
Pulmonary Drug Delivery of Antimicrobials and Anticancer Drugs Using Solid Dispersions



Abstract
1. Introduction
Physiology of the Lungs and Factors Affecting Particles Deposition
2. The Necessity to Deliver Larger Doses to Treat Lung Infections and Cancer
3. Challenges Associated with Drug Delivery to the Lungs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
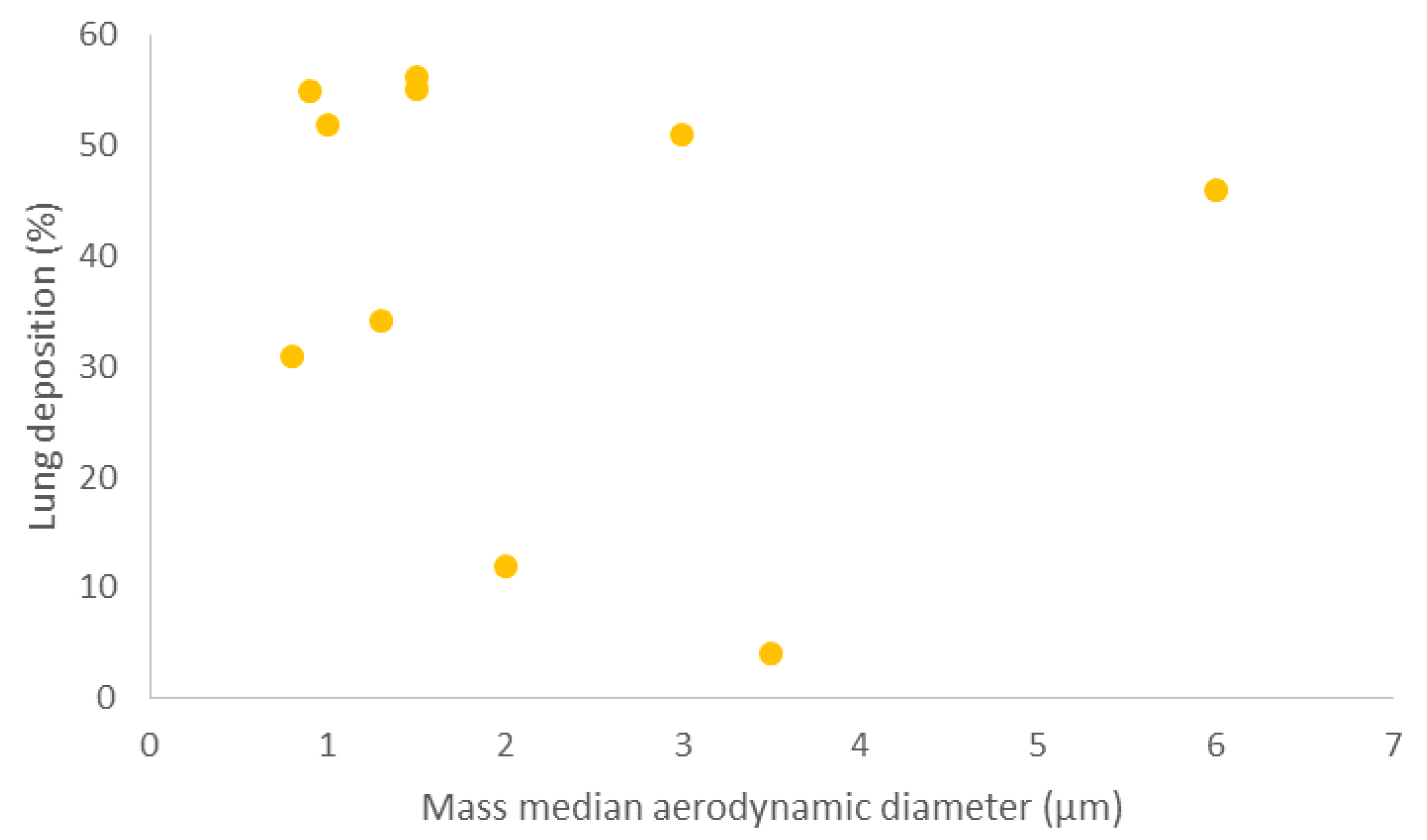
| API | MMAD (μm) | Lung Deposition (%) | Preparation of API | Ref |
|---|---|---|---|---|
| Formoterol | 0.8 | 31 ± 11 | Labelled with technetium-99 and dissolved in hydrofluoroalkane (HFA) in an pMDI | [64] |
| Beclomethasone dipropionate | 0.9 | 53 ± 7 | Labelled with technetium-99 and dissolved in HFA in an pMDI | [63] |
| Fluticasone propionate | 2 | 12 ± 7 | Labelled with technetium-99 and dissolved in chlorofluorocarbon (CFC) in an pMDI | |
| Beclomethasone dipropionate | 3.5 | 4 ± 11 | ||
| Albuterol (salbutamol) | 1.5 | 56.3 ± 9.2 | Labelled with technetium-99 in an pMDI | [62] |
| 3 | 51 ± 8.9 | |||
| 6 | 46 ± 13.6 | |||
| Beclomethasone dipropionate and formoterol | 1.3 | 34.08 ± 9.3 | Labelled with technetium-99 and dissolved in HFA in a pMDI | [60] |
| Ciclesonide | 1 | 52 ± 11 | Labelled with technetium-99 and dissolved in HFA in a pMDI | [65] |
| Beclomethasone dipropionate and formoterol fumarate | 1.5 | 55.2 ± 3.7 | Labelled with technetium-99 in a NEXThaler® DPI | [61] |
4. Impact of Drug Delivery Devices on the Extent of Pulmonary Drug Delivery
5. Pulmonary Drug Delivery Using Carrier Free Technology
6. Pulmonary Drug Delivery on the Nanoscale
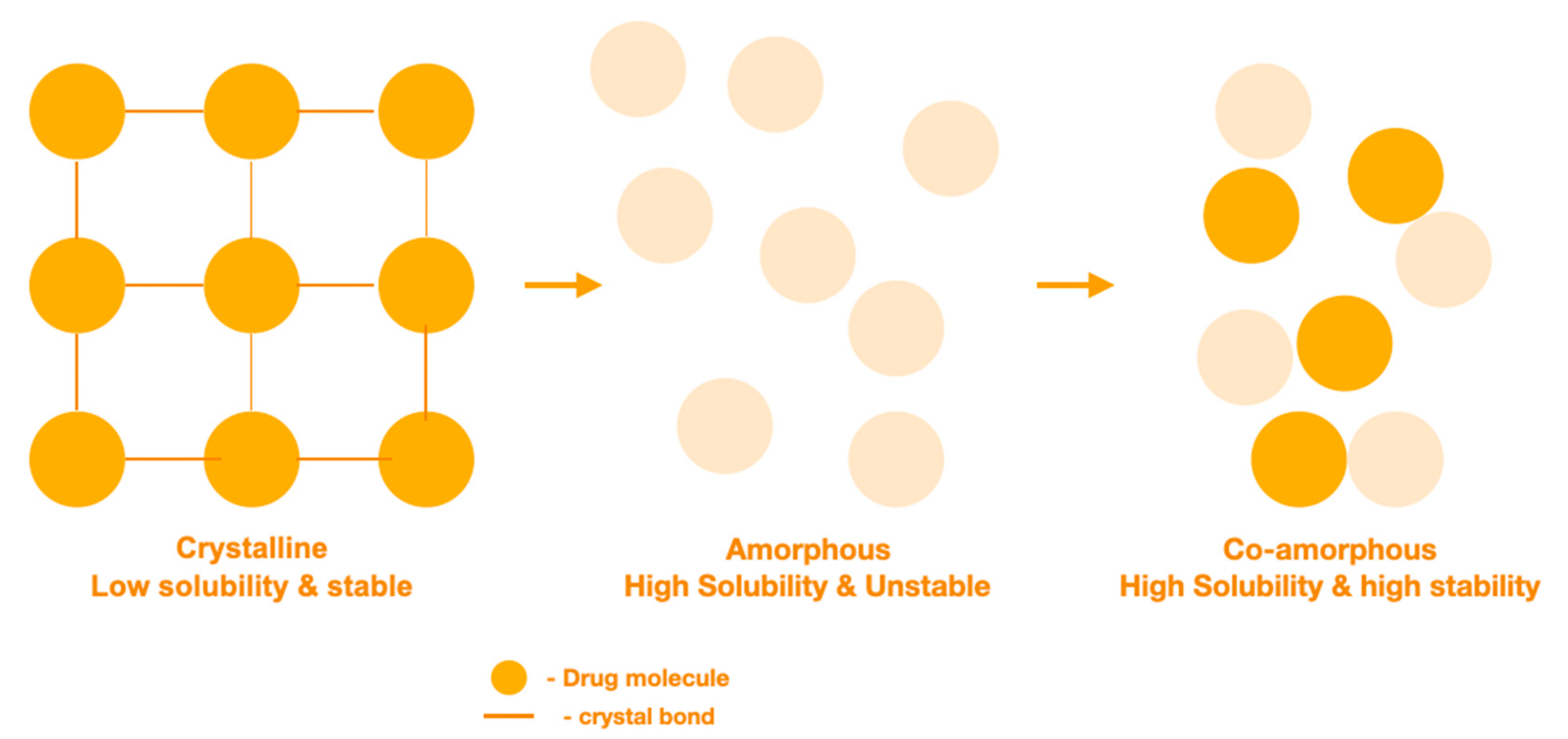
7. The Design of Carrier Free Formulations Using Coamorphous Solid Dispersions (CACDs)
8. Examples of Coformers as Components of CAMs
9. The Design of Carrier Free Formulations Using Cocrystals
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Moss, N.H.; Thompson, S.D.; Bauer, H.C.; Clark, J.J.; Clark, W.R. Diseases of the Respiratory System. New Complet. Med. Health Encycl. 1997, 2, 474–476. [Google Scholar]
- Rupp, J.; Droemann, D.; Goldmann, T.; Zabel, P.; Solbach, W.; Vollmer, E.; Branscheid, D.; Dalhoff, K.; Maass, M. Alveolar epithelial cells type II are major target cells for C. pneumoniae in chronic but not in acute respiratory infection. FEMS Immunol. Med. Microbiol. 2004, 41, 197–203. [Google Scholar] [CrossRef]
- Goyal, K.A.; Garg, T.; Bhandari, S.; Rath, G. Advancement in pulmonary drug delivery systems for treatment of tuberculosis. In Nanostructures for Drug Delivery; Andronescu, E., Grumezescu, M.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 669–695. [Google Scholar]
- Smith, I. Mycobacterium tuberculosis pathogenesis and molecular determinants of virulence. Clin. Microbiol. Rev. 2003, 16, 463–496. [Google Scholar] [CrossRef] [PubMed]
- Forum of International Respiratory Societies. Global Impact of Respiratory Disease; Forum of International Respiratory Societies: Sheffield, UK, 2017; pp. 12–26. [Google Scholar]
- Goel, A.; Baboota, S.; Sahni, J.K.; Ali, J. Exploring targeted pulmonary delivery for treatment of lung cancer. Int. J. Pharm. Investig. 2013, 3, 8–14. [Google Scholar] [CrossRef]
- Pearce, A.; Haas, M.; Viney, R.; Pearson, S.A.; Haywood, P.; Brown, C.; Ward, R. Incidence and severity of self-reported chemotherapy side effects in routine care: A prospective cohort study. PLoS ONE 2017, 12, e0184360. [Google Scholar] [CrossRef]
- Nurgali, K.; Jagoe, R.T.; Abalo, R. Editorial: Adverse Effects of Cancer Chemotherapy: Anything New to Improve Tolerance and Reduce Sequelae? Front. Pharmacol. 2018, 9, 245. [Google Scholar] [CrossRef]
- Rangaraj, N.; Pailla, S.; Sampathi, S. Insight into pulmonary drug delivery: Mechanism of drug deposition to device characterization and regulatory requirements. Pulm. Pharmacol. Ther. 2019, 54, 1–21. [Google Scholar] [CrossRef]
- Chaurasiya, B.; Zhao, Y.-Y. Dry powder for pulmonary delivery: A comprehensive review. Pharmaceutics 2021, 13, 31. [Google Scholar] [CrossRef]
- Al-Obaidi, H.; Buckton, G. The characterization and dissolution properties of griseofulvin solid dispersions with HPMCAS. J. Pharm. Pharmacol. 2006, 1, A18–A19. [Google Scholar]
- Newman, S. Drug delivery to the lungs: Challenges and opportunities. Ther. Deliv. 2017, 8. [Google Scholar] [CrossRef]
- Islam, N.; Ferro, V. Recent Advances in Chitosan-Based Nanoparticulate Pulmonary Drug Delivery. Nanoscale 2016, 8, 14341–14358. [Google Scholar] [CrossRef]
- Luczak-Wozniak, K.; Dabrowska, M.; Domagala, I.; Miszczuk, M.; Lubanski, W.; Leszczynski, A.; Krenke, R. Mishandling of pMDI and DPI inhalers in asthma and COPD- repetitive and non-repetitive errors. Pulm. Pharmacol. Ther. 2018, 51, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Parumasivam, T.; Chang, R.; Abdelghany, S.; Ye, T.; Britton, W.; Chan, H. Dry powder inhalable formulations for anti-tubercular therapy. Adv. Drug Deliv. Rev. 2016, 102, 83–101. [Google Scholar] [CrossRef] [PubMed]
- Patwa, A.; Shah, A. Anatomy and physiology of respiratory system relevant to anaesthesia. Indian J. Anaesth 2015, 59, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Hakim, A.; Usmani, O.S. Structure of the Lower Respiratory Tract. In Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Carvalho, T.; Peters, J.; Williams, R.r. Influence of particle size on regional lung deposition- What evidence is there? Int. J. Pharm. 2011, 406, 1–10. [Google Scholar] [CrossRef]
- DeCarlo, P.; Slowik, J.; Worsnop, D.; Davidovits, P.; Jimenez, J. Particle Morphology and Density Characterization by Combined Mobility and Aerodynamic Diameter Measurements. Part 1: Theory. Aerosol Sci. Technol. 2004, 38, 1185–1205. [Google Scholar] [CrossRef]
- Laube, B.; Janssens, H.; de Jongh, F.; Devadason, S.; Dhand, R.; Diot, P.; Everard, M.; Horvath, I.; Navalesi, P.; Voshaar, T.; et al. What the pulmonary specialist should know about the new inhalation therapies. Eur. Respir. J. 2011, 37, 1308–1417. [Google Scholar] [CrossRef]
- Benke, E.; Farkas, Á.; Szabó-Révész, P.; Ambrus, R. Development of an Innovative, Carrier-Based Dry Powder Inhalation Formulation Containing Spray-Dried Meloxicam Potassium to Improve the In Vitro and In Silico Aerodynamic Properties. Pharmaceutics 2020, 12, 535. [Google Scholar] [CrossRef]
- Labiris, N.R.; Dolovich, M.B. Pulmonary drug delivery. Part I: Physiological factors affecting therapeutic effectiveness of aerosolized medications. Br. J. Clin. Pharmacol. 2003, 56, 588–599. [Google Scholar] [CrossRef]
- Verma, K.R.; Ibrahim, M.; Garcia-Contreras, L. Lung Anatomy and Physiology and Their Implications for Pulmonary Drug Delivery; Wiley: Chichester, UK, 2015; pp. 2–14. [Google Scholar]
- Kim, C.S.; Rosati, J.A. Comparison of Monodisperse and Polydisperse Aerosol Deposition in a Packed Bed. Available online: https://cfpub.epa.gov/si/si_public_record_Report.cfm?Lab=NHEERL&dirEntryID=62538 (accessed on 18 April 2021).
- Patton, J.S. Mechanisms of macromolecule absorption by the lungs. Adv. Drug Deliv. Rev. 1996, 19, 3–36. [Google Scholar] [CrossRef]
- Heyder, J. Deposition of inhaled particles in the human respiratory tract and consequences for regional targeting in respiratory drug delivery. Proc. Am. Thorac. Soc. 2004, 1, 315–320. [Google Scholar] [CrossRef]
- Cheng, Y.S. Mechanisms of pharmaceutical aerosol deposition in the respiratory tract. AAPS PharmSciTech 2014, 15, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Weers, J.; Clark, A. The Impact of Inspiratory Flow Rate on Drug Delivery to the Lungs with Dry Powder Inhalers. Pharm. Res. 2017, 34, 507–528. [Google Scholar] [CrossRef]
- Ibrahim, M.; Verma, R.; Garcia-Contreras, L. Inhalation drug delivery devices: Technology update. Med. Devices 2015, 8, 131–139. [Google Scholar] [CrossRef]
- Momin, M.A.M.; Tucker, I.G.; Das, S.C. High dose dry powder inhalers to overcome the challenges of tuberculosis treatment. Int. J. Pharm. 2018, 550, 398–417. [Google Scholar] [CrossRef] [PubMed]
- Sallam, A.S.; Nazzal, S.; AlKhatib, S.H.; Darwazeh, N. Quality by Design: Concept for Product Development of Dry-Powder Inhalers, 1st ed.; Wiley: Chichester, UK, 2015; pp. 322–324. [Google Scholar]
- Alhaddad, B.; Smith, F.; Robertson, T.; Watman, G.; Taylor, K. Patients’ practices and experiences of using nebuliser therapy in the management of COPD at home. BMJ Open Respir. Res. 2015, 2, e000076. [Google Scholar] [CrossRef]
- Desgrouas, M.; Ehrmann, S. Inhaled antibiotics during mechanical ventilation—Why it will work. Ann. Transl. Med. 2020, 9, 598. [Google Scholar] [CrossRef]
- NICE. Salbutamol. Available online: https://bnf.nice.org.uk/medicinal-forms/salbutamol.html (accessed on 5 November 2020).
- NICE. Fluticasone. Available online: https://bnf.nice.org.uk/drug/fluticasone.html (accessed on 4 November 2020).
- Kalil, A.; Metersky, M.; Klompas, M.; Muscedere, J.; Sweeney, D.; Palmer, L.; Napolitano, L.; O’Grady, N.; Bartlett, J.; Carratalà, J.; et al. Management of Adults with Hospital-Acquired and Ventilator-Associated Pneumonia: 2016 Clinical Practice Guidelines by the Infectious Diseases Society of America and the American Thoracic Society. Clin. Infect. Dis. 2016, 63, e61–e111. [Google Scholar] [CrossRef]
- NICE. Tiotropium. Available online: https://bnf.nice.org.uk/medicinal-forms/tiotropium.html (accessed on 5 November 2020).
- NICE. Nedocromil Sodium. Available online: https://bnf.nice.org.uk/drug/nedocromil-sodium.html#medicinalForms (accessed on 5 November 2020).
- NICE. Zanamivir. Available online: https://bnf.nice.org.uk/medicinal-forms/zanamivir.html (accessed on 5 November 2020).
- NICE. Mannitol. Available online: https://bnf.nice.org.uk/drug/mannitol.html#medicinalForms (accessed on 5 November 2020).
- NICE. Budesonide with Formoterol. Available online: https://bnf.nice.org.uk/drug/budesonide-with-formoterol.html (accessed on 28 March 2021).
- NICE. Ciclesonide. Available online: https://bnf.nice.org.uk/drug/ciclesonide.html (accessed on 28 March 2021).
- Rogers, D.F. Physiology of Airway Mucus Secretion and Pathophysiology of Hypersecretion. Respir. Care 2007, 52, 1134–1149. [Google Scholar]
- Paul, K. Haemophilus influenzae and the lung (Haemophilus and the lung). Clin. Transl. Med. 2012, 1, 10. [Google Scholar] [CrossRef]
- Bjarnsholt, T.; Jensen, P.; Fiandaca, M.; Pedersen, J.; Hansen, C.; Andersen, C.; Pressler, T.; Givskov, M.; Høiby, N. Pseudomonas aeruginosa biofilms in the respiratory tract of cystic fibrosis patients. Paediatr. Pulmonol. 2009, 44, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.; Nichols, B.; Edgar, K.; Murgia, X.; Loretz, B.; Lehr, C. Challenges and strategies in drug delivery systems for treatment of pulmonary infections. Eur. J. Pharm. Biopharm. 2019, 144, 110–124. [Google Scholar] [CrossRef] [PubMed]
- Cramer, N.; Wiehlmann, L.; Tümmler, B. Clonal epidemiology of Pseudomonas aeruginosa in cystic fibrosis. Int. J. Med. Microbiol. 2010, 300, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J. Colonization and infection of the respiratory tract: What do we know? Paediatr Child. Health 2004, 9, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Carpagnano, G.E.; Lacedonia, D.; Palladino, G.P.; Logrieco, G.; Crisetti, E.; Susca, A.; Logrieco, A.; Foschino-Barbaro, M.P. Aspergillus spp. colonization in exhaled breath condensate of lung cancer patients from Puglia Region of Italy. BMC Pulm Med. 2014, 14, 22. [Google Scholar] [CrossRef] [PubMed]
- Cancer Research UK. Survival. Available online: https://www.cancerresearchuk.org/about-cancer/lung-cancer/survival (accessed on 8 April 2021).
- Sardeli, C.; Zarogoulidis, P.; Kosmidis, C.; Amaniti, A.; Katsaounis, A.; Giannakidis, D.; Koulouris, C.; Hohenforst-Schmidt, W.; Huang, H.; Bai, C.; et al. Inhaled chemotherapy adverse effects: Mechanisms and protection methods. Lung Cancer Manag. 2020, 8, LMT19. [Google Scholar] [CrossRef]
- Garbuzenko, O.; Saad, M.; Pozharov, V.; Reuhl, K.; Mainelis, G.; Minko, T. Inhibition of lung tumor growth by complex pulmonary delivery of drugs with oligonucleotides as suppressors of cellular resistance. Proc. Natl. Acad. Sci. USA 2010, 107, 10737–10742. [Google Scholar] [CrossRef]
- Garbuzenko, O.; Mainelis, G.; Taratula, O.; Minko, T. Inhalation treatment of lung cancer: The influence of composition, size and shape of nanocarriers on their lung accumulation and retention. Cancer Biol. Med. 2014, 11, 44–55. [Google Scholar] [CrossRef]
- Lee, W.; Loo, C.; Ghadiri, M.; Leong, C.; Young, P.; Traini, D. The potential to treat lung cancer via inhalation of repurposed drugs. Adv. Drug Deliv. Rev. 2018, 133, 107–130. [Google Scholar] [CrossRef]
- Gandhimathi, C.; Venugopal, J.; Sundarrajan, S.; Sridhar, R.; Tay, S.; Ramakrishna, S.; Kumar, S. Breathable Medicine: Pulmonary Mode of Drug Delivery. J. Nanosci. Nanotechnol. 2015, 15, 2591–2604. [Google Scholar] [CrossRef]
- Reychler, G.; San Miguel-Pagola, M.; Aubriot, A.; Herrero-Cortina, B.; Lecocq, V.; Hesse, M.; Liistro, G.; Jamar, F. Targeted Lung Deposition from Nebulization Is Not Improved in the Lateral Decubitus Position in Healthy Volunteers. Respir. Care 2019, 64, 1537–1544. [Google Scholar] [CrossRef]
- Healy, A.M.; Amaro, M.I.; Paluch, K.J.; Tajber, L. Dry powders for oral inhalation free of lactose carrier particles. Adv. Drug Deliv. Rev. 2014, 75, 32–52. [Google Scholar] [CrossRef]
- Kaialy, W.; Martin, G.P.; Larhrib, H.; Ticehurst, M.D.; Kolosionek, E.; Nokhodchi, A. The influence of physical properties and morphology of crystallised lactose on delivery of salbutamol sulphate from dry powder inhalers. Colloids Surf. B Biointerfaces 2012, 89, 29–39. [Google Scholar] [CrossRef]
- Sibum, I.; Hagedoorn, P.; de Boer, A.; Frijlink, H.; Grasmeijer, F. Challenges for pulmonary delivery of high powder doses. Int. J. Pharm. 2018, 548. [Google Scholar] [CrossRef] [PubMed]
- De Backer, W.; Devolder, A.; Poli, G.; Acerbi, D.; Monno, R.; Herpich, C.; Sommerer, K.; Meyer, T.; Mariotti, F. Lung Deposition of BDP/Formoterol HFA pMDI in Healthy Volunteers, Asthmatic, and COPD Patients. J. Aerosol Med. Pulm. Drug Deliv. 2010, 23, 137–148. [Google Scholar] [CrossRef]
- Virchow, J.; Poli, G.; Herpich, C.; Kietzig, C.; Ehlich, H.; Braeutigam, D.; Sommerer, K.; Häussermann, S.; Mariotti, F. Lung Deposition of the Dry Powder Fixed Combination Beclometasone Dipropionate Plus Formoterol Fumarate Using NEXThaler Device in Healthy Subjects, Asthmatic Patients and, COPD Patients. J. Aerosol Med. Pulm. Drug Deliv. 2018, 31, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Usmani, O.; Biddiscombe, M.; Barnes, P. Regional Lung Deposition and Bronchodilator Response as a Function of β2-Agonist Particle Size. Am. J. Respir. Crit. Care Med. 2005, 172, 1497–1504. [Google Scholar] [CrossRef]
- Leach, C.; Davidson, P.; Hasselquist, B.; Boudreau, R. Lung Deposition of Hydrofluoroalkane-134a Beclomethasone is Greater Than That of Chlorofluorocarbon Fluticasone and Chlorofluorocarbon Beclomethasone: A Cross-Over Study in Healthy Volunteers. Chest 2002, 122, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Häussermann, S.; Acerbi, D.; Brand, P.; Herpich, C.; Poli, G.; Sommerer, K.; Meyer, T. Lung deposition of formerol HFA (Atimos/Forair) in healthy volunteers, asthmatic and COPD patients. J. Aerosol Med. 2007, 20, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Leach, C.; Bethke, T.; Boudreau, R.; Hasselquist, B.; Drollmann, A.; Davidson, P.; Wurst, W. Two-dimensional and three-dimensional imaging show ciclesonide has high lung deposition and peripheral distribution: A nonrandomized study in healthy volunteers. J. Aerosol Med. 2006, 19, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Javadzadeh, Y.; Yaqoubi, S. Therapeutic nanostructures for pulmonary drug delivery. In Nanostructures for Drug Delivery; Andronescu, E., Grumezescu, A.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 619–638. [Google Scholar]
- Peng, T.; Lin, S.; Niu, B.; Wang, X.; Huang, Y.; Zhang, X.; Li, G.; Pan, X.; Wu, C. Influence of physical properties of carrier on the performance of dry powder inhalers. Acta Pharm. Silica B 2016, 4, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Finlay, W. Nebulizers for Drug Delivery to the Lungs. Expert Opin. Drug Deliv. 2015, 12, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Hamed, K.; Debonnett, L. Tobramycin inhalation powder for the treatment of pulmonary Pseudomonas aeruginosa in patients with cystic fibrosis: A review based on clinical evidence. Ther. Adv. Respir. Dis. 2017, 11, 193–209. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.; Tan, T.; Nakamura, J.; Malcolmson, R.; Tarara, T.; Weers, J. Physical Characterization of Tobramycin Inhalation Powder. II. State Diagram of an Amorphous Engineered Particle Formulation. Mol. Pharm. 2017, 14, 1950–1960. [Google Scholar] [CrossRef]
- Emc. Tobi Podhaler 28 mg Inhalation Powder, Hard Capsules. Available online: https://www.medicines.org.uk/emc/product/4757 (accessed on 19 November 2020).
- Quon, B.; Goss, C.; Ramsey, B. Inhaled Antibiotics for Lower Airway Infections. Annu. Am. Thorac. Soc. 2014, 11, 425–434. [Google Scholar] [CrossRef]
- Miller, D.; Tan, T.; Tarara, T.; Nakamura, J.; Malcolmson, R.; Weers, J. Physical Characterization of Tobramycin Inhalation Powder. I. Rational Design of a Stable-Engineered Particle Formulation for Delivery to the Lungs. Mol. Pharm. 2015, 12. [Google Scholar] [CrossRef]
- Conole, D.; Keating, G. Colistimethate Sodium Dry Powder for Inhalation: A Review of Its Use in the Treatment of Chronic Pseudomonas aeruginosa Infection in Patients with Cystic Fibrosis. Drugs 2014, 74, 377–387. [Google Scholar] [CrossRef]
- Emc. Colobreathe. Available online: https://www.medicines.org.uk/emc/product/3063 (accessed on 19 November 2020).
- Schwarz, C. Colobreathe for the Treatment of Cystic Fibrosis-Associated Pulmonary Infections. Pulm. Ther. 2015, 1, 19–30. [Google Scholar] [CrossRef]
- Cai, X.; Yang, Y.; Xie, X.; Yu, F.; Yang, Y.; Yang, Z.; Zhang, T.; Mei, X. Preparation, characterization and pulmonary pharmacokinetics of a new inhalable zanamivir dry powder. Drug Deliv. 2016, 23, 1962–1971. [Google Scholar] [CrossRef][Green Version]
- NICE. Tobramycin. Available online: https://bnf.nice.org.uk/medicinal-forms/tobramycin.html (accessed on 5 November 2020).
- Emc. Colomycin 1 Million International Units (IU) Powder for Solution, Injection, Infusion or Inhalation. Available online: https://www.medicines.org.uk/emc/product/1094/smpc (accessed on 5 November 2020).
- NICE. Colistimethate Sodium. Available online: https://bnf.nice.org.uk/medicinal-forms/colistimethate-sodium.html (accessed on 5 November 2020).
- NICE. Aztreonam. Available online: https://bnf.nice.org.uk/medicinal-forms/aztreonam.html (accessed on 5 November 2020).
- Pharma, D. Dry Powder Inhalation. Available online: https://dfepharma.com/Excipients/Expertise/Dry-Powder-Inhalation (accessed on 31 March 2021).
- Demoly, P.; Hagedoorn, P.; De Boer, A.H.; Frijlink, H.W. The clinical relevance of dry powder inhaler performance for drug delivery. Respir. Med. 2014, 108, 1195–1203. [Google Scholar] [CrossRef]
- Tan, B.M.J.; Chan, L.W.; Heng, P.W.S. Improving Dry Powder Inhaler Performance by Surface Roughening of Lactose Carrier Particles. Pharm. Res. 2016, 33. [Google Scholar] [CrossRef]
- Lee, H.; Lee, H.; Kwon, Y.; Kim, J.; Rhee, Y.; Chon, J.; Park, E.; Kim, D.; Park, C. The role of lactose carrier on the powder behaviour and aerodynamic performance of bosentan microparticles for dry powder inhalation. Eur. J. Pharm. Sci. 2018, 117, 279–289. [Google Scholar] [CrossRef]
- Yeung, S.; Traini, D.; Tweedie, A.; Lewis, D.; Church, T.; Young, P. Limitations of high dose carrier based formulations. Int. J. Pharm. 2018, 544, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Pacławski, A.; Szlęk, J.; Lau, R.; Jachowicz, R.; Mendyk, A. Empirical modeling of the fine particle fraction for carrier-based pulmonary delivery formulations. Int J. Nanomed. 2015, 10, 801–810. [Google Scholar] [CrossRef]
- Ambrus, R.; Benke, E.; Farkas, Á.; Balásházy, I.; Szabó-Révész, P. Novel dry powder inhaler formulation containing antibiotic using combined technology to improve aerodynamic properties. Eur. J. Pharm. Sci. 2018, 123, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Ceschan, N.; Bucalá, V.; Mateos, M.; Smyth, H.; Ramírez-Rigo, M. Carrier free indomethacin microparticles for dry powder inhalation. Int. J. Pharm. 2018, 549, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Momin, M.; Sinha, S.; Tucker, I.; Das, S. Carrier-free combination dry powder inhaler formulation of ethionamide and moxifloxacin for treating drug resistant tuberculosis. Drug Dev. Ind. Pharm. 2019, 45, 1321–1331. [Google Scholar] [CrossRef] [PubMed]
- Azari, F.; Ghanbarzadeh, S.; Safdari, R.; Yaqoubi, S.; Adibkia, K.; Hamishehkar, H. Development of a Carrier Free Dry Powder Inhalation Formulation of Ketotifen for Pulmonary Drug Delivery. Drug Res. 2020, 70, 26–32. [Google Scholar] [CrossRef]
- Nguyen, T.; Yi, E.; Hwang, K.; Cho, C.; Park, C.; Kim, J.; Rhee, Y.; Park, E. Formulation and evaluation of carrier-free dry powder inhaler containing sildenafil. Drug Deliv. Transl. Res. 2019, 9, 319–333. [Google Scholar] [CrossRef]
- Yazdi, A.; Smyth, H. Carrier-free high-dose dry powder inhaler formulation of ibuprofen: Physicochemical characterization and in vitro aerodynamic performance. Int. J. Pharm. 2016, 511, 403–414. [Google Scholar] [CrossRef]
- Edwards, A.; Chambers, A. Comparison of a lactose-free formulation of sodium cromoglycate and sodium cromoglycate plus lactose in the treatment of asthma. Curr. Med. Res. Opin. 1989, 11, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Pilcer, G.; De Bueger, V.; Traina, K.; Traore, H.; Sebti, T.; Vanderbist, F.; Amighi, K. Carrier-free combination for dry powder inhalation of antibiotics in the treatment of lung infections in cystic fibrosis. Int. J. Pharm. 2013, 451, 112–120. [Google Scholar] [CrossRef]
- Raula, J.; Lahde, A.; Kauppinen, E. Aerosolization behaviour of carrier-free L-leucine coated salbutamol sulphate powders. Int. J. Pharm. 2009, 365, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Haghi, M.; van den Oetelaar, W.; Moir, L.; Zhu, B.; Phillips, G.; Crapper, J.; Young, P.; Traini, D. Inhalable tranexamic acid for haemoptysis treatment. Eur. J. Pharm. Biopharm. 2015, 93, 311–319. [Google Scholar] [CrossRef]
- Suzuki, É.; Amaro, M.; de Almeida, G.; Cabral, L.; Healy, A.; de Sousa, V. Development of a new formulation of roflumilast for pulmonary drug delivery to treat inflammatory lung conditions. Int. J. Pharm. 2018, 550, 89–99. [Google Scholar] [CrossRef]
- Zhu, B.; Padroni, M.; Colombo, G.; Phillips, G.; Crapper, J.; Young, P.; Traini, D. The development of a single-use, capsule-free multi-breath tobramycin dry powder inhaler for the treatment of cystic fibrosis. Int. J. Pharm. 2016, 514, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Chvatal, A.; Farkas, Á.; Balásházy, I.; Szabó-Révész, P.; Ambrus, R. Aerodynamic properties and in silico deposition of meloxicam potassium incorporated in a carrier-free DPI pulmonary system. Int. J. Pharm. 2017, 520, 70–78. [Google Scholar] [CrossRef]
- Shetty, N.; Ahn, P.; Park, H.; Bhujbal, S.; Zemlyanov, D.; Cavallaro, A.; Mangal, S.; Li, J.; Zhou, Q. Improved Physical Stability and Aerosolization of Inhalable Amorphous Ciprofloxacin Powder Formulations by Incorporating Synergistic Colistin. Mol. Pharm. 2018, 15, 4004–4020. [Google Scholar] [CrossRef]
- Cui, Y.; Zhang, X.; Wang, W.; Huang, Z.; Zhao, Z.; Wang, G.; Cai, S.; Jing, H.; Huang, Y.; Pan, X.; et al. Moisture-Resistant Co-Spray-Dried Netilmicin with l-Leucine as Dry Powder Inhalation for the Treatment of Respiratory Infections. Pharmaceutics 2018, 10, 252. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Dalvi, S.V.; Siril, P.F. Nanoparticle-Based Drugs and Formulations: Current Status and Emerging Applications. ACS Appl. Nano Mater. 2020, 3, 4944–4961. [Google Scholar] [CrossRef]
- Mangal, S.; Gao, W.; Li, T.; Zhou, Q. Pulmonary delivery of nanoparticle chemotherapy for the treatment of lung cancers: Challenges and opportunities. Acta Pharmacol. Sin. 2017, 38, 782–797. [Google Scholar] [CrossRef]
- Huang, Z.; Kłodzińska, S.N.; Wan, F.; Nielsen, H.M. Nanoparticle-mediated pulmonary drug delivery: State of the art towards efficient treatment of recalcitrant respiratory tract bacterial infections. Drug Deliv. Transl. Res. 2021, 11, 1634–1654. [Google Scholar] [CrossRef]
- Zhu, C.; Chen, J.; Yu, S.; Que, C.; Taylor, L.S.; Tan, W.; Wu, C.; Zhou, Q.T. Inhalable Nanocomposite Microparticles with Enhanced Dissolution and Superior Aerosol Performance. Mol. Pharm. 2020, 17, 3270–3280. [Google Scholar] [CrossRef]
- Roa, W.H.; Azarmi, S.; Al-Hallak, M.H.D.K.; Finlay, W.H.; Magliocco, A.M.; Löbenberg, R. Inhalable nanoparticles, a non-invasive approach to treat lung cancer in a mouse model. J. Control. Release 2011, 150, 49–55. [Google Scholar] [CrossRef]
- Azarmi, S.; Tao, X.; Chen, H.; Wang, Z.; Finlay, W.H.; Löbenberg, R.; Roa, W.H. Formulation and cytotoxicity of doxorubicin nanoparticles carried by dry powder aerosol particles. Int. J. Pharm. 2006, 319, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Indermun, S.; Govender, M.; Kumar, P.; Choonara, Y.E.; Pillay, V. Porous particulate platforms for enhanced pulmonary delivery of bioactives. In Targeting Chronic Inflammatory Lung Diseases Using Advanced Drug Delivery Systems; Elsevier: Amsterdam, The Netherlands, 2020; pp. 359–373. [Google Scholar]
- Chvatal, A.; Ambrus, R.; Party, P.; Katona, G.; Jójárt-Laczkovich, O.; Szabó-Révész, P.; Fattal, E.; Tsapis, N. Formulation and comparison of spray dried non-porous and large porous particles containing meloxicam for pulmonary drug delivery. Int. J. Pharm. 2019, 559, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Ogienko, A.G.; Bogdanova, E.G.; Trofimov, N.A.; Myz, S.A.; Ogienko, A.A.; Kolesov, B.A.; Yunoshev, A.S.; Zubikov, N.V.; Manakov, A.Y.; Boldyrev, V.V.; et al. Large porous particles for respiratory drug delivery. Glycine-based formulations. Eur. J. Pharm. Sci. 2017, 110, 148–156. [Google Scholar] [CrossRef]
- Al-Obaidi, H.; Majumder, M.; Bari, F. Amorphous and crystalline particulates: Challenges and perspectives in drug delivery. Curr. Pharm. Des. 2016, 23, 350–361. [Google Scholar] [CrossRef]
- Al-Obaidi, H.; Lawrence, M.J.; Buckton, G. Atypical effects of incorporated surfactants on stability and dissolution properties of amorphous polymeric dispersions. J. Pharm. Pharmacol. 2016, 68, 1373–1383. [Google Scholar] [CrossRef]
- Hancock, B.C.; Parks, M. What is the true solubility advantage for amorphous pharmaceuticals? Pharm. Res. 2000, 17, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Bhanderi, A.; Bari, F.; Al-Obaidi, H. Evaluation of the impact of surfactants on miscibility of griseofulvin in spray dried amorphous solid dispersions. J. Drug Deliv. Sci. Technol. 2021, 64, 102606. [Google Scholar] [CrossRef]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Polymeric Amorphous Solid Dispersions: A Review of Amorphization, Crystallization, Stabilization, Solid-State Characterization, and Aqueous Solubilization of Biopharmaceutical Classification System Class II Drugs. J. Pharm. Sci. 2016, 105, 2527–2544. [Google Scholar] [CrossRef]
- Grzybowska, K.; Paluch, M.; Wlodarczyk, P.; Grzybowski, A.; Kaminski, K.; Hawelek, L.; Zakowiecki, D.; Kasprzycka, A.; Jankowska-Sumara, I. Enhancement of amorphous celecoxib stability by mixing it with octaacetylmaltose: The molecular dynamics study. Mol. Pharmacol. 2012, 9, 894–904. [Google Scholar] [CrossRef]
- Al-Obaidi, H.; Brocchini, S.; Buckton, G. Anomalous properties of spray dried solid dispersions. J. Pharm. Sci. 2009, 98, 4724–4737. [Google Scholar] [CrossRef]
- Al-Obaidi, H.; Buckton, G. Evaluation of griseofulvin binary and ternary solid dispersions with HPMCAS. AAPS PharmSciTech 2009, 10, 1172–1177. [Google Scholar] [CrossRef]
- AboulFotouh, K.; Zhang, Y.; Maniruzzaman, M.; Williams, R.O.; Cui, Z. Amorphous solid dispersion dry powder for pulmonary drug delivery: Advantages and challenges. Int. J. Pharm. 2020, 587, 119711. [Google Scholar] [CrossRef]
- Karimi, K.; Katona, G.; Csóka, I.; Ambrus, R. Physicochemical stability and aerosolization performance of dry powder inhalation system containing ciprofloxacin hydrochloride. J. Pharm. Biomed. Anal. 2018, 148, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Moes, J.; Koolen, S.; Huitema, A.; Schellens, J.; Beijnen, J.; Nuijen, B. Development of an oral solid dispersion formulation for use in low-dose metronomic chemotherapy of paclitaxel. Eur. J. Pharm. Biopharm. 2013, 83, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Purohit, H.S.; Taylor, L.S. Phase Behavior of Ritonavir Amorphous Solid Dispersions during Hydration and Dissolution. Pharm. Res. 2017, 34, 2842–2861. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, R.; Waraya, H.; Hirakura, Y. Application of Co-Amorphous Technology for Improving the Physicochemical Properties of Amorphous Formulations. Mol. Pharm. 2019, 16, 2142–2152. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.; Zografi, G. Commentary: Considerations in the Measurement of Glass Transition Temperatures of Pharmaceutical Amorphous Solids. Am. Assoc. Pharm. Sci. 2019, 21, 26. [Google Scholar] [CrossRef]
- Wostry, M.; Plappert, H.; Grohganz, H. Preparation of Co-Amorphous Systems by Freeze-Drying. Pharmaceutics 2020, 12, 941. [Google Scholar] [CrossRef]
- Lim, A.W.; Löbmann, K.; Grohganz, H.; Rades, T.; Chieng, N. Investigation of physical properties and stability of indomethacin-cimetidine and naproxen-cimetidine co-amorphous systems prepared by quench cooling, coprecipitation and ball milling. J. Pharm. Pharmacol. 2016, 68, 36–45. [Google Scholar] [CrossRef]
- Karagianni, A.; Kachrimanis, K.; Nikolakakis, I. Co-Amorphous Solid Dispersions for Solubility and Absorption Improvement of Drugs: Composition, Preparation, Characterization and Formulations for Oral Delivery. Pharmaceutics 2018, 10, 98. [Google Scholar] [CrossRef]
- Lababidi, N.; Ofosu Kissi, E.; Elgaher, W.A.M.; Sigal, V.; Haupenthal, J.; Schwarz, B.C.; Hirsch, A.K.H.; Rades, T.; Schneider, M. Spray-drying of inhalable, multifunctional formulations for the treatment of biofilms formed in cystic fibrosis. J. Control. Release 2019, 314, 62–71. [Google Scholar] [CrossRef]
- Zhou, Q.T.; Loh, Z.H.; Yu, J.; Sun, S.P.; Gengenbach, T.; Denman, J.A.; Li, J.; Chan, H.K. How Much Surface Coating of Hydrophobic Azithromycin Is Sufficient to Prevent Moisture-Induced Decrease in Aerosolisation of Hygroscopic Amorphous Colistin Powder? Am. Assoc. Pharm. Sci. 2016, 18, 1213–1224. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, M.; Liu, T.; Gao, Z.; Ye, Q.; Tan, X.; Hou, Y.; Sun, J.; Wang, D.; He, Z. Co-amorphous solid dispersion systems of lacidipine-spironolactone with improved dissolution rate and enhanced physical stability. Asian J. Pharm. Sci. 2019, 14, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Mangal, S.; Nie, H.; Xu, R.; Guo, R.; Cavallaro, A.; Zemlyanov, D.; Zhou, Q.T. Physico-Chemical Properties, Aerosolization and Dissolution of Co-Spray Dried Azithromycin Particles with L-Leucine for Inhalation. Pharm. Res. 2018, 35. [Google Scholar] [CrossRef] [PubMed]
- Kasten, G.; Löbmann, K.; Grohganz, H.; Rades, T. Co-former selection for co-amorphous drug-amino acid formulations. Int. J. Pharm. 2019, 557, 366–373. [Google Scholar] [CrossRef]
- Su, M.; Xia, Y.; Shen, Y.; Heng, W.; Wei, Y.; Zhang, L.; Gao, Y.; Zhang, J.; Qian, S. A novel drug–drug coamorphous system without molecular interactions: Improve the physicochemical properties of tadalafil and repaglinide. RSC Adv. 2020, 10, 565–583. [Google Scholar] [CrossRef]
- Carvahlo, R.S.; Watts, B.A.; Peters, I.J.; Williams, O.R. Dry Powder Inhalation for Pulmonary Delivery: Recent Advances and Continuing Challenges; Wiley: Chichester, UK, 2015; pp. 36–48. [Google Scholar]
- Chen, Z.; Yang, K.; Huang, C.; Zhu, A.; Yu, L.; Qian, F. Surface Enrichment and Depletion of the Active Ingredient in Spray Dried Amorphous Solid Dispersions. Pharm. Res. 2018, 35, 38. [Google Scholar] [CrossRef]
- Al-Obaidi, H.; Ke, P.; Brocchini, S.; Buckton, G. Characterization and stability of ternary solid dispersions with PVP and PHPMA. Int. J. Pharm. 2011, 419, 20–27. [Google Scholar] [CrossRef]
- Vehring, R. Pharmaceutical particle engineering via spray drying. Pharm. Res. 2008, 25, 999–1022. [Google Scholar] [CrossRef]
- Onoue, S.; Sato, H.; Kawabata, Y.; Mizumoto, T.; Hashimoto, N.; Yamada, S. In vitro and in vivo characterization on amorphous solid dispersion of cyclosporine A for inhalation therapy. J. Control. Release 2009, 138, 16–23. [Google Scholar] [CrossRef]
- Jong, T.; Li, J.; Morton, D.A.; Zhou, Q.T.; Larson, I. Investigation of the Changes in Aerosolization Behavior Between the Jet-Milled and Spray-Dried Colistin Powders Through Surface Energy Characterization. J. Pharm. Sci. 2016, 105, 1156–1163. [Google Scholar] [CrossRef]
- Shetty, N.; Park, H.; Zemlyanov, D.; Mangal, S.; Bhujbal, S.; Zhou, Q.T. Influence of excipients on physical and aerosolization stability of spray dried high-dose powder formulations for inhalation. Int. J. Pharm. 2018, 544, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, Y.; Tang, X. Characterization of a new inhalable thymopentin formulation. Int. J. Pharm. 2009, 375, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Zijlstra, G.S.; Rijkeboer, M.; Jan van Drooge, D.; Sutter, M.; Jiskoot, W.; van de Weert, M.; Hinrichs, W.L.; Frijlink, H.W. Characterization of a cyclosporine solid dispersion for inhalation. AAPS J. 2007, 9, E190–E199. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-B.; Watts, A.B.; Peters, J.I.; Liu, S.; Batra, A.; Williams, R.O., 3rd. In vitro and in vivo performance of dry powder inhalation formulations: Comparison of particles prepared by thin film freezing and micronization. AAPS PharmSciTech 2014, 15, 981–993. [Google Scholar] [CrossRef]
- Watts, A.B.; Wang, Y.-B.; Johnston, K.P.; Williams, R.O. Respirable Low-Density Microparticles Formed In Situ from Aerosolized Brittle Matrices. Pharm. Res. 2013, 30, 813–825. [Google Scholar] [CrossRef]
- Momin, M.A.M.; Sinha, S.; Tucker, G.I.; Doyle, C.; Das, C.S. Dry powder formulation of kanamycin with enhanced aerosolization efficiency for drug-resistant tuberculosis. Int. J. Pharm. 2017, 528, 107–117. [Google Scholar] [CrossRef]
- McShane, P.J.; Weers, J.G.; Tarara, T.E.; Haynes, A.; Durbha, P.; Miller, D.P.; Mundry, T.; Operschall, E.; Elborn, J.S. Ciprofloxacin Dry Powder for Inhalation (ciprofloxacin DPI): Technical design and features of an efficient drug-device combination. Pulm. Pharmacol. Ther. 2018, 50, 72–79. [Google Scholar] [CrossRef]
- Mohammed, A.; Zurek, J.; Madueke, S.; Al-Kassimy, H.; Yaqoob, M.; Houacine, C.; Ferraz, A.; Kalgudi, R.; Zariwala, M.G.; Hawkins, N.; et al. Generation of High Dose Inhalable Effervescent Dispersions against Pseudomonas aeruginosa Biofilms. Pharm. Res. 2020, 37, 150. [Google Scholar] [CrossRef]
- Porowska, A.; Dosta, M.; Fries, L.; Gianfrancesco, A.; Heinrich, S.; Palzer, S. Predicting the surface composition of spray-dried particle by modelling component reorganisation in a drying droplet. Chem. Eng. Res. Des. 2016, 110, 131–140. [Google Scholar] [CrossRef]
- Li, X.; Vogt, F.G.; Hayes, D.; Mansour, H.M. Design, characterization, and aerosol dispersion performance modeling of advanced co-spray dried antibiotics with mannitol as respirable microparticles/nanoparticles for targeted pulmonary delivery as dry powder inhalers. Pharm. Drug Deliv. Pharm. Technol. 2014, 103, 2937–2949. [Google Scholar] [CrossRef] [PubMed]
- Padhi, B.K.; Chougule, M.B.; Misra, A. Optimization of formulation components and characterization of large respirable powders containing high therapeutic payload. Pharm. Dev. Technol. 2006, 11, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.; Farkas, D.; Longest, W.; Hindle, M. Characterization of excipient enhanced growth (EEG) tobramycin dry powder aerosol formulations. Int. J. Pharm. 2020, 591, 120027. [Google Scholar] [CrossRef]
- Longest, P.W.; Farkas, D.; Hassan, A.; Hindle, M. Computational Fluid Dynamics (CFD) Simulations of Spray Drying: Linking Drying Parameters with Experimental Aerosolization Performance. Pharm. Res. 2020, 37, 101. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Chan, H.K.; Gengenbach, T.; Denman, J.A. Protection of hydrophobic amino acids against moisture-induced deterioration in the aerosolization performance of highly hygroscopic spray-dried powders. Eur. J. Pharm. Biopharm. 2017, 119, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Maa, Y.F.; Costantino, H.R.; Nguyen, P.A.; Hsu, C.C. The effect of operating and formulation variables on the morphology of spray-dried protein particles. Pharm. Dev. Technol. 1997, 2, 213–223. [Google Scholar] [CrossRef]
- Guenette, E.; Barrett, A.; Kraus, D.; Brody, R.; Harding, L.; Magee, G. Understanding the effect of lactose particle size on the properties of DPI formulations using experimental design. Int. J. Pharm. 2009, 380, 80–88. [Google Scholar] [CrossRef]
- Tulane University. Mucolytics. Available online: https://tmedweb.tulane.edu/pharmwiki/doku.php/mucolytics (accessed on 14 April 2021).
- British National Formulary (Online). Cystic Fibrosis. Available online: https://bnf.nice.org.uk/treatment-summary/cystic-fibrosis.html (accessed on 18 April 2021).
- Lamy, B.; Tewes, F.; Serrano, D.R.; Lamarche, I.; Gobin, P.; Couet, W.; Healy, A.M.; Marchand, S. New aerosol formulation to control ciprofloxacin pulmonary concentration. J. Control. Release 2018, 271, 118–126. [Google Scholar] [CrossRef]
- Tewes, F.; Brillault, J.; Lamy, B.; O’Connell, P.; Olivier, J.-C.; Couet, W.; Healy, A.M. Ciprofloxacin-Loaded Inorganic–Organic Composite Microparticles To Treat Bacterial Lung Infection. Mol. Pharm. 2016, 13, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Barazesh, A.; Gilani, K.; Rouini, M.; Barghi, M.A. The effect of metal salts on aerosol performance of spray dried carrier-free formulations of levofloxacin. Daru 2020, 28, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.; Serrano, D.; Worku, Z.; Norris, B.; Healy, A. Production of cocrystals in an excipient matrix by spray drying. Int. J. Pharm. 2018, 536, 467–477. [Google Scholar] [CrossRef]
- Karashima, M.; Sano, N.; Yamamoto, S.; Arai, Y.; Yamamoto, K.; Amano, N.; Ikeda, Y. Enhanced pulmonary absorption of poorly soluble itraconazole by micronized cocrystal dry powder formulations. Eur. J. Pharm. Biopharm. Off. J. Arb. Pharm. Verfahr. EV 2017, 115, 65–72. [Google Scholar] [CrossRef]
- Emami, S.; Adibkia, K.; Barzegar-Jalali, M.; Siahi-Shadbad, M. Piroxicam cocrystals with phenolic coformers: Preparation, characterisation and dissolution properties. Pharm. Dev. Technol. 2018, 24, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Wicker, J.; Crowley, L.; Robshaw, O.; Little, E.; Stokes, S.; Cooper, R.; Lawrence, S. Will they cocrystallize? CrysEngComm 2017, 19, 5336–5340. [Google Scholar] [CrossRef]
- Fukte, S.; Wagh, M.; Rawat, S. Coformer selection: An important tool in cocrystal formation. Int. J. Pharm. Pharm. Sci. 2014, 6, 9–14. [Google Scholar]
- Cvetkovski, A.; Ferretti, V.; Bertolasi, V. New Pharmaceutical Salts Containing Pyridoxine. Acta Crystallogr. Sect. C Struct. Chem. 2017, 73, 1064–1070. [Google Scholar] [CrossRef]
- Chadha, R.; Bhalla, Y.; Nandan, A.; Chadha, K.; Karan, M. Chrysin Cocrystals: Characterization and Evaluation. J. Pharm. Biomed. Anal. 2017, 134, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Perlovich, G.L. Thermodynamic characteristics of cocrystal formation and melting points for rational design of pharmaceutical two-component systems. CrysEngComm 2015, 17, 7019–7028. [Google Scholar] [CrossRef]
- Panzade, P.; Shendarkar, G. Superior Solubility and Dissolution of Zaltoprofen via Pharmaceutical Cocrystals. Turk. J. Pharm. Sci. 2019, 16, 310–316. [Google Scholar] [CrossRef]
- Ogienko, A.G.; Myz, S.A.; Ogienko, A.A.; Nefedov, A.A.; Stoporev, A.S.; Mel’gunov, M.S.; Yunoshev, A.S.; Shakhtshneider, T.P.; Boldyrev, V.V.; Boldyreva, E.V. Cryosynthesis of Co-Crystals of Poorly Water-Soluble Pharmaceutical Compounds and Their Solid Dispersions with Polymers. The “Meloxicam–Succinic Acid” System as a Case Study. Cryst Growth Des. 2018, 18, 7401–7409. [Google Scholar] [CrossRef]
- Zellnitz, S.; Roblegg, E.; Pinto, J.; Fröhlich, E. Delivery of Dry Powders to the Lungs: Influence of Particle Attributes from a Biological and Technological Point of View. Curr. Drug Deliv. 2019, 16, 180–194. [Google Scholar] [CrossRef]
- Alhalaweh, A.; Kaialy, W.; Buckton, G.; Gill, H.; Nokhodchi, A.; Velaga, S.P. Theophylline cocrystals prepared by spray drying: Physicochemical properties and aerosolization performance. AAPS PharmSciTech 2013, 14, 265–276. [Google Scholar] [CrossRef]
- Tanaka, R.; Hattori, Y.; Otsuka, M.; Ashizawa, K. Application of spray freeze drying to theophylline-oxalic acid cocrystal engineering for inhaled dry powder technology. Drug Dev. Ind. Pharm. 2020, 46, 179–187. [Google Scholar] [CrossRef]
- Gautam, M.; Besan, M.; Pandit, D.; Mandal, S.; Chadha, R. Cocrystal of 5-Fluorouracil: Characterization and Evaluation of Biopharmaceutical Parameters. AAPS PharmSciTech 2019, 20, 149. [Google Scholar] [CrossRef]
- Tomar, S.; Chakraborti, S.; Jindal, A.; Grewal, M.; Chadha, R. Cocrystals of diacerein: Towards the development of improved biopharmaceutical parameters. Int. J. Pharm. 2020, 574, 118942. [Google Scholar] [CrossRef]
- Bhalla, Y.; Chadha, K.; Chadha, R.; Karan, M. Daidzein cocrystals: An opportunity to improve its biopharmaceutical parameters. Heliyon 2019, 5, e02669. [Google Scholar] [CrossRef]
- Shinozaki, T.; Ono, M.; Higashi, K.; Moribe, K. A Novel Drug-Drug Cocrystal of Levofloxacin and Metacetamol: Reduced Hygroscopicity and Improved Photostability of Levofloxacin. J. Pharm. Sci. 2019, 108, 2383–2390. [Google Scholar] [CrossRef]
- Wang, Q.; Xue, J.; Hong, Z.; Du, Y. Pharmaceutical Cocrystal Formation of Pyrazinamide with 3-Hydroxybenzoic Acid: A Terahertz and Raman Vibrational Spectroscopies Study. Molecules 2019, 24, 488. [Google Scholar] [CrossRef]
- do Amaral, L.; do Carmo, F.; Amaro, M.; de Sousa, V.; da Silva, L.; de Almeida, G.; Rodrigues, C.; Healy, A.; Cabral, L. Development and Characterization of Dapsone Cocrystal Prepared by Scalable Production Methods. AAPS PharmSciTech 2018, 19, 2687–2699. [Google Scholar] [CrossRef]
- Khan, E.; Shukla, A.; Jhariya, A.; Tandon, P.; Vangala, V. Nitrofurantoin-melamine monohydrate (cocrystal hydrate): Probing the role of H-bonding on the structure and properties using quantum chemical calculations and vibrational spectroscopy. Spectrochim. Acta Part. A Mol. Biomol. Spectrosc. 2019, 221, 117170. [Google Scholar] [CrossRef]
- Stavropoulos, K.; Johnston, S.; Zhang, Y.; Rao, B.; Hurrey, M.; Hurter, P.; Topp, E.; Kadiyala, I. Cocrystalline Solids of Telaprevir with Enhanced Oral Absorption. J. Pharm. Sci. 2015, 104, 3343–3350. [Google Scholar] [CrossRef]
- Ton, Q.; Egert, E. Cocrystals of the antibiotic trimethoprim with glutarimide and 3,3-dimethylglutarimide held together by three hydrogen bonds. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 75–79. [Google Scholar] [CrossRef]
- Haneef, J.; Arora, P.; Chadha, R. Implication of Coformer Structural Diversity on Cocrystallization Outcomes of Telmisartan with Improved Biopharmaceutical Performance. AAPS PharmSciTech 2019, 21, 10. [Google Scholar] [CrossRef] [PubMed]
- Serrano, D.; Persoons, T.; D’Arcy, D.; Galiana, C.; Dea-Ayuela, M.; Healy, A. Modelling and shadowgraph imaging of cocrystal dissolution and assessment of in vitro antimicrobial activity for sulfadimidine/4-aminosalicylic acid cocrystals. Eur. J. Pharm. Sci. 2016, 89, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Swapna, B.; Maddileti, D.; Nangia, A. Cocrystals of the Tuberculosis Drug Isoniazid: Polymorphism, Isostructurality, and Stability. Cryst. Growth Des. 2014, 14, 5991–6005. [Google Scholar] [CrossRef]
- Seo, J.; Hwang, K.; Lee, S.; Kim, D.; Park, E. Preparation and characterization of adefovir dipivoxil-stearic acid cocrystal with enhanced physicochemical properties. Pharm. Dev. Technol. 2017, 23, 890–899. [Google Scholar] [CrossRef]
- Cai, Q.; Xue, J.; Wang, Q.; Du, Y. Solid-state cocrystal formation between acyclovir and fumaric acid: Terahertz and Raman vibrational spectroscopic studies. Spectrochim. Acta Part. A Mol. Biomol. Spectrosc. 2017, 186, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Cheney, M.; Weyna, D.; Shan, N.; Hanna, M.; Wojtas, L.; Zaworotko, M. Coformer selection in pharmaceutical cocrystal development: A case study of a meloxicam aspirin cocrystal that exhibits enhanced solubility and pharmacokinetics. J. Pharm. Sci. 2011, 100, 2172–2181. [Google Scholar] [CrossRef] [PubMed]
- Ray, E.; Vaghasiya, K.; Sharma, A.; Shukla, R.; Khan, R.; Kumar, A.; Verma, R. Autophagy-Inducing Inhalable Co-crystal Formulation of Niclosamide-Nicotinamide for Lung Cancer Therapy. AAPS PharmSciTech 2020, 21, 260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, Q.; Zhang, Q.; Chen, C.; He, M.; Chen, Q.; Song, G.; Xuan, X.; Huang, X. From a binary salt to salt co-crystals of antibacterial agent lomefloxacin with improved solubility and bioavailability. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2015, 71, 437–446. [Google Scholar] [CrossRef]
- Liu, L.; Zou, D.; Zhang, Y.; Zhang, Q.; Feng, Y.; Guo, Y.; Liu, Y.; Zhang, X.; Cheng, G.; Wang, C.; et al. Pharmaceutical salts/cocrystals of enoxacin with dicarboxylic acids: Enhancing in vitro antibacterial activity of enoxacin by improving the solubility and permeability. Eur. J. Pharm. Biopharm. 2020, 154, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Abidi, S.; Azim, Y.; Khan, S.; Khan, A. Sulfaguanidine cocrystals: Synthesis, structural characterization and their antibacterial and hemolytic analysis. J. Pharm. Biomed. Anal. 2018, 149, 351–357. [Google Scholar] [CrossRef]




| Property Type | Parameter |
|---|---|
| Aerosol | Air/Particle velocity Mass median aerodynamic diameter Fine particle fraction |
| Particle | Bulk density Tap density Shape Charge Surface energy * Surface texture * Surface composition * |
| Physicochemical | Solubility Hygroscopicity |
| Drug | Quantity of API per Dose | Indication | Ref |
|---|---|---|---|
| Salbutamol | 100–200 μg | Asthma | [34] |
| Fluticasone propionate | 50–500 μg | Prophylaxis of asthma | [35] |
| Colistimethate sodium | 80–125 mg | Treatment of pneumonia | [36] |
| Tiotropium | 10–18 μg | Maintenance of COPD | [37] |
| Nedocromil sodium | 2 mg | Prophylaxis of asthma | [38] |
| Zanamivir | 5 mg | Treatment of influenza | [39] |
| Mannitol | 5–40 mg | Treatment of cystic fibrosis as add-on therapy to standard care | [40] |
| Budesonide with formoterol | 100–400 μg with 4.5–12 μg | Maintenance of asthma | [41] |
| Ciclesonide | 80–160 μg | Prophylaxis of asthma | [42] |
| Organism | Type | Location/Generation | Reference |
|---|---|---|---|
| Bacteria | Pseudomonas aeruginosa | Non-mucoid strain located mainly in the conduction airways Mucoid strain present throughout the respiratory zone | [47] |
| Staphylococcus aureus | Nasal cavity Generation 0 | [48] | |
| Mycobacterium tuberculosis Chlamydia pneumonia | Alveolar surfaces Macrophages in lungs Generation 20–22 Generation 21–23 Alveolar type 2 cells | [4] [2] | |
| Fungi | Aspergillus spp. | Terminal bronchioles, Terminal airways Generation 16–23 | [49] |
| Viruses | Herpes Simplex Virus | Oropharynx–Generation 0 | [48] |
| Antibacterial Drug | Form | Available Strength | Ref |
|---|---|---|---|
| Tobramycin | Nebuliser liquid | 300 mg/5 mL, 300 mg/4 mL, 170 mg/1.7 mL | [78] |
| Inhalation powder | 28 mg (1 dose = 4 × 28 mg inhalations) | ||
| Colistimethate sodium | Inhalation powder | 1,662,500 IU ≈ 125 mg | [79,80] |
| Powder for nebuliser solution | 1,000,000 IU ≈ 80 mg | ||
| Aztreonam | Powder and solvent for nebuliser solution | 75 mg | [81] |
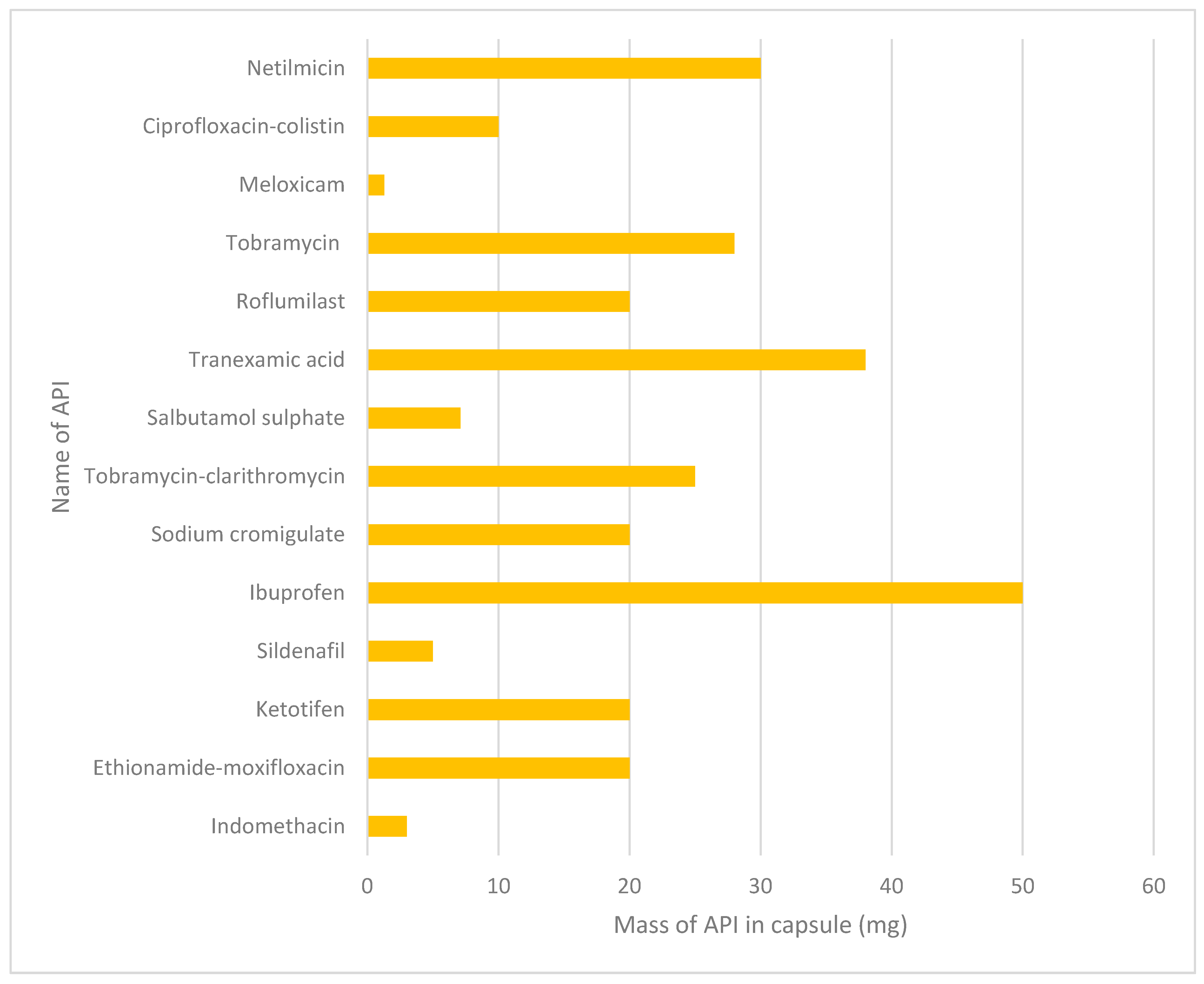
| API | Dose in Capsule | Conditions Used | Ref |
|---|---|---|---|
| Indomethacin | 3 mg | Spray drying an aqueous-based feed to form microparticles | [89] |
| Ethionamide + moxifloxacin | 20 mg | Spray drying using a mini spray dryer | [90] |
| Ketotifen | 20 mg | Spray drying with different solvents (water, ethanol and water-ethanol mix) | [91] |
| Sildenafil | 5 mg | Spray drying using a mini spray dryer | [92] |
| Ibuprofen | 50 mg | Air-jet milling to produce micronised samples | [93] |
| Sodium cromoglycate | 20 mg | Pelletised | [94] |
| Tobramycin + clarithromycin | 22.72 mg tobramycin, 2.27 mg clarithromycin | Spray drying | [95] |
| Salbutamol sulphate | 5.1–7.1 mg | Gas-phase coating method to produce L-leucine coated powders | [96] |
| Tranexamic acid | 38 mg | Spray drying | [97] |
| Roflumilast | 20 mg | Spray drying with hydroxypropyl-β-cyclodextrin | [98] |
| Tobramycin | 28 mg | Micronised using a Labomill jet milling system | [99] |
| Meloxicam potassium | 1.3 mg | Cospray drying | [100] |
| Ciprofloxacin + colistin | 10 mg | Cospray drying | [101] |
| Netilmicin | 30 mg | Cospray drying | [102] |
| API | Prime Excipient | Preparation Method | Fine Particle Fraction (FPF) (%) | Ref |
|---|---|---|---|---|
| Cyclosporin A (CsA) | Lactose, methylcellulose and erythritol | Jet-milling and freeze drying | 54 | [139] |
| Ciprofloxacin | Polyvinyl alcohol PVA | Spray drying | 25 ± 2.1 after 6 months | [121] |
| Ciprofloxacin | No excipient | Spray drying | 67.35 ± 1.1 after 6 months | [121] |
| Ciprofloxacin | Leucine | Spray drying | 79.78 ± 1.2 after 6 months | [121] |
| Ciprofloxacin | Hydroxypropyl-beta-cyclodextrin | Spray drying | 36.32 ± 1.3 after 6 months | [121] |
| Colistin | No excipient | Spray drying | 43.8 ± 4.6% | [140] |
| Colistin | No excipient | Jet milling | 28.4 ± 6.7 % | [140] |
| Colistin | L-leucine | Spray drying | 43.8 ± 4.6% (no difference from Spray dried alone) | [140] |
| Ciprofloxacin | No excipient | Spray drying | 28.0 ± 3.2% | [141] |
| Ciprofloxacin | Lactose, sucrose, trehalose, L-leucine | Spray drying | Lactose (43.5 ± 3.3%), sucrose (44.0 ± 4.3%), trehalose (44.0 ± 1.9%), L-leucine (73.5 ± 7.1%) | [141] |
| Thymopentin | Lactose/mannitol, Leucine, poloxamer 188 | Spray drying | 44.8%, 45.6%, 44.9%, 43.8% | [142] |
| CsA | Inulin | Spray freeze drying | >50 | [143] |
| Tacrolimus | Mannitol | Thin-film freeze-drying | 83.3 | [144] |
| Tacrolimus | Raffinose | Thin-film freeze-drying | 69.2 | [145] |
| Tacrolimus | Lactose | Thin-film freeze-drying | 68.7 | [145] |
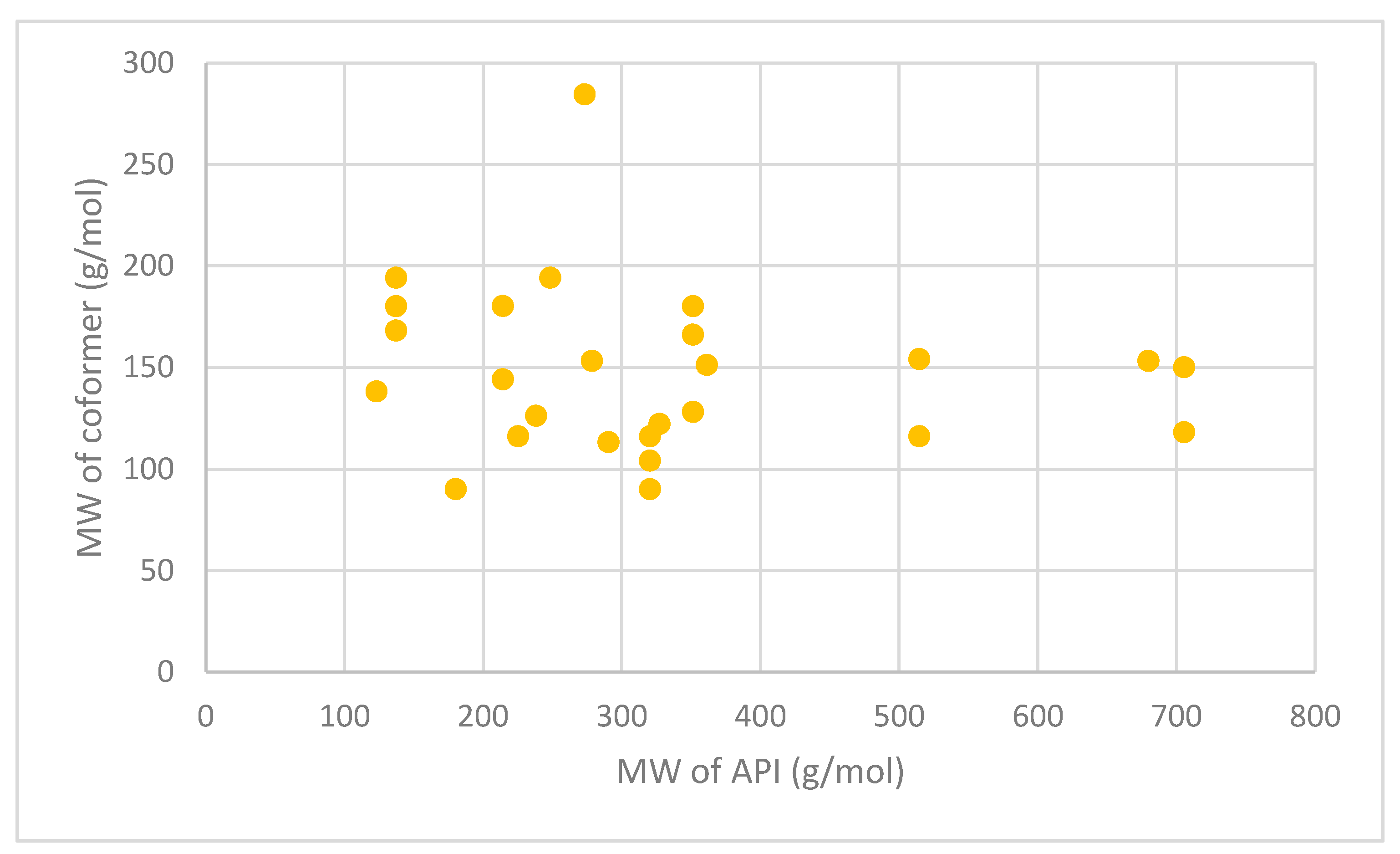
| API | Coformer | Technology Used | Molecular Weight (g/mol) | ||
|---|---|---|---|---|---|
| API | Coformer | Ref | |||
| Itraconazole | Succinic acid | Jet-milling | 705.63 | 118.09 | [163] |
| L-tartaric acid | 150.09 | ||||
| Levofloxacin | Metacetamol | Grinding and heating | 361.37 | 151.16 | [178] |
| Pyrazinamide | 3-Hydroxy benzoic acid | Slow evaporation and neat grinding | 123.11 | 138.12 | [179] |
| Dapsone | Caffeine | Slow evaporation, liquid-assisted grinding, spray drying | 248.30 | 194.19 | [180] |
| Nitrofurantoin | Melamine | Slow evaporation | 238.16 | 126.12 | [181] |
| Telaprevir | 4-aminosalicylic acid | Ball milling | 679.85 | 153.14 | [182] |
| Trimethoprim | Glutarimide | Slow evaporation | 290.32 | 113.11 | [183] |
| Telmisartan | Gentisic acid | Slurry approach | 514.62 | 154.12 | [184] |
| Maleic acid | 116.07 | ||||
| Sulfadimidine | 4-aminosalicylic acid | Liquid-assisted comilling | 278.33 | 153.14 | [185] |
| Isoniazid | Ferulic acid | Liquid-assisted grinding | 137.14 | 194.18 | [186] |
| Caffeic acid | 180.16 | ||||
| Vanillic acid | 168.15 | ||||
| Adefovir | Stearic acid | Antisolvent precipitation | 273.19 | 284.50 | [187] |
| Acyclovir | Fumaric acid | Liquid-assisted grinding | 225.20 | 116.07 | [188] |
| Meloxicam | Aspirin | Solution, slurry and solvent drop methods | 351.40 | 180.16 | [189] |
| Theophylline | Oxalic acid | Spray freeze drying | 180.16 | 90.03 | [174] |
| Niclosamide | Nicotinamide | Spray drying | 327.12 | 122.12 | [190] |
| Lomefloxacin | Barbituric acid | Slow evaporation | 351.35 | 128.09 | [191] |
| Isophthalic acid | 166.13 | ||||
| Enoxacin | Oxalic acid | Slow evaporation | 320.32 | 90.03 | [192] |
| Malonic acid | 104.06 | ||||
| Fumaric acid | 116.07 | ||||
| Sulfaguanidine | Thiobarbutaric acid | Slow evaporation | 214.24 | 144.15 | [193] |
| 1,10-phenanthroline | 180.20 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Obaidi, H.; Granger, A.; Hibbard, T.; Opesanwo, S. Pulmonary Drug Delivery of Antimicrobials and Anticancer Drugs Using Solid Dispersions. Pharmaceutics 2021, 13, 1056. https://doi.org/10.3390/pharmaceutics13071056
Al-Obaidi H, Granger A, Hibbard T, Opesanwo S. Pulmonary Drug Delivery of Antimicrobials and Anticancer Drugs Using Solid Dispersions. Pharmaceutics. 2021; 13(7):1056. https://doi.org/10.3390/pharmaceutics13071056
Chicago/Turabian StyleAl-Obaidi, Hisham, Amy Granger, Thomas Hibbard, and Sefinat Opesanwo. 2021. "Pulmonary Drug Delivery of Antimicrobials and Anticancer Drugs Using Solid Dispersions" Pharmaceutics 13, no. 7: 1056. https://doi.org/10.3390/pharmaceutics13071056
APA StyleAl-Obaidi, H., Granger, A., Hibbard, T., & Opesanwo, S. (2021). Pulmonary Drug Delivery of Antimicrobials and Anticancer Drugs Using Solid Dispersions. Pharmaceutics, 13(7), 1056. https://doi.org/10.3390/pharmaceutics13071056