 2) | ||
| NaCl | 1.98 | 34 |
| Adjust to pH 2.0 by diluting 12.1 N HCl to 0.01 N HCl | ||
| Concentrated intestinal medium (pH 5 and pH 2 gastric tests) | ||
| Na2HPO4∙7H2O | 10.72 | 40 |
| KH2PO4 | 12.72 | 94 |
| NaCl | 9.6 | 164 |
| FaSSIF/FeSSIF/FaSSGF | 10 | --- |
| Adjust to pH 6.55 dropwise using 10 N NaOH | ||
| Concentrated intestinal medium (pH 6 gastric tests) | ||
| Na2HPO4∙7H2O | 5.36 | 20 |
| KH2PO4 | 6.36 | 47 |
| NaCl | 4.8 | 82 |
| FaSSIF/FeSSIF/FaSSGF | 10 | --- |
| Adjust to pH 6.9 dropwise using 10 N NaOH | ||
| Dose/Volume in Gastric Medium (mg/mL) | Do pH 2 Gastric Medium (ASD/Crystalline Acalabrutinib) a | Do pH 5 Gastric Medium (ASD/Crystalline Acalabrutinib) a | Do pH 6 Gastric Medium (ASD/Crystalline Acalabrutinib) a | Dose/Volume in Intestinal Medium (mg/mL) | Do intestinal Medium (ASD/Crystalline Acalabrutinib) a |
|---|---|---|---|---|---|
| 2 | 0.03/0.04 b | 2.6/5.9 c | 4.6/26.0 d | 1 | 1.4 b/9.1 c |
| UV Probe Path Length (mm) | pH 5 Gastric Medium UV Analysis Wavelength (nm)/Calibration Range (mg/mL) | pH 6 Gastric Medium UV Analysis Wavelength (nm)/Calibration Range (mg/mL) | pH 2 Gastric Medium UV Analysis Wavelength (nm)/Calibration Range (mg/mL) | Intestinal Medium UV Analysis Wavelength (nm)/Calibration Range (mg/mL) |
|---|---|---|---|---|
| 1 | 298–308/0–0.6 | 340–344/0–0.6 | 370–376/0–0.6 | 340–344/0–0.6 |
Appendix A.3.2. In Vitro Dissolution Testing of Dosage Forms
| Ingredient | Mass Concentration (g/L) | Molar Concentration (mM) |
|---|---|---|
| Gastric medium (pH 6) | ||
| Adjust to pH 6.0 dropwise using 0.1 N HCl | ||
| Gastric medium (pH 2) | ||
| Adjust to pH 2.0 by diluting 12.1 N HCl to 0.01 N HCl | ||
| Concentrated intestinal medium | ||
| Na2HPO4∙7H2O | 10.72 | 40 |
| KH2PO4 | 12.72 | 94 |
| NaCl | 9.6 | 164 |
| FaSSIF/FeSSIF/FaSSGF | 10 | --- |
| Adjust to pH 6.55 dropwise using 10 N NaOH | ||
| Dose/Volume in Gastric Medium (mg/mL) | Do pH 2 Gastric Medium (ASD Tablet/Calquence) a | Do pH 6 Gastric Medium (ASD Tablet/Calquence) a | Dose/Volume in Intestinal Medium (mg/mL) | Do intestinal Medium (ASD Tablet/Calquence) a |
|---|---|---|---|---|
| 0.4 | 0.002 b/0.003 b | 1.0 c/5.2 c | 0.2 | 0.3 b/1.8 c |
| Dose/Volume in Gastric Medium (mg/mL) | UV Probe Path Length (mm) | pH 2 Gastric Medium UV Analysis Wavelength (nm)/Calibration Range (mg/mL) | pH 6 Gastric Medium UV Analysis Wavelength (nm)/Calibration Range (mg/mL) | Intestinal Medium UV Analysis Wavelength (nm)/Calibration Range (mg/mL) |
|---|---|---|---|---|
| 0.4 | 1 | 330–340/0–0.7 | 330–340/0–0.7 | 330–340/0–0.7 |
Appendix A.4. Amorphous Acalabrutinb Slurry pH and pH-Solubility Method Details
| pH | Concentration of Acalabrutinib Added (mg/mL) a | Molarity of HCl Titration Medium (M) |
|---|---|---|
| 4.0 | 25 | 0.1 |
| 4.5 | 10 | 0.01 |
| 5.0 | 3.5 | 0.001 |
| 6.0 | 2.5 | 0.001 |
Appendix A.5. Bulk and Surface pH Values for In Silico Predictions
| Compartment | GastroPlus Fasted Dog Physiology Bulk pH | Adjusted (i.e. Surface) pH ASD Tablet | Adjusted (i.e. Surface) pH Calquence Capsule |
|---|---|---|---|
| Stomach (fasted) | 2.0 a | 4.6 | 4.0 |
| Stomach (ARA) | 6.0 a | 6.3 | 6.0 |
| Duodenum | 6.2 | 6.2 | 6.2 |
| Jejunum 1 | 6.2 | 6.2 | 6.2 |
| Jejunum 2 | 6.2 | 6.2 | 6.2 |
| Ileum 1 | 6.4 | 6.4 | 6.4 |
| Ileum 2 | 6.6 | 6.6 | 6.6 |
| Ileum 3 | 6.68 | 6.68 | 6.68 |
| Caecum | 6.75 | 6.75 | 6.75 |
| Asc Colon | 6.45 | 6.45 | 6.45 |
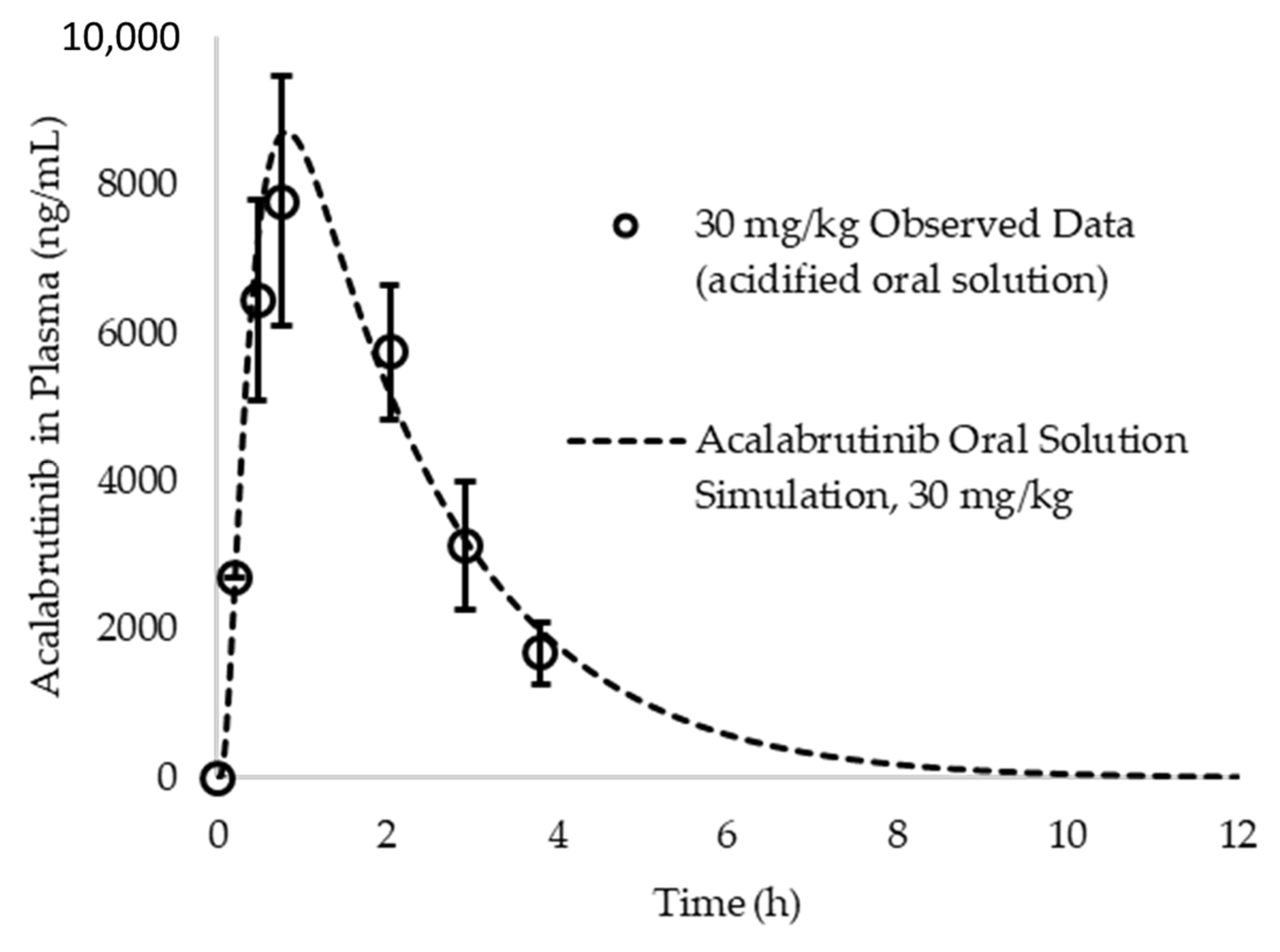
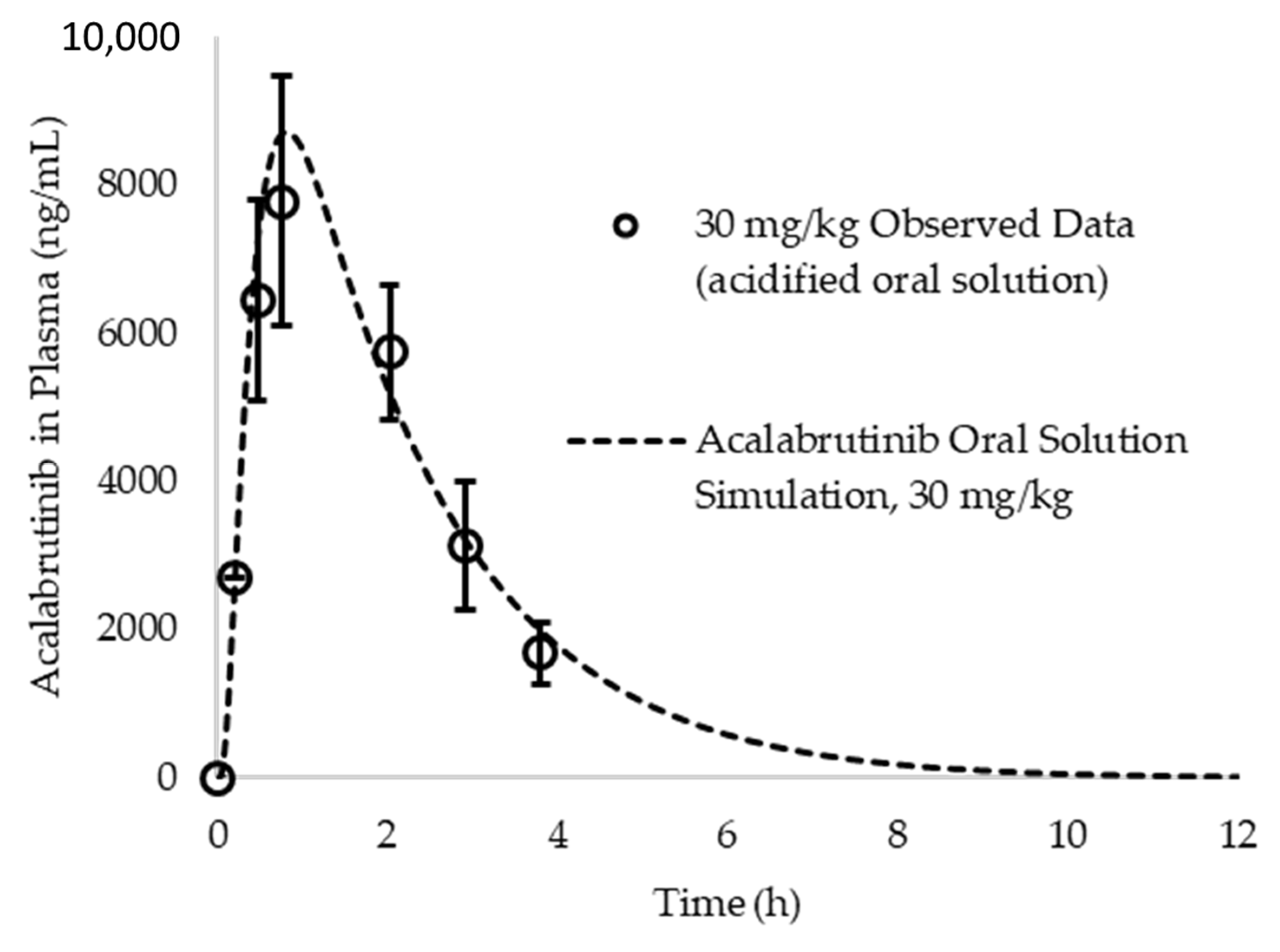
Appendix A.6. PK Parameter Calculations and In Silico Prediction Accuracy of Observed Data from Acalabrutinib Acidified Oral Solution
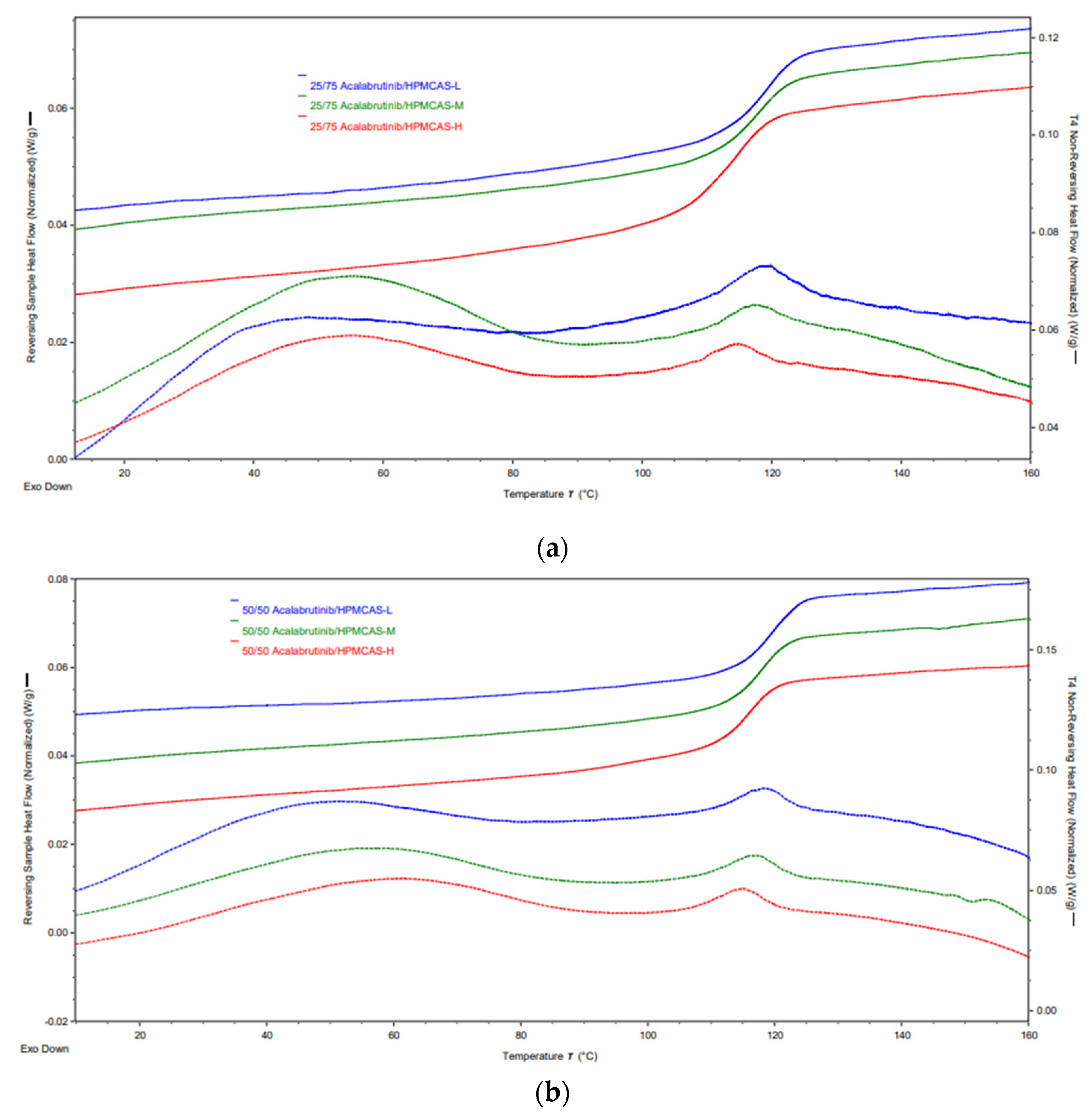
Appendix A.7. ASD Characterization Results
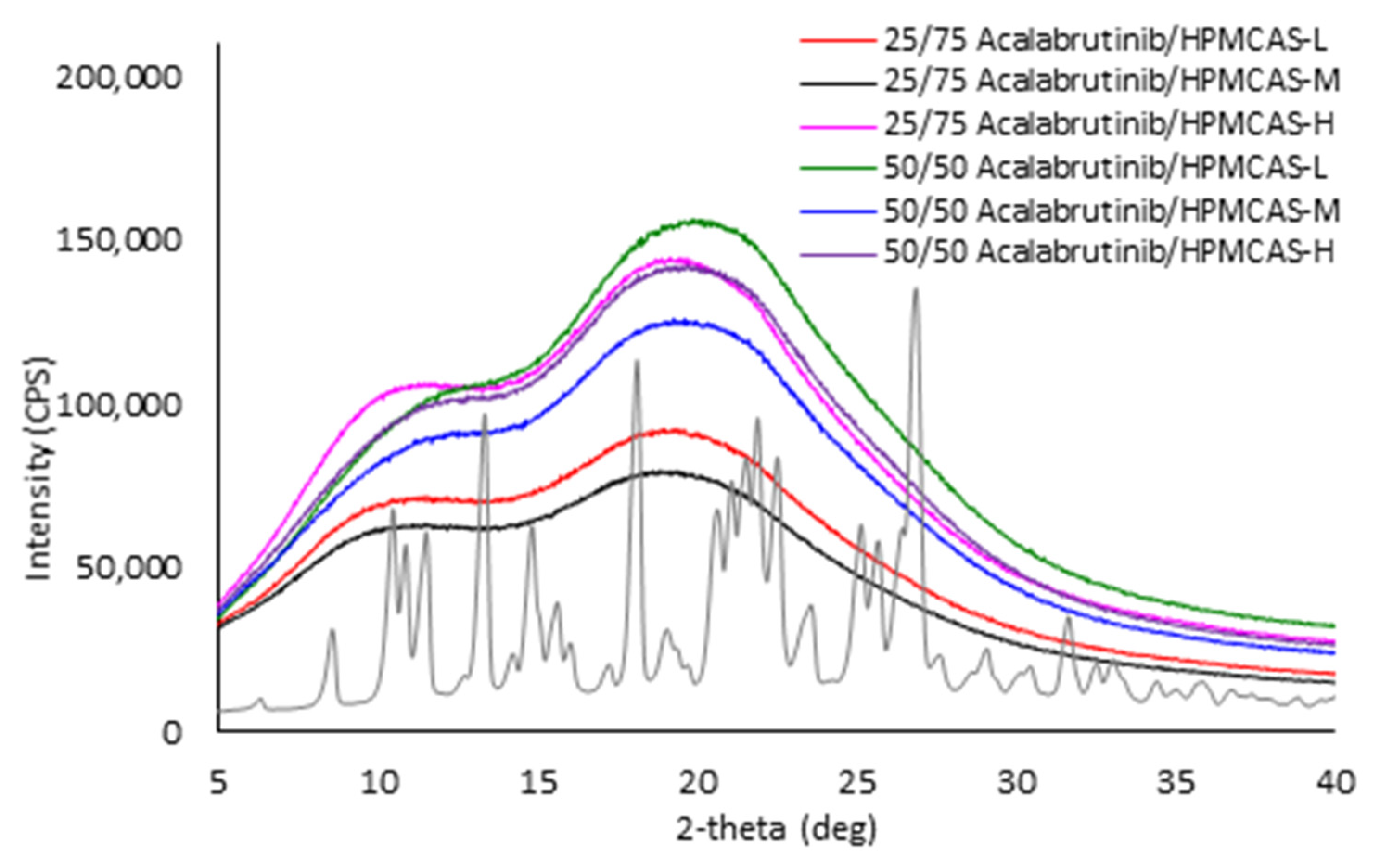
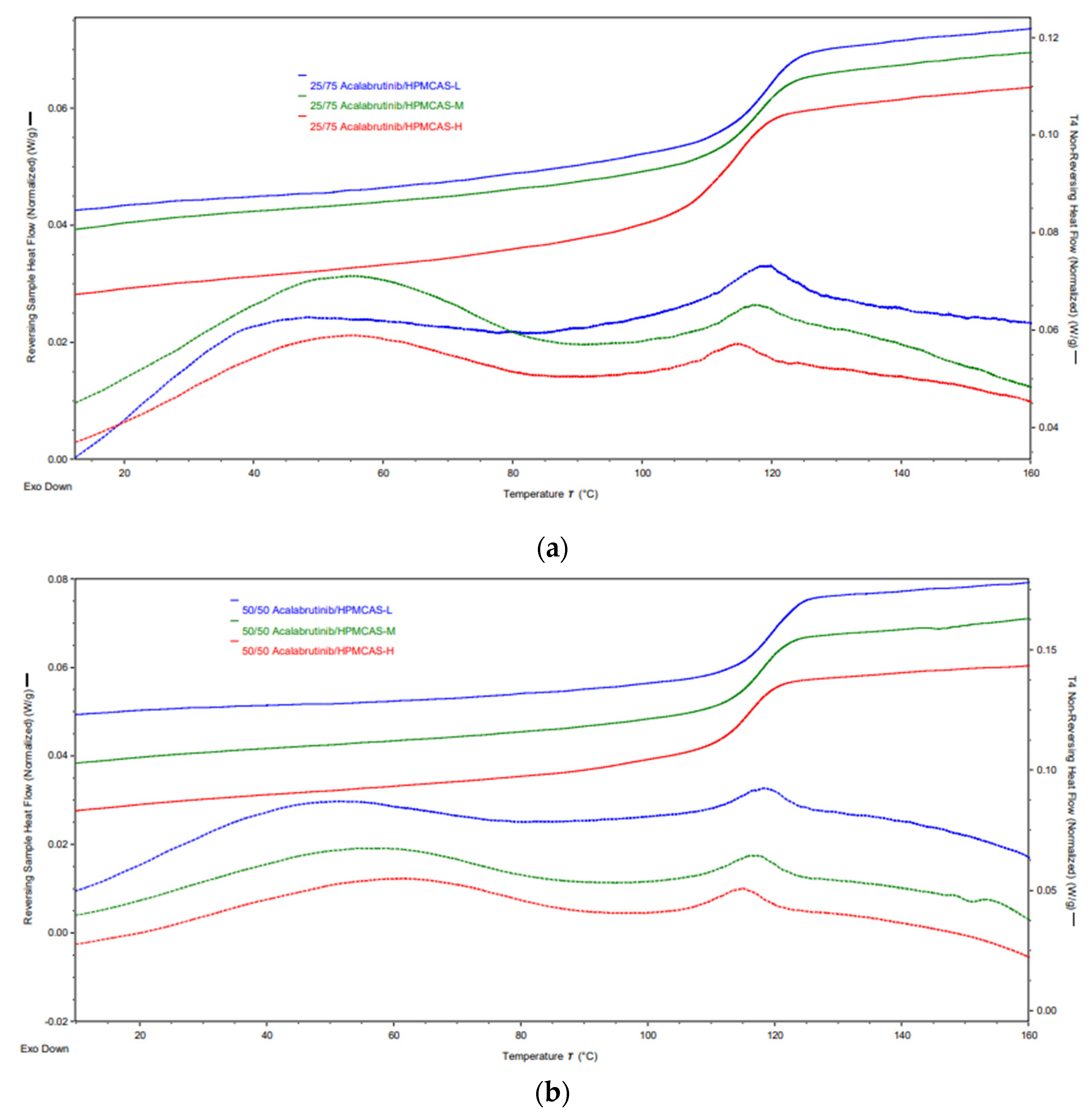
| Formulation | Tg (°C) |
|---|---|
| 25/75 Acalabrutinib/HPMCAS-L | 118.7 ± 0.1 |
| 25/75 Acalabrutinib/HPMCAS-M | 117.6 ± 0.2 |
| 25/75 Acalabrutinib/HPMCAS-H | 114.1 ± 0.9 |
| 50/50 Acalabrutinib/HPMCAS-L | 119.0 ± 0.0 |
| 50/50 Acalabrutinib/HPMCAS-M | 118.0 ± 0.0 |
| 50/50 Acalabrutinib/HPMCAS-H | 116.0 ± 1.0 |
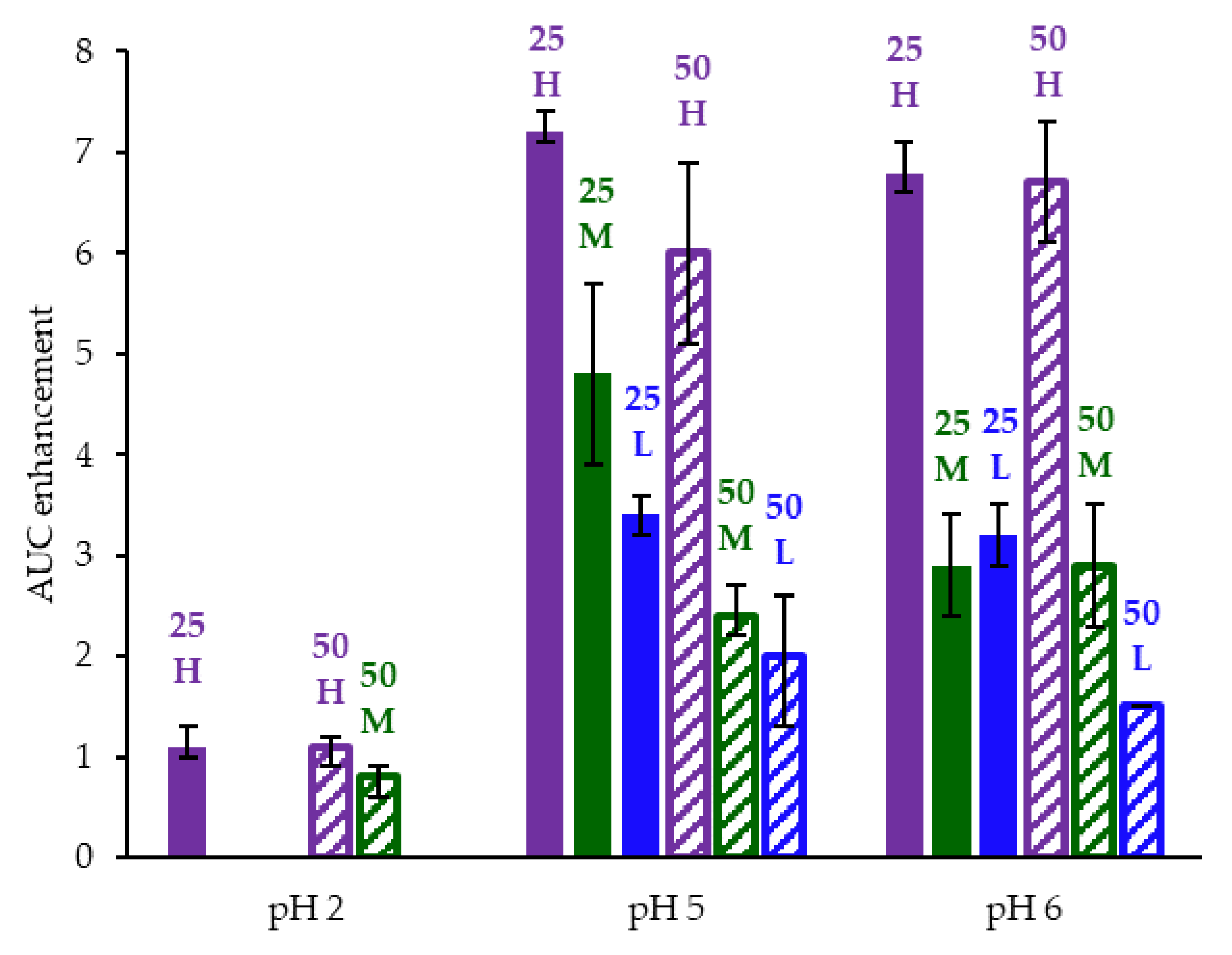
Appendix A.8. In Vitro Dissolution Testing of ASD Intermediates Tabulated Results
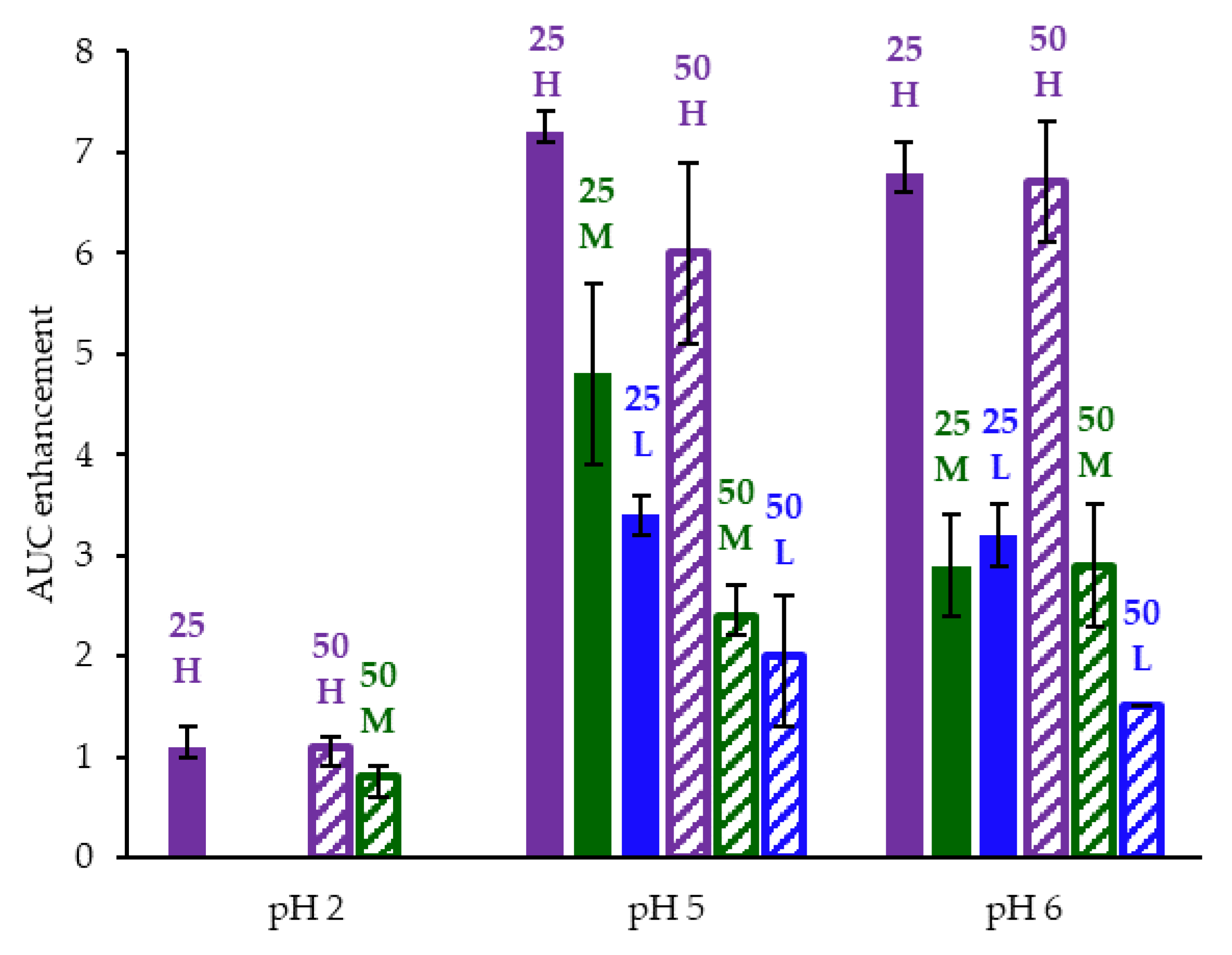
| ASD | AUC Enhancement a Gastric pH = 5 | AUC Enhancement a Gastric pH = 6 | AUC Enhancement a Gastric pH = 2 |
|---|---|---|---|
| 25/75 acalabrutinib/HPMCAS-H | 7.2 (7.1–7.4) | 6.8 (6.6–7.1) | 1.1 (1.0–1.3) |
| 25/75 acalabrutinib /HPMCAS-M | 4.8 (3.9–5.7) | 2.9 (2.4–3.4) | |
| 25/75 acalabrutinib /HPMCAS-L | 3.4 (3.2–3.6) | 3.2 (2.9–3.5) | |
| 50/50 acalabrutinib /HPMCAS-H | 6.0 (5.1–6.9) | 6.7 (6.1–7.3) | 1.1 (0.9–1.2) |
| 50/50 acalabrutinib /HPMCAS-M | 2.4 (2.2–2.7) | 2.9 (2.3–3.5) | 0.8 (0.6–0.9) |
| 50/50 acalabrutinib /HPMCAS-L | 2.0 (1.3–2.6) | 1.5 (1.5–1.5) |
References
- Brown, D.G.; Wobst, H.J. A decade of FDA-approved drugs (2010–2019): Trends and future directions. J. Med. Chem. 2021, 64, 2312–2338. [Google Scholar] [CrossRef]
- Sawicki, E.; Schellens, J.H.; Beijnen, J.H.; Nuijen, B. Inventory of oral anticancer agents: Pharmaceutical formulation aspects with focus on the solid dispersion technique. Cancer Treat. Rev. 2016, 50, 247–263. [Google Scholar] [CrossRef] [Green Version]
- Patel, D.; Bertz, R.; Ren, S.; Boulton, D.W.; Någård, M. A systematic review of gastric acid-reducing agent-mediated drug–drug interactions with orally administered medications. Clin. Pharmacokinet. 2020, 59, 447–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deisseroth, A.B. Calquence FDA Label; U.S. Food and Drug Administration: Silver Spring, MD, USA, 2019; Volume 2020. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/210259s006s007lbl.pdf (accessed on 15 March 2021).
- Smelick, G.S.; Heffron, T.P.; Chu, L.; Dean, B.; West, D.A.; DuVall, S.L.; Lum, B.L.; Budha, N.; Holden, S.N.; Benet, L.Z.; et al. Prevalence of acid-reducing agents (ara) in cancer populations and ara drug–drug interaction potential for molecular targeted agents in clinical development. Mol. Pharm. 2013, 10, 4055–4062. [Google Scholar] [CrossRef] [PubMed]
- Pepin, X.J.H.; Moir, A.J.; Mann, J.C.; Sanderson, N.J.; Barker, R.; Meehan, E.; Plumb, A.P.; Bailey, G.R.; Murphy, D.S.; Krejsa, C.M.; et al. Bridging in vitro dissolution and in vivo exposure for acalabrutinib. Part ii. A mechanistic pbpk model for ir formulation comparison, proton pump inhibitor drug interactions, and administration with acidic juices. Eur. J. Pharm. Biopharm. 2019, 142, 435–448. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, F.; Lee, S.C.; Zhao, H.; Zhang, L. Ph-dependent drug–drug interactions for weak base drugs: Potential implications for new drug development. Clin. Pharmacol. Ther. 2014, 96, 266–277. [Google Scholar] [CrossRef]
- Mudie, D.M.; Stewart, A.M.; Rosales, J.A.; Biswas, N.; Adam, M.S.; Smith, A.; Craig, C.D.; Morgen, M.M.; Vodak, D.T. Amorphous solid dispersion tablets overcome acalabrutinib ph effect in dogs. Pharmaceutics 2021, 13, 557. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, A.A.T.; Silva, P.A.I.A.; Lopes, M.S.M.; Yen, C.T.; Ricardo, E.D.; Mutão, T.; Pimenta, J.R.; Machado, L.M.; Shimba, D.S.; Peixoto, R.D. Proton pump inhibitors and oncologic treatment efficacy: A practical review of the literature for oncologists. Curr. Oncol. 2021, 28, 783–799. [Google Scholar] [CrossRef] [PubMed]
- van Leeuwen, R.W.; van Gelder, T.; Mathijssen, R.H.; Jansman, F.G. Drug-drug interactions with tyrosine-kinase inhibitors: A clinical perspective. Lancet Oncol. 2014, 15, e315–e326. [Google Scholar] [CrossRef]
- Blatter, F.; Ingallinera, T.; Barf, T.; Aret, E.; Krejsa, C.; Evarts, J. Solid Forms and Formuations of (s)-4->-Amino-3-(l-(but-2-ynoyl)pyrrolidin-2-yl)imidazo[1,5-ajpyrazin-1-yl)-n-(pyridin-2-yl)benzamide. 2016. Available online: https://patentimages.storage.googleapis.com/5e/4d/3f/0e3e4fb9e1a906/WO2017002095A1.pdf (accessed on 1 March 2021).
- Almeida e Sousa, L.; Reutzel-Edens, S.M.; Stephenson, G.A.; Taylor, L.S. Assessment of the amorphous “solubility” of a group of diverse drugs using new experimental and theoretical approaches. Mol. Pharm. 2015, 12, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.; Yates, I.; Mudie, D.; Pivette, P.; Goodwin, A.; Sarmiento, A.; Winter, M.; Morgen, M.; Vodak, D. Mechanistic study of belinostat oral absorption from spray-dried dispersions. J. Pharm. Sci. 2019, 108, 326–336. [Google Scholar] [CrossRef] [Green Version]
- Friesen, D.T.; Shanker, R.; Crew, M.; Smithey, D.T.; Curatolo, W.J.; Nightingale, J.A.S. Hydroxypropyl methylcellulose acetate succinate-based spray-dried dispersions: An overview. Mol. Pharm. 2008, 5, 1003–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mudie, D.M.; Stewart, A.M.; Biswas, N.; Brodeur, T.J.; Shepard, K.B.; Smith, A.; Morgen, M.M.; Baumann, J.M.; Vodak, D.T. Novel high-drug-loaded amorphous dispersion tablets of posaconazole; in vivo and in vitro assessment. Mol. Pharm. 2020, 17, 4463–4472. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.M.; Grass, M.E.; Brodeur, T.J.; Goodwin, A.K.; Morgen, M.M.; Friesen, D.T.; Vodak, D.T. Impact of drug-rich colloids of itraconazole and hpmcas on membrane flux in vitro and oral bioavailability in rats. Mol. Pharm. 2017, 14, 2437–2449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosquera-Giraldo, L.I.; Borca, C.H.; Meng, X.; Edgar, K.J.; Slipchenko, L.V.; Taylor, L.S. Mechanistic design of chemically diverse polymers with applications in oral drug delivery. Biomacromolecules 2016, 17, 3659–3671. [Google Scholar] [CrossRef]
- Litou, C.; Vertzoni, M.; Goumas, C.; Vasdekis, V.; Xu, W.; Kesisoglou, F.; Reppas, C. Characteristics of the human upper gastrointestinal contents in the fasted state under hypo- and a-chlorhydric gastric conditions under conditions of typical drug–drug interaction studies. Pharm. Res. 2016, 33, 1399–1412. [Google Scholar] [CrossRef]
- Prichard, P.J.; Yeomans, N.D.; Mihaly, G.W.; Jones, D.B.; Buckle, P.J.; Smallwood, R.A.; Louis, W.J. Omeprazole: A study of its inhibition of gastric ph and oral pharmacokinetics after morning or evening dosage. Gastroenterology 1985, 88, 64–69. [Google Scholar] [CrossRef]
- Tutuian, R.; Katz, P.O.; Bochenek, W.; Castell, D.O. Dose-dependent control of intragastric ph by pantoprazole, 10, 20 or 40 mg, in healthy volunteers. Aliment. Pharmacol. Ther. 2002, 16, 829–836. [Google Scholar] [CrossRef]
- Fancher, R.M.; Zhang, H.; Sleczka, B.; Derbin, G.; Rockar, R.; Marathe, P. Development of a canine model to enable the preclinical assessment of ph-dependent absorption of test compounds. J. Pharm. Sci. 2011, 100, 2979–2988. [Google Scholar] [CrossRef]
- Koziolek, M.; Grimm, M.; Bollmann, T.; Schäfer, K.J.; Blattner, S.M.; Lotz, R.; Boeck, G.; Weitschies, W. Characterization of the gi transit conditions in beagle dogs with a telemetric motility capsule. Eur. J. Pharm. Biopharm. 2019, 136, 221–230. [Google Scholar] [CrossRef]
- Arndt, M.; Chokshi, H.; Tang, K.; Parrott, N.J.; Reppas, C.; Dressman, J.B. Dissolution media simulating the proximal canine gastrointestinal tract in the fasted state. Eur. J. Pharm. Biopharm. 2013, 84, 633–641. [Google Scholar] [CrossRef]
- Dressman, J.B. Comparison of canine and human gastrointestinal physiology. Pharm. Res. 1986, 3, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Smeets-Peeters, M.; Watson, T.; Minekus, M.; Havenaar, R. A review of the physiology of the canine digestive tract related to the development of in vitro systems. Nutr. Res. Rev. 1998, 11, 45–69. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Zhai, B.; Guo, H.; Wang, P.; Liu, Z.; Gu, H.; Ho, H.; Langguth, P.; Li, K.; Wang, C.; et al. In vivo measurement of gastric fluid volume in anesthetized dogs. J. Drug Deliv. Sci. Technol. 2020, 55, 101488. [Google Scholar] [CrossRef]
- Takano, R.; Sugano, K.; Higashida, A.; Hayashi, Y.; Machida, M.; Aso, Y.; Yamashita, S. Oral absorption of poorly water-soluble drugs: Computer simulation of fraction absorbed in humans from a miniscale dissolution test. Pharm. Res. 2006, 23, 1144–1156. [Google Scholar] [CrossRef]
- Serajuddin, A.T.; Jarowski, C.I. Effect of diffusion layer ph and solubility on the dissolution rate of pharmaceutical bases and their hydrochloride salts. I: Phenazopyridine. J. Pharm. Sci. 1985, 74, 142–147. [Google Scholar] [CrossRef]
- Pepin, X.J.H.; Sanderson, N.J.; Blanazs, A.; Grover, S.; Ingallinera, T.G.; Mann, J.C. Bridging in vitro dissolution and in vivo exposure for acalabrutinib. Part i. Mechanistic modelling of drug product dissolution to derive a p-psd for pbpk model input. Eur. J. Pharm. Biopharm. 2019, 142, 421–434. [Google Scholar] [CrossRef]
- Mudie, D.M.; Samiei, N.; Marshall, D.J.; Amidon, G.E.; Bergström, C.A.S. Selection of in vivo predictive dissolution media using drug substance and physiological properties. AAPS J. 2020, 22, 34. [Google Scholar] [CrossRef] [Green Version]
- Ueda, K.; Moseson, D.E.; Pathak, V.; Taylor, L.S. Effect of polymer species on maximum aqueous phase supersaturation revealed by quantitative nuclear magnetic resonance spectroscopy. Mol. Pharm. 2021, 18, 1344–1355. [Google Scholar] [CrossRef]
- Pepin, X.J.H.; Huckle, J.E.; Alluri, R.V.; Basu, S.; Dodd, S.; Parrott, N.; Emami Riedmaier, A. Understanding mechanisms of food effect and developing reliable pbpk models using a middle-out approach. AAPS J. 2021, 23, 12. [Google Scholar] [CrossRef]
- Matsumura, N.; Hayashi, S.; Akiyama, Y.; Ono, A.; Funaki, S.; Tamura, N.; Kimoto, T.; Jiko, M.; Haruna, Y.; Sarashina, A.; et al. Prediction characteristics of oral absorption simulation software evaluated using structurally diverse low-solubility drugs. J. Pharm. Sci. 2020, 109, 1403–1416. [Google Scholar] [CrossRef]
- Podoll, T.; Pearson, P.G.; Evarts, J.; Ingallinera, T.; Bibikova, E.; Sun, H.; Gohdes, M.; Cardinal, K.; Sanghvi, M.; Slatter, J.G. Bioavailability, biotransformation, and excretion of the covalent bruton tyrosine kinase inhibitor acalabrutinib in rats, dogs, and humans. Drug Metab. Dispos. 2019, 47, 145–154. [Google Scholar] [CrossRef]
- Gala, U.H.; Miller, D.A.; Williams, R.O. Harnessing the therapeutic potential of anticancer drugs through amorphous solid dispersions. Biochim. Biophys. Acta BBA Rev. Cancer 2020, 1873, 188319. [Google Scholar] [CrossRef]
- Chen, Y.; Pui, Y.; Chen, H.; Wang, S.; Serno, P.; Tonnis, W.; Chen, L.; Qian, F. Polymer-mediated drug supersaturation controlled by drug–polymer interactions persisting in an aqueous environment. Mol. Pharm. 2019, 16, 205–213. [Google Scholar] [CrossRef]
- Ueda, K.; Higashi, K.; Yamamoto, K.; Moribe, K. The effect of hpmcas functional groups on drug crystallization from the supersaturated state and dissolution improvement. Int. J. Pharm. 2014, 464, 205–213. [Google Scholar] [CrossRef]
- Kesisoglou, F.; Wang, M.; Galipeau, K.; Harmon, P.; Okoh, G.; Xu, W. Effect of amorphous nanoparticle size on bioavailability of anacetrapib in dogs. J. Pharm. Sci. 2019, 108, 2917–2925. [Google Scholar] [CrossRef]
- Mitra, A.; Zhu, W.; Kesisoglou, F. Physiologically based absorption modeling for amorphous solid dispersion formulations. Mol. Pharm. 2016, 13, 3206–3215. [Google Scholar] [CrossRef]
- Stewart, A.M.; Grass, M.E. Practical approach to modeling the impact of amorphous drug nanoparticles on the oral absorption of poorly soluble drugs. Mol. Pharm. 2020, 17, 180–189. [Google Scholar] [CrossRef] [Green Version]
| Drug Loading (wt %) | Dispersion Polymer | Spray Solvent | Total Dissolved Drug and Polymer in Spray Solvent (wt %) |
|---|---|---|---|
| 25 | HPMCAS-H | Methanol | 6 |
| 25 | HPMCAS-M | Methanol | 6 |
| 25 | HPMCAS-L | Methanol | 6 |
| 50 | HPMCAS-H | Methanol | 5.4 |
| 50 | HPMCAS-M | Methanol | 5.4 |
| 50 | HPMCAS-L | Methanol | 5.4 |
| Starting pH | Slurry (Ending) pH | Amorphous Acalabrutinib Solubility (mg/mL) a |
|---|---|---|
| 1.6 | 4.4 | - |
| 2.0 | 4.6 | - |
| 3.0 | 5.4 | - |
| 4.0 | 6.2 | 6.47 (6.43–6.51) |
| 4.5 | - | 2.62 (2.48–2.76) |
| 5.0 | 6.3 | 0.81 (0.80–0.82) |
| 6.0 | 6.3 | 0.43 (0.43–0.44) |
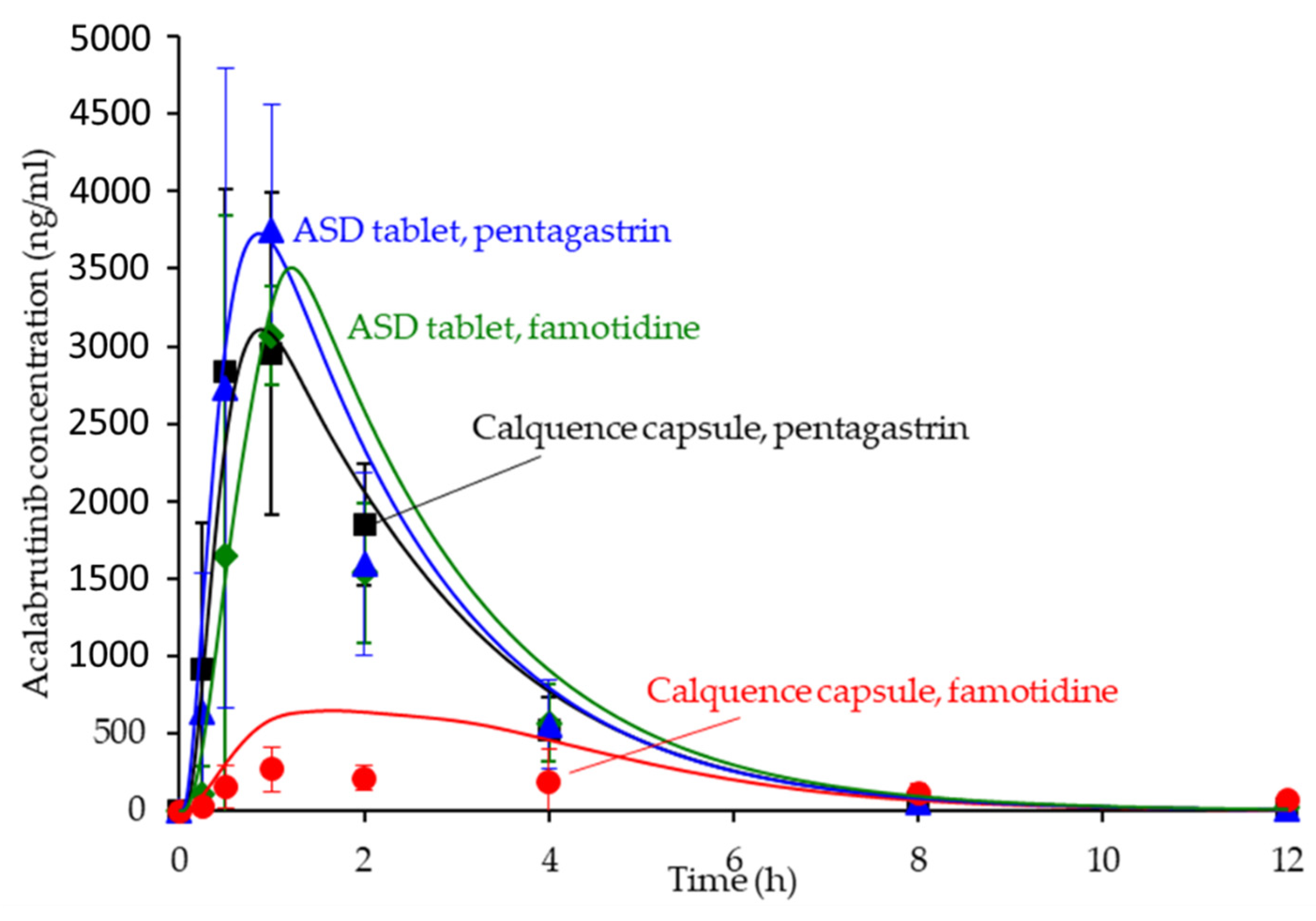
| Formulation | AUC0-inf (ng h/mL) | Cmax (ng/mL) | Tmax (h) | AAFE | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Obs | Sim | Obs | Sim | Obs | Sim | AUC0-inf | Cmax | Tmax | Cp vs. Time | |
| ASD tablet, pentagastrin | 8161 | 9766 | 3332 | 3727 | 0.9 | 0.9 | 1.2 | 1.2 | 1.6 | 1.3 |
| ASD tablet, famotidine | 7579 | 9555 | 3443 | 3508 | 0.9 | 1.6 | 1.3 | 1.2 | 1.8 | 1.6 |
| Calquence capsule, pentagastrin | 8365 | 8607 | 4480 | 3110 | 0.8 | 0.9 | 1.1 | 1.4 | 1.3 | 1.3 |
| Calquence capsule, famotidine | 3112 | 3096 | 355 | 648 | 1.6 | 1.2 | 1.6 | 1.9 | 1.7 | 3.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mudie, D.M.; Stewart, A.M.; Rosales, J.A.; Adam, M.S.; Morgen, M.M.; Vodak, D.T. In Vitro-In Silico Tools for Streamlined Development of Acalabrutinib Amorphous Solid Dispersion Tablets. Pharmaceutics 2021, 13, 1257. https://doi.org/10.3390/pharmaceutics13081257
Mudie DM, Stewart AM, Rosales JA, Adam MS, Morgen MM, Vodak DT. In Vitro-In Silico Tools for Streamlined Development of Acalabrutinib Amorphous Solid Dispersion Tablets. Pharmaceutics. 2021; 13(8):1257. https://doi.org/10.3390/pharmaceutics13081257
Chicago/Turabian StyleMudie, Deanna M., Aaron M. Stewart, Jesus A. Rosales, Molly S. Adam, Michael M. Morgen, and David T. Vodak. 2021. "In Vitro-In Silico Tools for Streamlined Development of Acalabrutinib Amorphous Solid Dispersion Tablets" Pharmaceutics 13, no. 8: 1257. https://doi.org/10.3390/pharmaceutics13081257
APA StyleMudie, D. M., Stewart, A. M., Rosales, J. A., Adam, M. S., Morgen, M. M., & Vodak, D. T. (2021). In Vitro-In Silico Tools for Streamlined Development of Acalabrutinib Amorphous Solid Dispersion Tablets. Pharmaceutics, 13(8), 1257. https://doi.org/10.3390/pharmaceutics13081257