3.1.1. Pre-Formulation Studies
Gaikwad et al. (2017) reported that within the pharmaceutical industry, HSPs are commonly used to determine the miscibility of an API with pharmaceutical excipients/carriers in SDs. It has also been reported that HSPs have been used within the pharmaceutical industry to determine the compatibility of pharmaceutical materials, and are therefore recommended in pre-formulation and in the development of SDs and tablets [
19].
Maniruzzaman et al. (2015) used the Flory–Huggins (F–H) lattice-based theory to explain intermolecular interactions between polymer and API is limited as it does not take into consideration the various multiple interactions in drug-polymer systems [
20]. Therefore, the HSPs developed by Van Krevelen and Hoftyzer group were used as a substitute for the Flory–Huggins theory to explore the nature of interactions that occur within drug–polymer systems and is mainly based upon the chemical structures of each of the pure components.
For an intermolecular interaction to occur between an API and polymer, the change in the free energy of mixing must be negative according to the laws of thermodynamics of mixing. This specific change in the free energy of mixing is related to enthalpy and entropy contributions according to the following equation (Equation (4)):
where Δ
Gmix, Δ
Hmix, Δ
Smix and
T are the Gibbs free energy, enthalpy of mixing, entropy of mixing and absolute temperature, respectively. The free energy change is spontaneous as a result of the increase in entropy of mixing and is therefore negative [
20]. However, the presence of repulsive and cohesive intermolecular and intramolecular forces (e.g., dispersion force, hydrogen bonding and dipole–dipole interaction) which are present within the amorphous SD, making the intermolecular interaction between drug and polymer more complex.
The drug–polymer interaction factor was calculated via the Hildebrand-Scott method which is a hypothetical method used to calculate F–H interaction parameters [
16]. It is well reported in the literature that unfavourable intermolecular interactions will result in phase separation and recrystallization if Δδ > 7 MPa
1/2, whilst favourable intermolecular interactions and a uniform phase will occur if Δδ < 7 MPa
1/2 [
21].
This is also the case for drug–polymer interactions if the value for χ is close to zero, as shown in
Table 2. Δδ and χ between INM and the polymers was less than 7 MPa
1/2 and close to zero respectively, therefore they are likely to be miscible [
16].
The molar attraction constant for INM was high (10.37 MPa
1/2) and was similar to the values calculated for the polymers (PVP VA64: 11.86 MPa
1/2 and PL-S630: 11.86 MPa
1/2) due to the interaction via hydrogen bonding. The molar attraction constant values are vital for intermolecular interactions between drug and polymer to occur during the transformation to the amorphous state. See
Table 3,
Table 4,
Table 5 and
Table 6 for HSPs calculations used to predict drug–polymer miscibility and drug–polymer interaction factor (χ).
Maniruzzaman et al. (2015) prepared amorphous solid dispersions using hot-melt extrusion using the BCS class II drug Verapamil HCL, which had a molar attraction constant of 6.95 MPa
1/2 and reported that due to the high molar attraction constants of the APIs and polymeric carriers, they were able to contribute to hydrogen bonding [
20]. As the HSPs are only theoretical, drug–polymer miscibility was further examined using various characterization, thermal analysis and solubility techniques to determine if the drug–polymer mixtures were miscible. In order for a drug and polymer to be miscible it requires the drug to be successfully converted to the amorphous form and to interact with the polymeric carrier. Conversion to the amorphous state was determined using XRPD and hyper DSC to not only determine if the drug is amorphous, but also to determine if only one glass transition temperature (
Tg) is present in the SD formulations. A single
Tg means successful integration of the drug into the polymer. Interaction between drug and polymer was examined using ATR-FTIR and Raman spectroscopy.
3.1.2. ATR-FTIR and Raman Studies
ATR-FTIR was used in conjunction with Raman studies to elucidate the mechanism of interaction between INM and each of the polymers which is necessary for drug–polymer miscibility. XRPD and hyper DSC was used to confirm the amorphous nature of INM in each of the SD formulations and to determine
Tg. PVP VA64 and PL-S630 showed two strong bands at 1731 cm
−1 and 1672 cm
−1 which correspond to the C=O stretch of the vinyl acetate and the C=O stretch of the amide carbonyl respectively. When INM was converted to the amorphous form via hot melt extrusion, the amide carbonyl C=O of PVP VA64 and PL-S630 shifted to 1680 cm
−1. This indicates that hydrogen bonding had taken place between the amide carbonyl of the polymer and the free C=O bond of the carboxylic acid of INM [
5]. This was the case for all SD formulations, as shown in
Figure 2a.
There was no change in the vinyl acetate C=O carbonyl due to weaker hydrogen bonding potential of the vinyl acetate carbonyl as reported by Yuan et al. (2015) as shown in
Figure 2b [
22]. Any P407 peaks within the pure P407 sample were completely absent in the ATR-FTIR spectra of the SD formulations as shown in
Figure 2a and
Figure 3. This indicates that P407 possibly has no intermolecular interaction with INM at the molecular level [
9].
Pure INM contains two strong non-hydrogen bonding C=O bands at 1690 cm
-1 (acid-acid dimer C=O stretch) and 1714 cm
−1 (free C=O of carboxylic acid) respectively [
23]. The hydrogen-bonded O–H stretch of the acid, shown in
Figure 4, is overlaid on the C–H sharp stretches, as literature has reported that the free carboxylic acid O–H stretch as a result of hydrogen bonding can exist as dimers [
23]. It is also important to note that in the ATR-FTIR reference spectrum of aINM, the two C=O bands at 1714 cm
−1 (free C=O of carboxylic acid) and 1690 cm
−1 (acid-acid dimer C=O stretch) shifted to 1707 cm
−1 and 1679 cm
−1, respectively, due to transformation to its amorphous form [
5], as shown in
Figure 2a, therefore do not align with the polymer peaks. The physical blends contained crystalline INM as expected as shown in
Figure 3 [
5].
Pharmaceutical drugs which are usually conjugated compounds have very strong Raman signals, while pharmaceutical excipients tend to have Raman signals which are weak [
8]. Like ATR-FTIR, Raman spectroscopy can identify potential mechanism of intermolecular interaction among the polymeric carriers and INM due to a shift in the PVP VA64 and PL-S630.
Hydrogen bonding was the predicted mechanism of interaction due to polarity of INM and polymeric carriers as a result of the relatively high INM molar attraction constant calculated using the HSPs. The amide carbonyl (
vC=O) peak at 1673 cm
−1 shifted to 1680 cm
−1 in all SD formulations due to hydrogen bond interaction with the –OH carboxylic acid of INM, as shown in
Figure 5. The intensity of the vinyl acetate carbonyl group is weak and appears at 1745 cm
-1 due to the weaker hydrogen bonding potential as reported by Yuan et al. (2015) [
22].
The acid
vC=O present at 1702 cm
−1 (free C=O of carboxylic acid) of INM disappears in the Raman spectra of the quaternary and ternary SD formulations due to the low intensity of INM. Based on the Raman spectra in
Figure 5, the peaks identified in the Raman spectra of aINM were not present in the Raman spectra of the amorphous SD formulations. The broad peak at 1482 cm
−1 in the Raman spectrum of solid-state P407 reflected the deformation vibration of the δCH
2 group.
The v(O–H) of P407 was present at 3200 cm−1 and v(C-O-C) was present at 850 cm−1. Careful analysis of the SD formulations indicates that the bands associated with P407 were not present in the SD formulations due to the lack of an intermolecular interaction. Raman spectroscopy similar to ATR-FTIR spectroscopy identified that hydrogen bonding occurred between PVP VA64/PL-S630 regardless of the cooling method used.
3.1.3. XRPD and Hyper DSC Studies
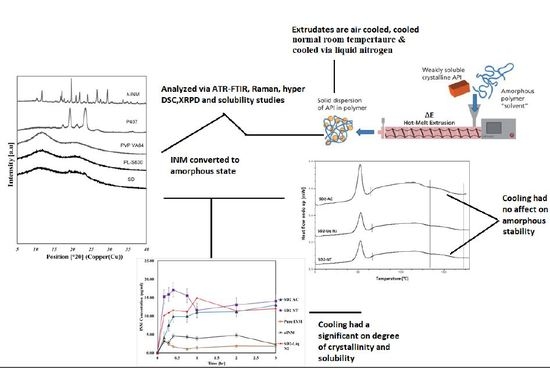
XRPD was used to examine the amorphous nature of the drug within the SD formulations. XRPD was performed on a PANalytical Empyrean X-ray diffractometer attached to a computer running High Score Plus to collect and process data. The XRPD of INM shows multiple Bragg peaks similar to the literature values reported for INM [
8], indicating the crystalline nature of the drug, as shown in
Figure 6a.
Figure 6a also shows the XRPD diffractograms of P407, PL-S630 and PVP VA64.
Figure 7 shows the XRPD diffractograms of the SD formulations used to investigate the effect of cooling.
The XRPD diffractograms of PL-S630 and PVP VA64 showed that they were amorphous in nature due to slight amorphous halo raised above the baseline. The presence of two Bragg peaks of P407 at 19 and 24° respectively indicates that P407 was not converted to the amorphous form as reported by Hurley et al. (2018) [
5]. The drug peaks associated with crystalline INM were completely absent in the melt extrudates with various drug–polymer ratios, indicating that INM was successfully converted to its amorphous form as shown in
Figure 7. Hyper DSC, like the XRPD diffractograms, also confirmed that INM was successfully converted to its amorphous form and P407 remained semi-crystalline.
Hyper DSC was performed to characterize the solid state of the SD formulations and to determine the
Tg of the SD formulations. The DSC thermograms of the pure components and SD formulations are shown in
Figure 6b,
Figure 8 and
Figure 9. The % crystallinity of P407 for each of the samples was also determined.
The thermogram of crystalline INM showed an endothermic peak at 165.47 °C corresponding to the melting endotherm of the stable γ INM form. Thermograms of all the SD formulations characterized showed the complete disappearance of the melting endotherm, indicating that INM was completely converted to its amorphous state, SD formulations existed as a two-phase system due to the presence of the semi-crystalline P407.
In the low-temperature region of the DSC scan, the glass transition temperature [
Tg] of aINM was detected at 45.5 °C [
2]. Pezzoli et al. (2019) also reported a
Tg of 45.5 °C for aINM when processed using HME [
2]. PVP VA64/PL-S630 were indeed amorphous with a
Tg at 109 and 107 °C respectively, as expected, based on the XRPD results.
A separate Tg attributable to INM was not present in the hyper DSC thermograms of the SD formulations, suggesting a complete absence of a separated phase rich in amorphous INM and a good level of mixing. P407 also displayed an endothermic peak at 58.16 °C due to its semi-crystalline structure with an enthalpy of fusion (ΔHm) of 117.1 J/g which corresponds to its crystalline fraction. Therefore, assuming that 100% semi-crystalline P407 has an enthalpy of fusion of 117.1 J/g, the degree of crystallinity of P407 within SD formulations was calculated. The P407 enthalpy of fusion (J/g) was calculated by dividing the area of the endothermic (melt) peak by the heating rate, which was 100°C/min. The % crystallinity of each of the SD formulations was calculated by dividing the P407 enthalpy of fusion (J/g) of each of the formulations by the theoretical enthalpy of fusion (J/g) of both the 5% and 15% P407 formulations.
For SD1-AC to SD6-AC, the P407 enthalpy of fusion (J/g) in
Table 7 was divided by 5.86 J/g for the 5% P407 formulations and 17.56 J/g for the 15% P407 formulations and multiplied by 100 to determine the % crystallinity in each of the formulations. For example, for SD1-AC, 0.593 J/g was divided by 5.86 J/g and multiplied by 100 to give 10.12 % P407 crystallinity.
Table 7 displays an overview of the degree of crystallinity exhibited by the various extrudates and glass transition temperatures (experimental vs predicted) relative to the pure crystalline drug. After careful analysis of the hyper DSC thermograms of the SD formulations, the endotherm attributed to the melting of the P407 fraction was detected in all formulations at 52.63 °C, with a mean ΔH
m of 0.862 J/g for the 5% P407 SD formulations and 6.68 J/g for the 15% P407 SD formulations. When calculated, ΔH
m of 100% crystalline P407 in 5% SD formulations was 5.86 J/g which corresponds to 14.7% crystalline P407. Similarly, 15% SD formulations contain in theory 38.0% P407.
Considering the concentration of plasticizer present in the amorphous SD formulations, it was estimated that the % crystallinity present in the 5% P407 formulations was within the range of 10.12% to 17.61% in its crystalline form and 27.33% to 54.39% for the 15% P407 formulations. It is important to note that the formulations with the higher P407 loadings had a degree of crystallinity that was significantly higher than formulations with low P407 loading since the APIs can be incorporated into the core of P407 micelle at high loads [
24].
It is important that cooling also had a significant effect on the degree of crystallinity. For example, SD2-NT had the highest % crystallinity, indicating that using a high P407 loading with a slow cooling process increases the % crystallinity. Whereas SD2-Liq N2 which was a rapid cooling method, had a low % crystallinity.
For all SD formulations, a single
Tg was observed which confirms that INM was converted to its amorphous form, indicating that the polymers and drug were present in a two-phase system as P407 was still semi-crystalline (see
Figure 8 and
Figure 9).
The experimental glass transition temperatures were significantly different to the predicted
Tgs calculated using the Couchman–Karasz equation, as shown in
Table 7, as drug–polymer loading can have a significant effect on the
Tg. The effect of drug and polymer loading on the glass transition of SD formulations was predicted using the model reported by Couchman and Karasz [
25,
26]. The theoretical glass transition of the SD formulations was calculated using the following Equation (5):
where
Tg1 and
Tg2, are the
Tgs of API and polymeric carriers, respectively,
X1 and
X2 correspond to the weight fractions of each of the pure materials and Δ
Cp1 and Δ
Cp2 corresponds to the change in the specific heat capacity at each of the glass transition temperature, respectively [
27].
This analysis confirmed that P407 was not solubilized and is present in its semi-crystalline form with some level of compatibility with PL-S630/PVP VA64-INM system due to the reduction of the enthalpy of fusion and melting temperature in the various SD formulations. This was consistent with higher Bragg peak intensities observed in the XRPD data from the 15% P407 sample with 30% drug loading.
The cooling method used had very little effect on the Tg. However, the degree of crystallinity was also affected by (1) the cooling method used (2) % P407 loading and (3) the polymeric carrier used. For example, SD4 had a higher % crystallinity compared to SD6. This is because PL-S630 and PVP VA64 possess different solubility properties with PVP VA64-INM SD formulations having a significantly greater than INM-PL-S630 SDs. There was a significant difference in the Tgs between SD4 and SD6 due to the different solid-state properties of PL-S630 and PVP VA64. However, the inclusion of P407 improved the processability and fragility of the extrudates, and also lowered the processing temperatures.
The HSPs alongside XRPD and hyper DSC confirmed that the drug and polymer were indeed miscible. XRPD and hyper DSC confirmed successful conversion of INM to the amorphous form as mentioned before, hyper DSC also confirmed that although P407 remained semi-crystalline, as only one single Tg was present in all SD formulations. According to Couchman and Kasasz, (1978) if a SD formulation only contains one Tg then the drug and polymer are indeed miscible. ATR-FTIR and Raman spectroscopy confirmed hydrogen bonding interaction between PVP VA64/PL-S630 and INM due to the interaction between the amide carbonyl C=O of the polymer and the free carboxylic acid C=O of INM. For successful conversion to the amorphous form interaction between drug and polymer is necessary.

 ,
, 


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


