Injectables and Depots to Prolong Drug Action of Proteins and Peptides

 ,
,  ,
,
Abstract

1. Introduction
2. General Challenges in Polypeptide Delivery
2.1. Aggregation
2.2. Pharmacokinetics
3. General Strategies to Increase Duration of Action
3.1. Half-Life Extension Strategies
3.1.1. Recycling
3.1.2. Increasing Size and Shielding Effects
Glycoengineering
Polypeptide Conjugation to a Water-Soluble Macromolecule
3.2. Depot Formulation Strategies to Prolong Duration of Action
3.2.1. Particulate Formulations
3.2.2. Gels
3.3. Targeting Tissue Components
Affinity-Based Drug Delivery
4. Polypeptide Delivery
4.1. Routes of Administration
4.1.1. IV Drug Delivery
General Concepts and Challenges
IV Formulation Strategies
4.1.2. SC Drug Delivery
General Concepts and Challenges
SC Formulation Strategies
4.1.3. Oral and Mucosal Drug Delivery
General Concepts and Challenges
Oral and Mucosal-Formulation Strategies
4.1.4. Ocular Drug Delivery
General Concepts and Challenges
Ocular Formulation Strategies
4.2. Targets
4.2.1. Targeting the Brain
General Concepts and Challenges
Brain Formulation Strategies
5. Conclusions
Funding
Conflicts of Interest
References
- Walsh, G. Biopharmaceutical benchmarks 2010. Nat. Biotechnol. 2010, 28, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Rader, R.A. FDA biopharmaceutical product approvals and trends in 2012. Bioprocess. Int. 2013, 11, 18–27. [Google Scholar]
- Mullard, A. 2011 FDA drug approvals. Nat. Rev. Drug Discov. 2012, 11, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Carter, P.J.; Lazar, G.A. Next generation antibody drugs: Pursuit of the “high-hanging fruit”. Nat. Rev. Drug Discov. 2018, 17, 197–223. [Google Scholar] [CrossRef] [PubMed]
- Breedveld, F. Therapeutic monoclonal antibodies. Lancet 2000, 355, 735–740. [Google Scholar] [CrossRef]
- Bruno, B.J.; Miller, G.D.; Lim, C.S. Basics and recent advances in peptide and protein drug delivery. Ther. Deliv. 2013, 4, 1443–1467. [Google Scholar] [CrossRef]
- Usmani, S.S.; Bedi, G.; Samuel, J.S.; Singh, S.; Kalra, S.; Kumar, P.; Ahuja, A.A.; Sharma, M.; Gautam, A.; Raghava, G.P.S. THPdb: Database of FDA-approved peptide and protein therapeutics. PLoS ONE 2017, 12, e0181748. [Google Scholar] [CrossRef]
- Market Analysis Report: Peptide Therapeutics Market. By Application (Cancer, Cardiovascular Disorder, Metabolic Disorder, Respiratory Disorder, Pain, Dermatology), by Type (Generic, Innovative) by Type of Manufacturers (In-House, Outsourced), And Segment Forecasts, 2014–2025; Grand View Research, Inc.: San Francisco, CA, USA, 2017.
- Global Therapeutic Proteins Market Report 2020: Market Was Valued at $93.14 Billion in 2018 and Is Expected to Grow to $172.87 Billion through 2022. Businesswire. Available online: https://www.businesswire.com/news/home/20191223005228/en/Global-Therapeutic-Proteins-Market-Report-2020-Market#:~:text=The global therapeutic proteins market,of sales of therapeutic proteins (accessed on 14 October 2020).
- Pisal, D.S.; Kosloski, M.P.; Balu-Iyer, S.V. Delivery of therapeutic proteins. J. Pharm. Sci. 2010, 99, 2557–2575. [Google Scholar] [CrossRef]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef]
- Lintner, K. Peptides and proteins. In Cosmetic Dermatology; Wiley-Blackwell: Oxford, UK, 2010; pp. 292–301. [Google Scholar] [CrossRef]
- Azhar, A.; Ahmad, E.; Zia, Q.; Rauf, M.A.; Owais, M.; Ashraf, G.M. Recent advances in the development of novel protein scaffolds based therapeutics. Int. J. Biol. Macromol. 2017, 102, 630–641. [Google Scholar] [CrossRef]
- Wurch, T.; Pierré, A.; Depil, S. Novel protein scaffolds as emerging therapeutic proteins: From discovery to clinical proof-of-concept. Trends Biotechnol. 2012, 30, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Mitragotri, S.; Burke, P.A.; Langer, R. Overcoming the challenges in administering biopharmaceuticals: Formulation and delivery strategies. Nat. Rev. Drug Discov. 2014, 13, 655–672. [Google Scholar] [CrossRef] [PubMed]
- Zapadka, K.L.; Becher, F.J.; Gomes dos Santos, A.L.; Jackson, S.E. Factors affecting the physical stability (aggregation) of peptide therapeutics. Interface Focus. 2017, 7, 20170030. [Google Scholar] [CrossRef] [PubMed]
- Dill, K.A.; MacCallum, J.L. The protein-folding problem, 50 years on. Science 2012, 338, 1042–1046. [Google Scholar] [CrossRef]
- Roberts, C.J. Therapeutic protein aggregation: Mechanisms, design, and control. Trends Biotechnol. 2014, 32, 372–380. [Google Scholar] [CrossRef]
- Lee, S.; Trinh, T.H.T.; Yoo, M.; Shin, J.; Lee, H.; Kim, J.; Hwang, E.; Lim, Y.; Ryou, C. Self-assembling peptides and their application in the treatment of diseases. Int. J. Mol. Sci. 2019, 20, 5850. [Google Scholar] [CrossRef]
- Valéry, C.; Pouget, E.; Pandit, A.; Verbavatz, J.-M.; Bordes, L.; Boisdé, I.; Cherif-Cheikh, R.; Artzner, F.; Paternostre, M. Molecular origin of the self-assembly of lanreotide into nanotubes: A mutational approach. Biophys. J. 2008, 94, 1782–1795. [Google Scholar] [CrossRef]
- Ambrosio, E.; Podmore, A.; Gomes Dos Santos, A.L.; Magarkar, A.; Bunker, A.; Caliceti, P.; Mastrotto, F.; van der Walle, C.F.; Salmaso, S. Control of Peptide Aggregation and Fibrillation by Physical PEGylation. Biomacromolecules 2018, 19, 3958–3969. [Google Scholar] [CrossRef]
- Zhang, J.; Mao, X.; Xu, W. Fibril Nucleation Kinetics of a Pharmaceutical Peptide: The Role of Conformation Stability, Formulation Factors, and Temperature Effect. Mol. Pharm. 2018, 15, 5591–5601. [Google Scholar] [CrossRef]
- Mahler, H.C.; Friess, W.; Grauschopf, U.; Kiese, S. Protein aggregation: Pathways, induction factors and analysis. J. Pharm. Sci. 2009, 98, 2909–2934. [Google Scholar] [CrossRef]
- Zayas, J.F. Solubility of Proteins. In Functionality of Proteins in Food; Springer: Berlin, Germany, 1997; pp. 6–75. [Google Scholar] [CrossRef]
- Meisl, G.; Yang, X.; Frohm, B.; Knowles, T.P.J.; Linse, S. Quantitative analysis of intrinsic and extrinsic factors in the aggregation mechanism of Alzheimer-associated Aβ-peptide. Sci. Rep. 2016, 6, 18728. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, A.; Mcgraw, N.R.; Schwartz, D.K.; Bee, J.S.; Carpenter, J.F.; Randolph, T.W. Protein Aggregation and Particle Formation in Prefilled Glass Syringes. J. Pharm. Sci. 2014, 1601–1612. [Google Scholar] [CrossRef] [PubMed]
- Khalili, H.; Sharma, G.; Froome, A.; Khaw, P.T.; Brocchini, S. Storage stability of bevacizumab in polycarbonate and polypropylene syringes. Eye 2015, 29, 820–827. [Google Scholar] [CrossRef]
- Manning, M.C.; Chou, D.K.; Murphy, B.M.; Payne, R.W.; Katayama, D.S. Stability of protein pharmaceuticals: An update. Pharm. Res. 2010, 27, 544–575. [Google Scholar] [CrossRef]
- Liu, L.; Ammar, D.A.; Ross, L.A.; Mandava, N.; Kahook, M.Y.; Carpenter, J.F. Silicone oil microdroplets and protein aggregates in repackaged bevacizumab and ranibizumab: Effects of long-term storage and product mishandling. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1023–1034. [Google Scholar] [CrossRef]
- Schargus, M.; Werner, B.P.; Geerling, G.; Winter, G. Contamination of anti-VEGF drugs for intravitreal injection how do repackaging and newly developed syringes affect the amount of silicone oil droplets and protein aggregates? Retina 2018, 38, 2088–2095. [Google Scholar] [CrossRef]
- O’Brien, C.J.; Blanco, M.A.; Costanzo, J.A.; Enterline, M.; Fernandez, E.J.; Robinson, A.S.; Roberts, C.J. Modulating non-native aggregation and electrostatic protein-protein interactions with computationally designed single-point mutations. Protein Eng. Des. Sel. 2016, 29, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.S. Effects of protein aggregates: An immunologic perspective. AAPS J. 2006, 8, E501–E507. [Google Scholar] [CrossRef]
- Angkawinitwong, U.; Sharma, G.; Khaw, P.T.; Brocchini, S.; Williams, G.R. Solid-state protein formulations. Ther. Deliv. 2015, 6, 59–82. [Google Scholar] [CrossRef]
- Chennamsetty, N.; Voynov, V.; Kayser, V.; Helk, B.; Trout, B.L. Prediction of aggregation prone regions of therapeutic proteins. J. Phys. Chem. B 2010, 114, 6614–6624. [Google Scholar] [CrossRef]
- Meric, G.; Robinson, A.S.; Roberts, C.J. Driving Forces for Nonnative Protein Aggregation and Approaches to Predict Aggregation-Prone Regions. Annu. Rev. Chem. Biomol. Eng. 2017, 8, 139–159. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.J. Protein aggregation and its impact on product quality. Curr. Opin. Biotechnol. 2014, 30, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Moore, W.V.; Leppert, P. Role of Aggregated Human Growth Hormone (hGH) in Development of Antibodies to hGH. J. Clin. Endocrinol. Metab. 1980, 51, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Baudouin, V.; Crusiaux, A.; Haddad, E.; Schandene, L.; Goldman, M.; Loirat, C.; Abramowicz, D. Anaphylactic shock caused by immunoglobulin E sensitization after retreatment with the chimeric anti-interleukin-2 receptor monoclonal antibody basiliximab. Transplantation 2003, 76, 459–463. [Google Scholar] [CrossRef]
- Iannuzzi, C.; Borriello, M.; Portaccio, M.; Irace, G.; Sirangelo, I. Insights into insulin fibril assembly at physiological and acidic ph and related amyloid intrinsic fluorescence. Int. J. Mol. Sci. 2017, 18, 2551. [Google Scholar] [CrossRef]
- Insulins (All Types): Risk of Cutaneous Amyloidosis at Injection Site. Drug Safety Update. Available online: https://www.gov.uk/drug-safety-update/insulins-all-types-risk-of-cutaneous-amyloidosis-at-injection-site (accessed on 14 October 2020).
- Rambaran, R.N.; Serpell, L.C. Amyloid fibrils. Prion 2008, 2, 112–117. [Google Scholar] [CrossRef]
- Datta-Mannan, A. Mechanisms influencing the pharmacokinetics and disposition of monoclonal antibodies and peptides. Drug Metab. Dispos. 2019, 47, 1100–1110. [Google Scholar] [CrossRef]
- Longmire, M.; Choyke, P.L.; Kobayashi, H. Clearance properties of nano-sized particles and molecules as imaging agents: Considerations and caveats. Nanomedicine 2008, 3, 703–717. [Google Scholar] [CrossRef]
- Di, L. Strategic Approaches to Optimizing Peptide ADME Properties. AAPS J. 2015, 17, 134–143. [Google Scholar] [CrossRef]
- Almeida, A.J.; Souto, E. Solid lipid nanoparticles as a drug delivery system for peptides and proteins. Adv. Drug Deliv. Rev. 2007, 59, 478–490. [Google Scholar] [CrossRef]
- Diao, L.; Meibohm, B. Pharmacokinetics and pharmacokinetic-pharmacodynamic correlations of therapeutic peptides. Clin. Pharmacokinet. 2013, 52, 855–868. [Google Scholar] [CrossRef] [PubMed]
- Pereira De Sousa, I.; Bernkop-Schnürch, A. Pre-systemic metabolism of orally administered drugs and strategies to overcome it. J. Control. Release 2014, 192, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Tibbitts, J.; Canter, D.; Graff, R.; Smith, A.; Khawli, L.A. Key factors influencing ADME properties of therapeutic proteins: A need for ADME characterization in drug discovery and development. MAbs 2016, 8, 229–245. [Google Scholar] [CrossRef]
- Sethu, S.; Govindappa, K.; Alhaidari, M.; Pirmohamed, M.; Park, K.; Sathish, J. Immunogenicity to Biologics: Mechanisms, Prediction and Reduction. Arch. Immunol. Ther. Exp. 2012, 60, 331–344. [Google Scholar] [CrossRef]
- Chirmule, N.; Jawa, V.; Meibohm, B. Immunogenicity to Therapeutic Proteins: Impact on PK/PD and Efficacy. AAPS J. 2012, 14, 296–302. [Google Scholar] [CrossRef]
- Kverneland, A.H.; Enevold, C.; Donia, M.; Bastholt, L.; Svane, I.M.; Nielsen, C.H. Development of anti-drug antibodies is associated with shortened survival in patients with metastatic melanoma treated with ipilimumab. Oncoimmunology 2018, 7, e1424674. [Google Scholar] [CrossRef]
- Jamnitski, A.; Bartelds, G.M.; Nurmohamed, M.T.; Schouwenburg, P.A.v.; Schaardenburg, D.v.; Stapel, S.O.; Dijkmans, B.A.C.; Aarden, L.; Wolbink1, G.J. The presence or absence of antibodies to infliximab or adalimumab determines the outcome of switching to etanercept. Ann. Rheum. Dis. 2011, 70, 284–288. [Google Scholar] [CrossRef]
- Menting, S.P.; Van Lümig, P.P.M.; De Vries, A.C.Q.; Van Den Reek, J.M.P.A.; Van Der Kleij, D.; De Jong, E.M.G.J.; Spuls, P.I.; Lecluse, L.L.A. Extent and consequences of antibody formation against adalimumab in patients with psoriasis one-year follow-up. JAMA Dermatol. 2014, 150, 130–136. [Google Scholar] [CrossRef]
- Farys, M.; Ginn, C.L.; Badescu, G.O.; Peciak, K.; Pawlisz, E.M.; Khalili, H.; Brocchini, S.J. Chemical and Genetic Modification. In Pharmaceutical Sciences Encyclopedia; John Wiley & Sons: Hoboken, NJ, USA, 2013; pp. 1–52. [Google Scholar] [CrossRef]
- Peciak, K.; Laurine, E.; Tommasi, R.; Choi, J.W.; Brocchini, S. Site-selective protein conjugation at histidine. Chem. Sci. 2019, 10, 427–439. [Google Scholar] [CrossRef]
- Carter, P.J. Introduction to current and future protein therapeutics: A protein engineering perspective. Exp. Cell Res. 2011, 317, 1261–1269. [Google Scholar] [CrossRef]
- Rath, T.; Baker, K.; Dumont, J.A.; Peters, R.T.; Jiang, H.; Qiao, S.-W.; Lencer, W.I.; Pierce, G.F.; Blumberg, R.S. Fc-fusion proteins and FcRn: Structural insights for longer-lasting and more effective therapeutics. Crit. Rev. Biotechnol. 2015, 35, 235–254. [Google Scholar] [CrossRef] [PubMed]
- Awwad, S.; Angkawinitwong, U. Overview of antibody drug delivery. Pharmaceutics 2018, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; d’Anjou, M. Pharmacological significance of glycosylation in therapeutic proteins. Curr. Opin. Biotechnol. 2009, 20, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Fares, F.; Azzam, N. Development of long-acting recombinant glycoprotein hormones by increasing the carbohydrate content. Drug Discov. Today 2019, 24, 1017–1022. [Google Scholar] [CrossRef]
- Moradi, S.V.; Hussein, W.M.; Varamini, P.; Simerska, P.; Toth, I. Glycosylation, an effective synthetic strategy to improve the bioavailability of therapeutic peptides. Chem. Sci. 2016, 7, 2492–2500. [Google Scholar] [CrossRef]
- Solá, R.J.; Griebenow, K. Glycosylation of therapeutic proteins: An effective strategy to optimize efficacy. BioDrugs 2011, 24, 9–21. [Google Scholar] [CrossRef]
- Kontermann, R.E. Strategies for extended serum half-life of protein therapeutics. Curr. Opin. Biotechnol. 2011, 22, 868–876. [Google Scholar] [CrossRef]
- Kontermann, R.E. Half-life extended biotherapeutics. Expert Opin. Biol. Ther. 2016, 16, 903–915. [Google Scholar] [CrossRef]
- Sinclair, A.M. Erythropoiesis stimulating agents: Approaches to modulate activity. Biol. Targets Ther. 2013, 7, 161–174. [Google Scholar] [CrossRef][Green Version]
- Kochendoerfer, G.G.; Chen, S.Y.; Mao, F.; Cressman, S.; Traviglia, S.; Shao, H.; Hunter, C.L.; Low, D.W.; Cagle, E.N.; Carnevali, M.; et al. Design and chemical synthesis of a homogeneous polymer-modified erythropoiesis protein. Science 2003, 299, 884–887. [Google Scholar] [CrossRef]
- van Witteloostuijn, S.B.; Pedersen, S.L.; Jensen, K.J. Half-Life Extension of Biopharmaceuticals using Chemical Methods: Alternatives to PEGylation. ChemMedChem 2016, 11, 2474–2495. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Yoon, I.S.; Kim, N.A.; Kim, D.-H.; Lee, J.; Lee, H.J.; Lee, S.; Choi, S.; Choi, M.-K.; Kim, H.H.; et al. Glycoengineering of Interferon-β 1a improves its biophysical and pharmacokinetic properties. PLoS ONE 2014, 9, e96967. [Google Scholar] [CrossRef]
- Veronese, F.; Pasut, G. PEGylation, successful approach to drug delivery. Drug Discov. Today 2005, 10, 1451–1458. [Google Scholar] [CrossRef]
- Picken, C.; Awwad, S.; Zloh, M.; Khalili, H.; Brocchini, S. 16—Protein modification by bis-alkylation. Polym. Protein Conjug. 2020. [Google Scholar] [CrossRef]
- Awwad, S.; Ginn, C.; Brocchini, S. The case for protein PEGylation. In Engineering of Biomaterials for Drug Delivery Systems; Elsevier: Cambridge, MA, USA, 2018; pp. 27–49. [Google Scholar] [CrossRef]
- Schellekens, H.; Hennink, W.E.; Brinks, V. The immunogenicity of polyethylene glycol: Facts and fiction. Pharm. Res. 2013, 30, 1729–1734. [Google Scholar] [CrossRef]
- Richter, A.W.; Åkerblom, E. Polyethylene glycol reactive antibodies in man: Titer distribution in allergic patients treated with monomethoxy polyethylene glycol modified allergens or placebo, and in healthy blood donors. Int. Arch. Allergy Immunol. 1984, 74, 36–39. [Google Scholar] [CrossRef]
- Sroda, K.; Rydlewski, J.; Langner, M.; Kozubek, A.; Grzybek, M.; Sikorski, A.F. Repeated injections of PEG-PE liposomes generate anti-PEG antibodies. Cell. Mol. Biol. Lett. 2005, 10, 37–47. [Google Scholar] [PubMed]
- Koide, H.; Asai, T.; Kato, H.; Ando, H.; Shiraishi, K.; Yokoyama, M.; Oku, N. Size-dependent induction of accelerated blood clearance phenomenon by repeated injections of polymeric micelles. Int. J. Pharm. 2012, 432, 75–79. [Google Scholar] [CrossRef]
- Abu Lila, A.S.; Kiwada, H.; Ishida, T. The accelerated blood clearance (ABC) phenomenon: Clinical challenge and approaches to manage. J. Control. Release 2013, 172, 38–47. [Google Scholar] [CrossRef]
- Müller, H.J.; Löning, L.; Horn, A.; Schwabe, D.; Gunkel, M.; Schrappe, M.; Schütz, V.V.; Henze, G.; da Palma, J.C.; Ritter, J.; et al. Pegylated asparaginase (Oncaspar(TM)) in children with ALL: Drug monitoring in reinduction according to the ALL/NHL-BFM 95 protocols. Br. J. Haematol. 2000, 110, 379–384. [Google Scholar] [CrossRef]
- Ganson, N.J.; Kelly, S.J.; Scarlett, E.; Sundy, J.S.; Hershfield, M.S. Control of hyperuricemia in subjects with refractory gout, and induction of antibody against poly(ethylene glycol) (PEG), in a phase I trial of subcutaneous PEGylated urate oxidase. Arthritis Res. Ther. 2005, 8, R12. [Google Scholar] [CrossRef]
- Webster, R.; Didier, E.; Harris, P.; Siegel, N. PEGylated proteins: Evaluation of their safety in the absence of definitive metabolism studies. Drug Metab. Dispos. 2007, 35, 9–16. [Google Scholar] [CrossRef]
- Bendele, A.; Seely, J.; Richey, C.; Sennello, G.; Shopp, G. Short communication: Renal tubular vacuolation in animals treated with polyethylene-glycol-conjugated proteins. Toxicol. Sci. 1998, 42, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Turecek, P.L.; Bossard, M.J.; Schoetens, F.; Ivens, I.A. PEGylation of Biopharmaceuticals: A Review of Chemistry and Nonclinical Safety Information of Approved Drugs. J. Pharm. Sci. 2016, 105, 460–475. [Google Scholar] [CrossRef]
- Ivens, I.A.; Achanzar, W.; Baumann, A.; Brändli-Baiocco, A.; Cavagnaro, J.; Dempster, M.; Depelchin, B.O.; Irizarry Rovira, A.R.; Dill-Morton, L.; Lane, J.H.; et al. PEGylated Biopharmaceuticals: Current Experience and Considerations for Nonclinical Development. Toxicol. Pathol. 2015, 43, 959–983. [Google Scholar] [CrossRef]
- Ivens, I.; Baumann, A.; McDonald, T.; Humphries, T.J.; Michaels, L.; Mathew, P. PEGylated therapeutic proteins for haemophilia treatment: A review for haemophilia caregivers. Haemophilia 2013, 19, 11–20. [Google Scholar] [CrossRef]
- Rudmann, D.G.; Alston, J.T.; Hanson, J.C.; Heidel, S. High molecular weight polyethylene glycol cellular distribution and PEG-associated cytoplasmic vacuolation is molecular weight dependent and does not require conjugation to proteins. Toxicol. Pathol. 2013, 41, 970–983. [Google Scholar] [CrossRef]
- Podust, V.N.; Sim, B.C.; Kothari, D.; Henthorn, L.; Gu, C.; Wang, C.-W.; McLaughlin, B.; Schellenberger, V. Extension of in vivo half-life of biologically active peptides via chemical conjugation to XTEN protein polymer. Protein Eng. Des. Sel. 2013, 26, 743–753. [Google Scholar] [CrossRef]
- Podust, V.N.; Balan, S.; Sim, B.-C.; Coyle, M.P.; Ernst, U.; Peters, R.T.; Schellenberger, V. Extension of in vivo half-life of biologically active molecules by XTEN protein polymers. J. Control. Release 2016, 240, 52–66. [Google Scholar] [CrossRef]
- Schlapschy, M.; Binder, U.; Börger, C.; Theobald, I.; Wachinger, K.; Kisling, S.; Haller, D.; Skerra, A. PASylation: A biological alternative to PEGylation for extending the plasma half-life of pharmaceutically active proteins. Protein Eng. Des. Sel. 2013, 26, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Kratz, F. Albumin as a drug carrier: Design of prodrugs, drug conjugates and nanoparticles. J. Control. Release 2008, 132, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Chuang, V.T.G.; Otagiri, M. Albumin Fusion Protein. In Albumin in Medicine; Springer: Singapore, 2016; pp. 71–89. [Google Scholar] [CrossRef]
- Pratley, R.E.; Nauck, M.A.; Barnett, A.H.; Feinglos, M.N.; Ovalle, F.; Harman-Boehm, I.; Ye, J.; BSc, R.S.; Johnson, S.; Stewart, M.; et al. Once-weekly albiglutide versus once-daily liraglutide in patients with type 2 diabetes inadequately controlled on oral drugs (HARMONY 7): A randomised, open-label, multicentre, non-inferiority phase 3 study. Lancet Diabetes Endocrinol. 2014, 2, 289–297. [Google Scholar] [CrossRef]
- Zorzi, A.; Linciano, S.; Angelini, A. Non-covalent albumin-binding ligands for extending the circulating half-life of small biotherapeutics. MedChemComm 2019, 10, 1068–1081. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, X. Simple bioconjugate chemistry serves great clinical advances: Albumin as a versatile platform for diagnosis and precision therapy. Chem. Soc. Rev. 2016, 45, 1432–1456. [Google Scholar] [CrossRef]
- Bech, E.M.; Pedersen, S.L.; Jensen, K.J. Chemical Strategies for Half-Life Extension of Biopharmaceuticals: Lipidation and Its Alternatives. ACS Med. Chem. Lett. 2018, 9, 577–580. [Google Scholar] [CrossRef]
- Muzykantov, V.R. Drug delivery by red blood cells: Vascular carriers designed by mother nature. Expert Opin. Drug Deliv. 2010, 7, 403–427. [Google Scholar] [CrossRef]
- Xia, Q.; Zhang, Y.; Li, Z.; Hou, X.; Feng, N. Red blood cell membrane-camouflaged nanoparticles: A novel drug delivery system for antitumor application. Acta Pharm. Sin. B 2019, 9, 675–689. [Google Scholar] [CrossRef]
- Rossi, L.; Fraternale, A.; Bianchi, M.; Magnani, M. Red Blood Cell Membrane Processing for Biomedical Applications. Front. Physiol. 2019, 10, 1070. [Google Scholar] [CrossRef]
- Kontos, S.; Kourtis, I.C.; Dane, K.Y.; Hubbell, J.A. Engineering antigens for in situ erythrocyte binding induces T-cell deletion. Proc. Natl. Acad. Sci. USA 2013, 110. [Google Scholar] [CrossRef]
- Villa, C.H.; Pan, D.C.; Zaitsev, S.; Cines, D.B.; Siegel, D.L.; Muzykantov, V.R. Delivery of drugs bound to erythrocytes: New avenues for an old intravascular carrier. Ther. Deliv. 2015, 6, 795–826. [Google Scholar] [CrossRef]
- Glassman, P.M.; Villa, C.H.; Ukidve, A.; Zhao, Z.; Smith, P.; Mitragotri, S.; Russell, A.J.; Brenner, J.S.; Muzykantov, V.R. Vascular drug delivery using carrier red blood cells: Focus on RBC surface loading and pharmacokinetics. Pharmaceutics 2020, 12, 440. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Seth, A.; Wibowo, N.; Zhao, C.-X.; Mitter, M.; Yu, C.; Middelberg, A.P.J. Nanoparticle vaccines. Vaccine 2014, 32, 327–337. [Google Scholar] [CrossRef]
- Bertrand, N.; Wu, J.; Xu, X.; Kamaly, N.; Farokhzad, O.C. Cancer nanotechnology: The impact of passive and active targeting in the era of modern cancer biology. Adv. Drug Deliv. Rev. 2014, 66, 2–25. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.D.; Whitehead, K.A.; Mitragotri, S. Materials for oral delivery of proteins and peptides. Nat. Rev. Mater. 2020, 5, 127–148. [Google Scholar] [CrossRef]
- Cheng, J.; Teply, B.A.; Jeong, S.Y.; Yim, C.H.; Ho, D.; Sherifi, I.; Jon, S.; Farokhzad, O.C.; Khademhosseini, A.; Langer, R.S. Magnetically responsive polymeric microparticles for oral delivery of protein drugs. Pharm. Res. 2006, 23, 557–564. [Google Scholar] [CrossRef]
- Joseph, J.W.; Kalitsky, J.; St-Pierre, S.; Brubaker, P.L. Oral delivery of glucagon-like peptide-1 in a modified polymer preparation normalizes basal glycaemia in diabetic db/db mice. Diabetologia 2000, 43, 1319–1328. [Google Scholar] [CrossRef]
- Wagner, A.M.; Gran, M.P.; Peppas, N.A. Designing the new generation of intelligent biocompatible carriers for protein and peptide delivery. Acta Pharm. Sin. B 2018, 8, 147–164. [Google Scholar] [CrossRef]
- Vila, A.; Sánchez, A.; Tobío, M.; Calvo, P.; Alonso, M.J. Design of biodegradable particles for protein delivery. J. Control. Release 2002, 78, 15–24. [Google Scholar] [CrossRef]
- He, P.; Liu, H.; Tang, Z.; Deng, M.; Yang, Y.; Pang, X.; Chen, X. Poly(ester amide) blend microspheres for oral insulin delivery. Int. J. Pharm. 2013, 455, 259–266. [Google Scholar] [CrossRef]
- Damgé, C.; Maincent, P.; Ubrich, N. Oral delivery of insulin associated to polymeric nanoparticles in diabetic rats. J. Control. Release 2007, 117, 163–170. [Google Scholar] [CrossRef]
- Damgé, C.; Socha, M.; Ubrich, N.; Maincent, P. Poly(ε-caprolactone)/eudragit nanoparticles for oral delivery of aspart-insulin in the treatment of diabetes. J. Pharm. Sci. 2010, 99, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Domb, A.J. Recent Advances in Polyanhydride Based Biomaterials. Adv. Mater. 2018, 30, 1706815. [Google Scholar] [CrossRef]
- Gaur, S.; Chen, L.; Yen, T.; Wang, Y.; Zhou, B.; Davis, M.; Yen, Y. Preclinical study of the cyclodextrin-polymer conjugate of camptothecin CRLX101 for the treatment of gastric cancer. Nanomed. Nanotechnol. Biol. Med. 2012, 8, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Hughes, P.; Ross, A.D.; Robinson, M.R. Biodegradable implants for sustained drug release in the eye. Pharm. Res. 2010, 27, 2043–2053. [Google Scholar] [CrossRef]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef]
- Miladi, K.; Ibraheem, D.; Iqbal, M.; Sfar, S.; Fessi, H.; Elaissari, A. Particles from preformed polymers as carriers for drug delivery. EXCLI J. 2014, 13, 28–57. [Google Scholar] [CrossRef]
- Tarhini, M.; Benlyamani, I.; Hamdani, S.; Agusti, G.; Fessi, H.; Greige-Gerges, H.; Bentaher, A.; Elaissari, A. Protein-based nanoparticle preparation via nanoprecipitation method. Materials 2018, 11, 394. [Google Scholar] [CrossRef]
- Tadros, T.F. Emulsion Formation, Stability, and Rheology. In Emulsion Formation and Stability; Wiley-VCH: Weinheim, Germany, 2013; pp. 1–75. [Google Scholar] [CrossRef]
- Yu, D.-G.; Zhu, L.-M.; White, K.; Branford-White, C. Electrospun nanofiber-based drug delivery systems. Health 2009, 67–75. [Google Scholar] [CrossRef]
- Song, H.H.; Gong, X.; Williams, G.R.; Quan, J.; Nie, L.I.; Zhu, L.M.; Nan, E.I.; Shao, S. Self-assembled magnetic liposomes from electrospun fibers. Mater. Res. Bull. 2014, 53, 280–289. [Google Scholar] [CrossRef]
- Schubert, S.; Delaney, J.T.; Schubert, U.S. Nanoprecipitation and nanoformulation of polymers: From history to powerful possibilities beyond poly(lactic acid). Soft Matter. 2011, 7, 1581–1588. [Google Scholar] [CrossRef]
- Jaiswal, M.; Dudhe, R.; Sharma, P.K. Nanoemulsion: An advanced mode of drug delivery system. 3 Biotechnology 2015, 5, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Bendjaballah, M.; Canselier, J.P.; Oumeddour, R. Optimization of oil-in-water emulsion stability: Experimental design, multiple light scattering, and acoustic attenuation spectroscopy. J. Dispers. Sci. Technol. 2010, 31, 1260–1272. [Google Scholar] [CrossRef]
- Fox, C.B. Squalene emulsions for parenteral vaccine and drug delivery. Molecules 2009, 14, 3286–3312. [Google Scholar] [CrossRef] [PubMed]
- Barrero, A.; Loscertales, I.G. Micro- and Nanoparticles via Capillary Flows. Annu. Rev. Fluid Mech. 2007, 39, 89–106. [Google Scholar] [CrossRef]
- Whitesides, G.M. The origins and the future of microfluidics. Nature 2006, 442, 368–373. [Google Scholar] [CrossRef]
- Damiati, S.; Kompella, U.B.; Damiati, S.A.; Kodzius, R. Microfluidic devices for drug delivery systems and drug screening. Genes 2018, 9, 103. [Google Scholar] [CrossRef]
- Rezvantalab, S.; Keshavarz Moraveji, M. Microfluidic assisted synthesis of PLGA drug delivery systems. RSC Adv. 2019, 9, 2055–2072. [Google Scholar] [CrossRef]
- Riahi, R.; Tamayol, A.; Shaegh, S.A.M.; Ghaemmaghami, A.; Dokmeci, M.R.; Khademshosseini, A. Microfluidics for Advanced Drug Delivery Systems. Curr. Opin. Chem. Eng. 2015, 7, 101–112. [Google Scholar] [CrossRef]
- Tran, K.T.M.; Nguyen, T.D. Lithography-based methods to manufacture biomaterials at small scales. J. Sci. Adv. Mater. Devices 2017, 2, 1–14. [Google Scholar] [CrossRef]
- Allahyari, M.; Mohit, E. Peptide/protein vaccine delivery system based on PLGA particles. Hum. Vaccines Immunother. 2016, 12, 806–828. [Google Scholar] [CrossRef]
- Xu, J.; Wong, D.H.C.; Byrne, J.D.; Chen, K.; Bowerman, C.; Desimone, J.M. Future of the particle replication in nonwetting templates (PRINT) technology. Angew. Chemie. Int. Ed. 2013, 52, 6580–6589. [Google Scholar] [CrossRef]
- Perry, J.L.; Herlihy, K.P.; Napier, M.E.; Desimone, J.M. PRINT: A novel platform toward shape and size specific nanoparticle theranostics. Acc. Chem. Res. 2011, 44, 990–998. [Google Scholar] [CrossRef] [PubMed]
- Galloway, A.L.; Murphy, A.; DeSimone, J.M.; Di, J.; Herrmann, J.P.; Hunter, M.E.; Kindig, J.P.; Malinoski, F.J.; Rumley, M.A.; Stoltz, D.M.; et al. Development of a nanoparticle-based influenza vaccine using the PRINT® technology. Nanomed. Nanotechnol. Biol. Med. 2013, 9, 523–531. [Google Scholar] [CrossRef]
- Waghule, T.; Singhvi, G.; Dubey, S.K.; Pandey, M.M.; Gupta, G.; Singh, M.; Dua, K. Microneedles: A smart approach and increasing potential for transdermal drug delivery system. Biomed. Pharmacother. 2019, 109, 1249–1258. [Google Scholar] [CrossRef] [PubMed]
- Santos, D.; Maurício, A.C.; Sencadas, V.; Santos, J.D.; Fernandes, M.H.; Gomes, P.S. Spray Drying: An Overview. In Biomaterials Physics and Chemistry New Edition; InTech: London, UK, 2018. [Google Scholar] [CrossRef]
- Sosnik, A.; Seremeta, K.P. Advantages and challenges of the spray-drying technology for the production of pure drug particles and drug-loaded polymeric carriers. Adv. Colloid Interface Sci. 2015, 223, 40–54. [Google Scholar] [CrossRef] [PubMed]
- Maury, M.; Murphy, K.; Kumar, S.; Shi, L.; Lee, G. Effects of process variables on the powder yield of spray-dried trehalose on a laboratory spray-dryer. Eur. J. Pharm. Biopharm. 2005, 59, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Wan, F.; Maltesen, M.J.; Andersen, S.K.; Bjerregaard, S.; Foged, C.; Rantanen, J.; Yang, M. One-Step Production of Protein-Loaded PLGA Microparticles via Spray Drying Using 3-Fluid Nozzle. Pharm. Res. 2014, 31, 1967–1977. [Google Scholar] [CrossRef]
- Wichterle, O.; Lím, D. Hydrophilic Gels for Biological Use. Nature 1960, 185, 117–118. [Google Scholar] [CrossRef]
- Chen, Q.; Chen, H.; Zhu, L.; Zheng, J. Fundamentals of double network hydrogels. J. Mater. Chem. B 2015, 3, 3654–3676. [Google Scholar] [CrossRef]
- Cooper, R.C.; Yang, H. Hydrogel-based ocular drug delivery systems: Emerging fabrication strategies, applications, and bench-to-bedside manufacturing considerations. J. Control. Release 2019, 306, 29–39. [Google Scholar] [CrossRef]
- Bi, X.; Liang, A. In Situ-Forming Cross-linking Hydrogel Systems: Chemistry and Biomedical Applications. In Emerging Concepts in Analysis and Applications of Hydrogels; InTech: London, UK, 2016; Volume 13. [Google Scholar] [CrossRef]
- Rizwan, M.; Yahya, R.; Hassan, A.; Yar, M.; Azzahari, A.D.; Selvanathan, V.; Sonsudin, F.; Abouloula, C.N. pH sensitive hydrogels in drug delivery: Brief history, properties, swelling, and release mechanism, material selection and applications. Polymers 2017, 9, 137. [Google Scholar] [CrossRef] [PubMed]
- Vermonden, T.; Censi, R.; Hennink, W.E. Hydrogels for protein delivery. Chem. Rev. 2012, 112, 2853–2888. [Google Scholar] [CrossRef] [PubMed]
- Bisht, R.; Jaiswal, J.K.; Chen, Y.-S.; Jin, J.; Rupenthal, I.D. Light-responsive in situ forming injectable implants for effective drug delivery to the posterior segment of the eye. Expert Opin. Drug Deliv. 2016, 5247, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Famili, A.; Kahook, M.Y.; Park, D. A combined micelle and poly(serinol hexamethylene urea)-Co-Poly(N-Isopropylacrylamide) reverse thermal gel as an injectable ocular drug delivery system. Macromol. Biosci. 2014, 14, 1719–1729. [Google Scholar] [CrossRef] [PubMed]
- Sapino, S.; Chirio, D.; Peira, E.; Rubio, E.A.; Brunella, V.; Jadhav, S.A.; Chindamo, G.; Gallarate, M. Ocular Drug Delivery: A Special Focus on the Thermosensitive Approach. Nanomaterials 2019, 9, 884. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Bae, Y.H.; Okano, T. Hydrogels: Swelling, Drug Loading, and Release. Pharm. Res. 1992, 9, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Gehrke, S.H.; Robeson, J.; Johnson, J.F.; Vaid, N. Protein isolation by solution-controlled gel sorption. Biotechnol. Prog. 1991, 7, 355–358. [Google Scholar] [CrossRef]
- Bromberg, L.E.; Ron, E.S. Temperature-responsive gels and thermogelling polymer matrices for protein and peptide delivery. Adv. Drug Deliv. Rev. 1998, 31, 197–221. [Google Scholar] [CrossRef]
- Vulic, K.; Shoichet, M.S. Affinity-based drug delivery systems for tissue repair and regeneration. Biomacromolecules 2014, 15, 3867–3880. [Google Scholar] [CrossRef]
- Wang, N.X.; von Recum, H.A. Affinity-Based Drug Delivery. Macromol. Biosci. 2011, 11, 321–332. [Google Scholar] [CrossRef]
- Ghosh, J.G.; Nguyen, A.A.; Bigelow, C.E.; Poor, S.; Qiu, Y.; Rangaswamy, N.; Ornberg, R.; Jackson, B.; Mak, H.; Ezell, T.; et al. Long-acting protein drugs for the treatment of ocular diseases. Nat. Commun. 2017, 8, 14837. [Google Scholar] [CrossRef]
- Morrison, P.W.J.; Khutoryanskiy, V.V. Advances in ophthalmic drug delivery. Ther. Deliv. 2014, 5, 1297–1315. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, D.J.; Hicks, B.C.; Parsons, S.; Sakiyama-Elbert, S.E. Development of rationally designed affinity-based drug delivery systems. Acta Biomater. 2005, 1, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Puoci, F.; Cirillo, G.; Curcio, M.; Parisi, O.I.; Iemma, F.; Picci, N. Molecularly imprinted polymers in drug delivery: State of art and future perspectives. Expert Opin. Drug Deliv. 2011, 8, 1379–1393. [Google Scholar] [CrossRef]
- Zaidi, S.A. Latest trends in molecular imprinted polymer based drug delivery systems. RSC Adv. 2016, 6, 88807–88819. [Google Scholar] [CrossRef]
- Sellergren, B.; Allender, C.J. Molecularly imprinted polymers: A bridge to advanced drug delivery. Adv. Drug Deliv. Rev. 2005, 57, 1733–1741. [Google Scholar] [CrossRef]
- Ali, M.; Byrne, M.E. Controlled release of high molecular weight hyaluronic acid from molecularly imprinted hydrogel contact lenses. Pharm. Res. 2009, 26, 714–726. [Google Scholar] [CrossRef]
- Zaidi, S.A. Molecular imprinting: A useful approach for drug delivery. Mater. Sci. Energy Technol. 2020, 3, 72–77. [Google Scholar] [CrossRef]
- Lakhin, A.V.; Tarantul, V.Z.; Gening, L.V. Aptamers: Problems, solutions and prospects. Acta Nat. 2013, 5, 34–43. [Google Scholar] [CrossRef]
- Soontornworajit, B.; Zhou, J.; Shaw, M.T.; Fan, T.H.; Wang, Y. Hydrogel functionalization with DNA aptamers for sustained PDGF-BB release. Chem. Commun. 2010, 46, 1857–1859. [Google Scholar] [CrossRef] [PubMed]
- Vugmeyster, Y.; Xu, X.; Theil, F.-P.; Khawli, L.; Leach, M.W. Pharmacokinetics and toxicology of therapeutic proteins: Advances and challenges. World J. Biol. Chem. 2012, 3, 73–92. [Google Scholar] [CrossRef] [PubMed]
- Keizer, R.J.; Huitema, A.D.R.; Schellens, J.H.M.; Beijnen, J.H. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin. Pharmacokinet. 2010, 49, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Luo, Y. Recent advances of polysaccharide-based nanoparticles for oral insulin delivery. Int. J. Biol. Macromol. 2018, 120, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Araújo, F.; das Neves, J.; Martins, J.P.; Granja, P.L.; Santos, H.A.; Sarmento, B. Functionalized materials for multistage platforms in the oral delivery of biopharmaceuticals. Prog. Mater. Sci. 2017, 89, 306–344. [Google Scholar] [CrossRef]
- Choi, H.J.; Ebersbacher, C.F.; Kim, M.C.; Kang, S.M.; Montemagno, C.D. A Mechanistic Study on the Destabilization of Whole Inactivated Influenza Virus Vaccine in Gastric Environment. PLoS ONE 2013, 8, e66316. [Google Scholar] [CrossRef]
- Choi, H.J.; Kim, M.C.; Kang, S.M.; Montemagno, C.D. The osmotic stress response of split influenza vaccine particles in an acidic environment. Arch. Pharm. Res. 2014, 37, 1607–1616. [Google Scholar] [CrossRef]
- Homayun, B.; Lin, X.; Choi, H.J. Challenges and recent progress in oral drug delivery systems for biopharmaceuticals. Pharmaceutics 2019, 11, 129. [Google Scholar] [CrossRef]
- Klotz, U.; Teml, A.; Schwab, M. Clinical pharmacokinetics and use of infliximab. Clin. Pharmacokinet. 2007, 46, 645–660. [Google Scholar] [CrossRef]
- Gläser, D.; Liniger, J.; Peter, D.C. Mini-pumps. In Challenges in Protein Product Development; Warne, N.W., Mahler, H.-C., Eds.; InTech: London, UK, 2018; pp. 235–256. [Google Scholar] [CrossRef]
- Pyzik, M.; Sand, K.M.K.; Hubbard, J.J.; Andersen, J.T.; Sandlie, I.; Blumberg, R.S. The neonatal Fc Receptor (FcRn): A misnomer? Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Patel, A.; Cholkar, K.; Mitra, A.K. Recent developments in protein and peptide parenteral delivery approaches. Ther. Deliv. 2014, 5, 337–365. [Google Scholar] [CrossRef]
- Sou, V.; McManus, C.; Mifflin, N.; Frost, S.A.; Ale, J.; Alexandrou, E. A clinical pathway for the management of difficult venous access. BMC Nurs. 2017, 16, 64. [Google Scholar] [CrossRef]
- Launay-Vacher, V. An appraisal of subcutaneous trastuzumab: A new formulation meeting clinical needs. Cancer Chemother. Pharmacol. 2013, 72, 1361–1367. [Google Scholar] [CrossRef] [PubMed]
- Mueller, C.; Altenburger, U.; Mohl, S. Challenges for the pharmaceutical technical development of protein coformulations. J. Pharm. Pharmacol. 2018, 70, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Villalonga, R.; Valdivia, A.; Pérez, Y.; Chongo, B. Improved pharmacokinetics and stability properties of catalase by chemical glycosidation with end-group activated dextran. J. Appl. Polym. Sci. 2006, 102, 4573–4576. [Google Scholar] [CrossRef]
- Pérez, Y.; Valdivia, A.; Ramírez, H.L.; Villalonga, R. Improved pharmacokinetics properties for catalase by site-specific glycosidation with aminated dextran. Macromol. Rapid. Commun. 2005, 26, 1304–1308. [Google Scholar] [CrossRef]
- Liebner, R.; Mathaes, R.; Meyer, M.; Hey, T.; Winter, G.; Besheer, A. Protein HESylation for half-life extension: Synthesis, characterization and pharmacokinetics of HESylated anakinra. Eur. J. Pharm. Biopharm. 2014, 87, 378–385. [Google Scholar] [CrossRef]
- Lindhout, T.; Iqbal, U.; Willis, L.M.; Reid, A.N.; Li, J.; Liu, X.; Moreno, M.; Wakarchuk, W.W. Site-specific enzymatic polysialylation of therapeutic proteins using bacterial enzymes. Proc. Natl. Acad. Sci. USA 2011, 108, 7397–7402. [Google Scholar] [CrossRef]
- Aghaabdollahian, S.; Ahangari Cohan, R.; Norouzian, D.; Davami, F.; Karam, M.R.A.; Torkashvand, F.; Vaseghi, G.; Moazzami, R.; Dizaji, S.L. Enhancing bioactivity, physicochemical, and pharmacokinetic properties of a nano-sized, anti-VEGFR2 Adnectin, through PASylation technology. Sci. Rep. 2019, 9, 2978. [Google Scholar] [CrossRef] [PubMed]
- Schlapschy, M.; Theobald, I.; Mack, H.; Schottelius, M.; Wester, H.J.; Skerra, A. Fusion of a recombinant antibody fragment with a homo-amino-acid polymer: Effects on biophysical properties and prolonged plasma half-life. Protein Eng. Des. Sel. 2007, 20, 273–284. [Google Scholar] [CrossRef]
- Despanie, J.; Dhandhukia, J.P.; Hamm-Alvarez, S.F.; MacKay, J.A. Elastin-like polypeptides: Therapeutic applications for an emerging class of nanomedicines. J. Control. Release. 2016, 240, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Strohl, W.R. Fusion Proteins for Half-Life Extension of Biologics as a Strategy to Make Biobetters. BioDrugs 2015, 29, 215–239. [Google Scholar] [CrossRef]
- Conrad, U.; Plagmann, I.; Malchow, S.; Sack, M.; Floss, D.M.; Kruglov, A.A.; Nedospasov, S.A.; Rose-John, S.; Scheller, J. ELPylated anti-human TNF therapeutic single-domain antibodies for prevention of lethal septic shock. Plant Biotechnol. J. 2011, 9, 22–31. [Google Scholar] [CrossRef]
- Zorzi, A.; Middendorp, S.J.; Wilbs, J.; Deyle, K.; Heinis, C. Acylated heptapeptide binds albumin with high affinity and application as tag furnishes long-acting peptides. Nat. Commun. 2017, 8, 16092. [Google Scholar] [CrossRef]
- Koehler, M.F.T.; Zobel, K.; Beresini, M.H.; Caris, L.D.; Combs, D.; Paasch, B.D.; Lazarus, R.A. Albumin affinity tags increase peptide half-life in vivo. Bioorg. Med. Chem. Lett. 2002, 12, 2883–2886. [Google Scholar] [CrossRef]
- Sasson, K.; Marcus, Y.; Lev-Goldman, V.; Rubinraut, S.; Fridkin, M.; Shechter, Y. Engineering prolonged-acting prodrugs employing an albumin-binding probe that undergoes slow hydrolysis at physiological conditions. J. Control. Release. 2010, 142, 214–220. [Google Scholar] [CrossRef]
- Steeland, S.; Vandenbroucke, R.E.; Libert, C. Nanobodies as therapeutics: Big opportunities for small antibodies. Drug Discov. Today 2016, 21, 1076–1113. [Google Scholar] [CrossRef]
- Jovčevska, I.; Muyldermans, S. The Therapeutic Potential of Nanobodies. BioDrugs 2020, 34, 11–26. [Google Scholar] [CrossRef]
- Tijink, B.M.; Laeremans, T.; Budde, M.; Walsum, M.S.-V.; Dreier, T.; de Haard, H.J.; Leemans, C.R.; van Dongen, G.A.M.S. Improved tumor targeting of anti-epidermal growth factor receptor Nanobodies through albumin binding: Taking advantage of modular Nanobody technology. Mol. Cancer Ther. 2008, 7, 2288–2297. [Google Scholar] [CrossRef]
- Plückthun, A. Designed Ankyrin Repeat Proteins (DARPins): Binding Proteins for Research, Diagnostics, and Therapy. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 489–511. [Google Scholar] [CrossRef]
- Stumpp, M.T.; Binz, H.K.; Amstutz, P. DARPins: A new generation of protein therapeutics. Drug Discov. Today 2008, 13, 695–701. [Google Scholar] [CrossRef]
- Steiner, D.; Merz, F.W.; Sonderegger, I.; Gulotti-Georgieva, M.; Villemagne, D.; Phillips, D.J.; Forrer, D.J.; Stumpp, M.T.; Zitt, C.; Binz, H.K. Half-life extension using serum albumin-binding DARPin® domains. Protein Eng. Des. Sel. 2017, 30, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Holt, L.J.; Basran, A.; Jones, K.; Chorlton, J.; Jespers, L.S.; Brewis, N.D.; Tomlinson, I.M. Anti-serum albumin domain antibodies for extending the half-lives of short lived drugs. Protein Eng. Des. Sel. 2008, 21, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.; Dunlevy, G.; Rycroft, D.; Topley, P.; Holt, L.J.; Herbert, T.; Davies, M.; Cook, F.; Holmes, S.; Jespers, L.; et al. Anti-serum albumin domain antibodies in the development of highly potent, efficacious and long-acting interferon. Protein Eng. Des. Sel. 2010, 23, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Goodall, L.J.; Ovecka, M.; Rycroft, D.; Friel, S.L.; Sanderson, A.; Mistry, P.; Davies, M.L. Pharmacokinetic and pharmacodynamic characterisation of an anti-mouse TNF receptor 1 domain antibody formatted for in vivo half-life extension. PLoS ONE 2015, 10, e0137065. [Google Scholar] [CrossRef] [PubMed]
- O’Connor-Semmes, R.L.; Lin, J.; Hodge, R.J.; Choudhury, A.; Nunez, D.J. GSK2374697, a Novel Albumin-Binding Domain Antibody (AlbudAb), Extends Systemic Exposure of Exendin-4: First Study in Humans-PK/PD and Safety. Clin. Pharmacol. Ther. 2014, 96, 704–712. [Google Scholar] [CrossRef]
- Lin, J.; Hodge, R.J.; O’Connor-Semmes, R.L.; Nunez, D.J. GSK2374697, a long duration glucagon-like peptide-1 (GLP-1) receptor agonist, reduces postprandial circulating endogenous total GLP-1 and peptide YY in healthy subjects. Diabetes Obes. Metab. 2015, 17, 1007–1010. [Google Scholar] [CrossRef]
- Kaplon, H.; Reichert, J.M. Antibodies to watch in 2019. MAbs 2019, 11, 219–238. [Google Scholar] [CrossRef]
- Miller, L.J.; Sexton, P.M.; Dong, M.; Harikumar, K.G. The class B G-protein-coupled GLP-1 receptor: An important target for the treatment of type-2 diabetes mellitus. Int. J. Obes. Suppl. 2014, 4, S9–S13. [Google Scholar] [CrossRef]
- Swartz, M.A. The physiology of the lymphatic system. Adv. Drug Deliv. Rev. 2001, 50, 3–20. [Google Scholar] [CrossRef]
- Richter, W.F.; Bhansali, S.G.; Morris, M.E. Mechanistic Determinants of Biotherapeutics Absorption Following SC Administration. AAPS J. 2012, 14, 559–570. [Google Scholar] [CrossRef]
- Fraser, J.R.E.; Laurent, T.C.; Laurent, U.B.G. Hyaluronan: Its nature, distribution, functions and turnover. J. Intern. Med. 1997, 242, 27–33. [Google Scholar] [CrossRef]
- Bookbinder, L.H.; Hofer, A.; Haller, M.F.; Zepeda, M.L.; Keller, G.-A.; Lim, J.E.; Edgington, T.S.; Shepard, H.M.; Patton, J.S.; Frost, G.I. A recombinant human enzyme for enhanced interstitial transport of therapeutics. J. Control. Release 2006, 114, 230–241. [Google Scholar] [CrossRef]
- Jackisch, C.; Müller, V.; Maintz, C.; Hell, S.; Ataseven, B. Subcutaneous Administration of Monoclonal Antibodies in Oncology. Geburtshilfe Frauenheilkd. 2014, 74, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Kinnunen, H.M.; Mrsny, R.J. Improving the outcomes of biopharmaceutical delivery via the subcutaneous route by understanding the chemical, physical and physiological properties of the subcutaneous injection site. J. Control. Release 2014, 182, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Kojic, N.; Goyal, P.; Lou, C.H.; Corwin, M.J. An Innovative Needle-free Injection System: Comparison to 1 ml Standard Subcutaneous Injection. AAPS PharmSciTech 2017, 18, 2965–2970. [Google Scholar] [CrossRef] [PubMed]
- Tomar, D.S.; Kumar, S.; Singh, S.K.; Goswami, S.; Li, L. Molecular basis of high viscosity in concentrated antibody solutions: Strategies for high concentration drug product development. MAbs 2016, 8, 216–228. [Google Scholar] [CrossRef]
- Wang, W.; Chen, N.; Shen, X.; Cunningham, P.; Fauty, S.; Michel, K.; Wang, B.; Hong, X.; Adreani, C.; Nunes, C.N.; et al. Lymphatic transport and catabolism of therapeutic proteins after subcutaneous administration to rats and dogs. Drug Metab. Dispos. 2012, 40, 952–962. [Google Scholar] [CrossRef]
- Hamuro, L.; Kijanka, G.; Kinderman, F.; Kropshofer, H.; Bu, D.X.; Zepeda, M.; Jawa, V. Perspectives on Subcutaneous Route of Administration as an Immunogenicity Risk Factor for Therapeutic Proteins. J. Pharm. Sci. 2017, 106, 2946–2954. [Google Scholar] [CrossRef]
- Supersaxo, A.; Hein, W.R.; Steffen, H. Effect of molecular weight on the lymphatic absorption of water-soluble compounds following subcutaneous administration. Pharm. Res. 1990, 7, 167–169. [Google Scholar] [CrossRef]
- Sleep, D.; Cameron, J.; Evans, L.R. Albumin as a versatile platform for drug half-life extension. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 5526–5534. [Google Scholar] [CrossRef]
- Schellenberger, V.; Wang, C.W.; Geething, N.C.; Spink, B.J.; Campbell, A.; To, W.; Scholle, M.D.; Yin, Y.; Yao, Y.; Bogin, O.; et al. A recombinant polypeptide extends the in vivo half-life of peptides and proteins in a tunable manner. Nat. Biotechnol. 2009, 27, 1186–1190. [Google Scholar] [CrossRef] [PubMed]
- Amiram, M.; Luginbuhl, K.M.; Li, X.; Feinglos, M.N.; Chilkoti, A. A depot-forming glucagon-like peptide-1 fusion protein reduces blood glucose for five days with a single injection. J. Control. Release 2013, 172, 144–151. [Google Scholar] [CrossRef][Green Version]
- Sanford, M. Dulaglutide: First global approval. Drugs 2014, 74, 2097–2103. [Google Scholar] [CrossRef] [PubMed]
- Menacho-Melgar, R.; Decker, J.S.; Hennigan, J.N.; Lynch, M.D. A review of lipidation in the development of advanced protein and peptide therapeutics. J. Control. Release 2019, 295, 1–12. [Google Scholar] [CrossRef]
- Wysham, C.; Grimm, M.; Chen, S. Once weekly exenatide: Efficacy, tolerability and place in therapy. Diabetes Obes. Metab. 2013, 15, 871–881. [Google Scholar] [CrossRef]
- Tunn, U.W. A 6-month depot formulation of leuprolide acetate is safe and effective in daily clinical practice: A non-interventional prospective study in 1273 patients. BMC Urol. 2011, 11. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, S.; Kim, H.J.; Jesena, A.; Parmar, V.; Sato, N.; Wang, H.-C.; Lokejaroenlarb, S.; Isidro, J.; Kim, K.S.; Itoh, Y.; et al. Phase 3, open-label, randomized study comparing 3-monthly with monthly goserelin in pre-menopausal women with estrogen receptor-positive advanced breast cancer. Breast Cancer 2016, 23, 771–779. [Google Scholar] [CrossRef]
- Hillery, A.M.; Lloyd, A.W.; Swarbrick, J. Drug Delivery and Targeting for Pharmacists and Pharmaceutical Scientists; InTech: London, UK, 2001. [Google Scholar]
- FDA. LANTUS (Insulin Glargine [RDNA Origin] Injection) Solution for Subcutaneous Injection; FDA: Silver Spring, MD, USA, 2000.
- FDA. Novolin R (Regular, Human Insulin [RDNA Origin] USP) Solution for Subcutaneous or Intravenous Use; FDA: Silver Spring, MD, USA, 1991. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/019938s066lbl.pdf (accessed on 13 October 2020).
- Rosenstock, J.; Balas, B.; Charbonnel, B.; Bolli, G.B.; Boldrin, M.; Ratner, R.; Balena, R. The fate of taspoglutide, a weekly GLP-1 receptor agonist, versus twice-daily exenatide for type 2 diabetes: The T-emerge 2 trial. Diabetes Care 2013, 36, 498–504. [Google Scholar] [CrossRef]
- Qi, Y.; Chilkoti, A. Protein-polymer conjugation-moving beyond PEGylation. Curr. Opin. Chem. Biol. 2015, 28, 181–193. [Google Scholar] [CrossRef]
- Egbu, R.; Brocchini, S.; Khaw, P.T.; Awwad, S. Antibody loaded collapsible hyaluronic acid hydrogels for intraocular delivery. Eur. J. Pharm. Biopharm. 2018, 124, 95–103. [Google Scholar] [CrossRef]
- Fletcher, N.A.; Babcock, L.R.; Murray, E.A.; Krebs, M.D. Controlled delivery of antibodies from injectable hydrogels. Mater. Sci. Eng. C 2016, 59, 801–806. [Google Scholar] [CrossRef]
- Kirschbrown, W.P.; Wynne, C.; Kågedal, M.; Wada, R.; Li, H.; Wang, B.; Nijem, I.; Crnjevic, T.B.; Gasser, H.; Eng-Wong, J.; et al. Development of a Subcutaneous Fixed-Dose Combination of Pertuzumab and Trastuzumab: Results from the Phase Ib Dose-Finding Study. J. Clin. Pharmacol. 2019, 59, 702–716. [Google Scholar] [CrossRef]
- Hatton, G.B.; Yadav, V.; Basit, A.W.; Merchant, H.A. Animal Farm: Considerations in Animal Gastrointestinal Physiology and Relevance to Drug Delivery in Humans. J. Pharm. Sci. 2015, 104, 2747–2776. [Google Scholar] [CrossRef]
- Antosova, Z.; Mackova, M.; Kral, V.; Macek, T. Therapeutic application of peptides and proteins: Parenteral forever? Trends Biotechnol. 2009, 27, 628–635. [Google Scholar] [CrossRef]
- Zhou, X.H.; Po, A.L.W. Peptide and protein drugs: II. Non-parenteral routes of delivery. Int. J. Pharm. 1991, 75, 117–130. [Google Scholar] [CrossRef]
- Ashley, G.W.; Henise, J.; Reid, R.; Santi, D.V. Hydrogel drug delivery system with predictable and tunable drug release and degradation rates. Proc. Natl. Acad. Sci. USA 2013, 110, 2318–2323. [Google Scholar] [CrossRef]
- Banerjee, A.; Mitragotri, S. Intestinal patch systems for oral drug delivery. Curr. Opin. Pharmacol. 2017, 36, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, M.; Evers, A.; Wagner, M. Recent progress and future options in the development of GLP-1 receptor agonists for the treatment of diabesity. Bioorganic. Med. Chem. Lett. 2013, 23, 4011–4018. [Google Scholar] [CrossRef] [PubMed]
- Werle, M.; Takeuchi, H. Chitosan-aprotinin coated liposomes for oral peptide delivery: Development, characterisation and in vivo evaluation. Int. J. Pharm. 2009, 370, 26–32. [Google Scholar] [CrossRef]
- Li, J.; Mooney, D.J. Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 2016, 1, 119–122. [Google Scholar] [CrossRef]
- Aungst, B.J. Intestinal permeation enhancers. J. Pharm. Sci. 2000, 89, 429–442. [Google Scholar] [CrossRef]
- Zheng, Y.; Qiu, Y.; Lu, M.Y.F.; Hoffman, D.; Reiland, T.L. Permeability and absorption of leuprolide from various intestinal regions in rabbits and rats. Int. J. Pharm. 1999, 185, 83–92. [Google Scholar] [CrossRef]
- Vllasaliu, D.; Thanou, M.; Stolnik, S.; Fowler, R. Recent advances in oral delivery of biologics: Nanomedicine and physical modes of delivery. Expert Opin. Drug Deliv. 2018, 15, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Schoellhammer, C.M.; Schroeder, A.; Maa, R.; Lauwers, G.Y.; Swiston, A.; Zervas, M.; Barman, R.; DiCiccio, A.M.; Brugge, W.R.; Anderson, D.G.; et al. Ultrasound-mediated gastrointestinal drug delivery. Sci. Transl. Med. 2015, 7, 310ra168. [Google Scholar] [CrossRef] [PubMed]
- Aran, K.; Chooljian, M.; Paredes, J.; Rafi, M.; Lee, K.; Kim, A.Y.; An, J.; Yau, J.F.; Chum, H.; Conboy, I.; et al. An oral microjet vaccination system elicits antibody production in rabbits. Sci. Transl. Med. 2017, 9, eaaf6413. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J. Advances in oral peptide therapeutics. Nat. Rev. Drug Discov. 2019. [Google Scholar] [CrossRef]
- Shen, Z.; Mitragotri, S. Intestinal patches for oral drug delivery. Pharm. Res. 2002, 19, 391–395. [Google Scholar] [CrossRef]
- Banerjee, A.; Lee, J.; Mitragotri, S. Intestinal mucoadhesive devices for oral delivery of insulin. Bioeng. Transl. Med. 2016, 1, 338–346. [Google Scholar] [CrossRef]
- Gupta, V.; Hwang, B.H.; Doshi, N.; Banerjee, A.; Anselmo, A.C.; Mitragotri, S. Delivery of Exenatide and Insulin Using Mucoadhesive Intestinal Devices. Ann. Biomed. Eng. 2016, 44, 1993–2007. [Google Scholar] [CrossRef]
- Eiamtrakarn, S.; Itoh, Y.; Kishimoto, J.; Yoshikawa, Y.; Shibata, N.; Murakami, M.; Takada, K. Gastrointestinal mucoadhesive patch system (GI-MAPS) for oral administration of G-CSF, a model protein. Biomaterials 2002, 23, 145–152. [Google Scholar] [CrossRef]
- Venkatesan, N.; Uchino, K.; Amagase, K.; Ito, Y.; Shibata, N.; Takada, K. Gastro-intestinal patch system for the delivery of erythropoietin. J. Control. Release 2006, 111, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Hwang, B.H.; Lee, J.; Anselmo, A.C.; Doshi, N.; Mitragotri, S. Mucoadhesive intestinal devices for oral delivery of salmon calcitonin. J. Control. Release 2013, 172, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.; Varum, F.; Bravo, R.; Furrer, E.; Basit, A.W. Gastrointestinal stability of therapeutic anti-TNF α IgG1 monoclonal antibodies. Int. J. Pharm. 2016, 502, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Ibekwe, V.C.; Khela, M.K.; Evans, D.F.; Basit, A.W. A new concept in colonic drug targeting: A combined pH-responsive and bacterially-triggered drug delivery technology. Aliment. Pharmacol. Ther. 2008, 28, 911–916. [Google Scholar] [CrossRef] [PubMed]
- Peppas, N.A.; Wood, K.M.; Blanchette, J.O. Hydrogels for oral delivery of therapeutic proteins. Expert Opin. Biol. Ther. 2004, 4, 881–887. [Google Scholar] [CrossRef]
- Li, Z.; Li, H.; Wang, C.; Xu, J.; Singh, V.; Chen, D.; Zhang, J. Sodium dodecyl sulfate/β-cyclodextrin vesicles embedded in chitosan gel for insulin delivery with pH-selective release. Acta Pharm. Sin. B 2016, 6, 344–351. [Google Scholar] [CrossRef]
- Kamei, N.; Morishita, M.; Chiba, H.; Kavimandan, N.J.; Peppas, N.A.; Takayama, K. Complexation hydrogels for intestinal delivery of interferon β and calcitonin. J. Control. Release 2009, 134, 98–102. [Google Scholar] [CrossRef]
- Edelman, E.R.; Nathan, A.; Katada, M.; Gates, J.; Karnovsky, M.J. Perivascular graft heparin delivery using biodegradable polymer wraps. Biomaterials 2000, 21, 2279–2286. [Google Scholar] [CrossRef]
- Ichikawa, H.; Peppas, N.A. Novel complexation hydrogels for oral peptide delivery: In vitro evaluation of their cytocompatibility and insulin-transport enhancing effects using Caco-2 cell monolayers. J. Biomed. Mater. Res. Part A 2003, 67, 609–617. [Google Scholar] [CrossRef]
- Toorisaka, E.; Watanabe, K.; Ono, H.; Hirata, M.; Kamiya, N.; Goto, M. Intestinal patches with an immobilized solid-in-oil formulation for oral protein delivery. Acta Biomater. 2012, 8, 653–658. [Google Scholar] [CrossRef]
- Carr, D.A.; Gómez-Burgaz, M.; Boudes, M.C.; Peppas, N.A. Complexation hydrogels for the oral delivery of growth hormone and salmon calcitonin. Ind. Eng. Chem. Res. 2010, 49, 11991–11995. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Conde, B.R.; Brewer Eman, A.; Peppas, N.A. Complexation Hydrogels as Oral Delivery Vehicles of Therapeutic Antibodies: An in Vitro and ex Vivo Evaluation of Antibody Stability and Bioactivity. Ind. Eng. Chem. Res. 2015, 54, 10197–10205. [Google Scholar] [CrossRef] [PubMed]
- Hintzen, F.; Perera, G.; Hauptstein, S.; Müller, C.; Laffleur, F.; Bernkop-Schnürch, A. In vivo evaluation of an oral self-microemulsifying drug delivery system (SMEDDS) for leuprorelin. Int. J. Pharm. 2014, 472, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Mahjub, R.; Dorkoosh, F.A.; Rafiee-Tehrani, M.; Bernkop Schnürch, A. Oral self-nanoemulsifying peptide drug delivery systems: Impact of lipase on drug release. J. Microencapsul. 2015, 32, 401–407. [Google Scholar] [CrossRef]
- Sheng, J.; He, H.; Han, L.; Qin, J.; Chen, S.; Ru, G.; Li, R.; Yang, P.; Wang, J.; Yang, V.C. Enhancing insulin oral absorption by using mucoadhesive nanoparticles loaded with LMWP-linked insulin conjugates. J. Control. Release 2016, 233, 181–190. [Google Scholar] [CrossRef]
- Su, F.Y.; Lin, K.J.; Sonaje, K.; Wey, S.-P.; Yen, T.-C.; Ho, Y.-C.; Panda, N.; Chuang, E.-Y.; Maiti, B.; Sung, H.-W. Protease inhibition and absorption enhancement by functional nanoparticles for effective oral insulin delivery. Biomaterials 2012, 33, 2801–2811. [Google Scholar] [CrossRef]
- Han, L.; Zhao, Y.; Yin, L.; Li, R.; Liang, Y.; Huang, H.; Pan, S.; Wu, C.; Feng, M. Insulin-loaded pH-sensitive hyaluronic acid nanoparticles enhance transcellular delivery. AAPS Pharm. Sci. Tech. 2012, 13, 836–845. [Google Scholar] [CrossRef]
- Bowman, K.; Leong, K.W. Chitosan nanoparticles for oral drug and gene delivery. Int. J. Nanomed. 2006, 1, 117–128. [Google Scholar] [CrossRef]
- George, M.; Abraham, T.E. Polyionic hydrocolloids for the intestinal delivery of protein drugs: Alginate and chitosan—A review. J. Control. Release 2006, 114, 1–14. [Google Scholar] [CrossRef]
- Pan, Y.; Li, Y.; Zhao, H.; Zheng, J.; Xu, H.; Wei, G.; Hao, J.; Cui, F. Bioadhesive polysaccharide in protein delivery system: Chitosan nanoparticles improve the intestinal absorption of insulin in vivo. Int. J. Pharm. 2002, 249, 139–147. [Google Scholar] [CrossRef]
- Sarmento, B.; Ribeiro, A.; Veiga, F.; Ferreira, D.; Neufeld, R. Oral bioavailability of insulin contained in polysaccharide nanoparticles. Biomacromolecules 2007, 8, 3054–3060. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Xu, S.; Wang, H.; Ling, Y.; Dong, J.; Xia, R.; Sun, X. Nanoparticles: Oral Delivery for Protein and Peptide Drugs. AAPS Pharm. Sci. Tech. 2019, 20, 190. [Google Scholar] [CrossRef]
- Gursoy, R.N.; Benita, S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed. Pharmacother. 2004, 58, 173–182. [Google Scholar] [CrossRef]
- Leonaviciute, G.; Bernkop-Schnürch, A. Self-emulsifying drug delivery systems in oral (poly)peptide drug delivery. Expert Opin. Drug Deliv. 2015, 12, 1703–1716. [Google Scholar] [CrossRef]
- Ma, E.; Ma, H.; Liu, Z.; Zheng, C.; Duan, M. In vitro and in vivo evaluation of a novel oral insulin formulation. Acta Pharmacol. Sin. 2006, 27, 1382–1388. [Google Scholar] [CrossRef]
- Mandal, A.; Pal, D.; Agrahari, V.; Trinh, H.M.; Joseph, M.; Mitra, A.K. Ocular delivery of proteins and peptides: Challenges and novel formulation approaches. Adv. Drug Deliv. Rev. 2018, 126, 67–95. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.; Park, K.; Famili, A. Hydrogels for sustained delivery of biologics to the back of the eye. Drug Discov. Today 2019, 24, 1470–1482. [Google Scholar] [CrossRef]
- Thway, T.M.; Magana, I.; Bautista, A.; Jawa, V.; Gu, W.; Ma, M. Impact of anti-drug antibodies in preclinical pharmacokinetic assessment. AAPS J. 2013, 15, 856–863. [Google Scholar] [CrossRef]
- Wakshull, E.; Quarmby, V.; Mahler, H.C.; Rivers, H.; Jere, D.; Ramos, M.; Szczesny, P.; Bechtold-Peters, K.; Masli, S.; Gupta, S. Advancements in Understanding Immunogenicity of Biotherapeutics in the Intraocular Space. AAPS J. 2017, 19, 1656–1668. [Google Scholar] [CrossRef] [PubMed]
- Wessels, U.; Zadak, M.; Reiser, A.; Brockhaus, J.; Ritter, M.; Abdolzade-Bavil, A.; Heinrich, J.; Stubenrauch, K. Immunogenicity testing of therapeutic antibodies in ocular fluids after intravitreal injection. Bioanalysis 2018, 10, 803–814. [Google Scholar] [CrossRef]
- EYLEATM (Aflibercept) Injection, 2011 US Package Insert; Regeneron Pharmaceuticals Inc.: New York, NY, USA, 2011; Available online: https://www.regeneron.com/sites/default/files/EYLEA_FPI.pdf (accessed on 13 October 2020).
- Steffensmeier, A.C.G.; Azar, A.E.; Fuller, J.J.; Muller, B.A.; Russell, S.R. Vitreous Injections of Pegaptanib Sodium Triggering Allergic Reactions. Am. J. Ophthalmol. 2007, 14, 512–513. [Google Scholar] [CrossRef] [PubMed]
- de Zafra, C.L.Z.; Sasseville, V.G.; Matsumoto, S.; Freichel, C.; Miltone, M.; MacLachlan, T.K.; Farman, C.; Raymond, I.; Gupta, S.; Newton, R.; et al. Inflammation and immunogenicity limit the utility of the rabbit as a nonclinical species for ocular biologic therapeutics. Regul. Toxicol. Pharmacol. 2017, 86, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Agrahari, V.; Agrahari, V.; Mandal, A.; Pal, D.; Mitra, A.K. How are we improving the delivery to back of the eye? Advances and challenges of novel therapeutic approaches. Expert Opin. Drug Deliv. 2017, 14, 1145–1162. [Google Scholar] [CrossRef]
- Marticorena, J.; Romano, V.; Gómez-Ulla, F. Sterile endophthalmitis after intravitreal injections. Mediators Inflamm. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Bakri, S.J.; Larson, T.A.; Edwards, A.O. Intraocular inflammation following intravitreal injection of bevacizumab. Graefe’s Arch. Clin. Exp. Ophthalmol. 2008, 246, 779–781. [Google Scholar] [CrossRef]
- Kampougeris, G.; Spyropoulos, D.; Mitropoulou, A. Intraocular pressure rise after anti-VEGF treatment: Prevalence, possible mechanisms and correlations. J. Curr. Glaucoma Pract. 2013, 7, 19–24. [Google Scholar] [CrossRef]
- Karabag, R.Y.; Parlak, M.; Cetin, G.; Yaman, A.; Osman Saatci, A. Retinal tears and rhegmatogenous retinal detachment after intravitreal injections: Its prevalence and case reports. Digit. J. Ophthalmol 2015, 21, 8–10. [Google Scholar] [CrossRef]
- Ghasemi Falavarjani, K.; Nguyen, Q.D. Adverse events and complications associated with intravitreal injection of anti-VEGF agents: A review of literature. Eye 2013, 27, 787–794. [Google Scholar] [CrossRef]
- D’Amico, D.J. Pegaptanib Sodium for Neovascular Age-Related Macular Degeneration. Two-Year Safety Results of the Two Prospective, Multicenter, Controlled Clinical Trials. Ophthalmology 2006, 113, 992–1001. [Google Scholar] [CrossRef]
- Laude, A.; Tan, L.E.; Wilson, C.G.; Lascaratos, M.; Elashrya, G.; Aslam, T.; Patton, N.; Dhillon, B. Intravitreal therapy for neovascular age-related macular degeneration and inter-individual variations in vitreous pharmacokinetics. Prog. Retin. Eye Res. 2010, 29, 466–475. [Google Scholar] [CrossRef]
- Awwad, S.; Henein, C.; Ibeanu, N.; Khaw, P.T.; Brocchini, S. Preclinical challenges for developing long acting intravitreal medicines. Eur. J. Pharm. Biopharm. 2020, 153, 130–149. [Google Scholar] [CrossRef]
- Campochiaro, P.A.; Marcus, D.M.; Awh, C.C.; Regillo, C.; Adamis, A.P.; Bantseev, V.; Chiang, Y.; Ehrlich, J.S.; Erickson, S.; Hanley, W.D.; et al. The Port Delivery System with Ranibizumab for Neovascular Age-Related Macular Degeneration: Results from the Randomized Phase 2 Ladder Clinical Trial. Ophthalmology 2019, 126, 1141–1154. [Google Scholar] [CrossRef]
- Lee, Y.C.; Yalkowsky, S.H. Effect of formulation on the systemic absorption of insulin from enhancer-free ocular devices. Int. J. Pharm. 1999, 185, 199–204. [Google Scholar] [CrossRef]
- Choonara, Y.E.; Pillay, V.; Danckwerts, M.P.; Carmichael, T.R.; du Toit, L.C. A review of implantable intravitreal drug delivery technologies for the treatment of posterior segment eye diseases. J. Pharm. Sci. 2010, 99, 2219–2239. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Cabrera-Ghayouri, S.; Christie, L.A.; Held, K.S.; Viswanath, V. Translational Preclinical Pharmacologic Disease Models for Ophthalmic Drug Development. Pharm. Res. 2019, 36. [Google Scholar] [CrossRef] [PubMed]
- Gote, V.; Pal, D. Ocular Implants in the Clinic and under Clinical Investigation for Ocular Disorders. EC Ophthalmol. 2019, 10, 660–666. [Google Scholar]
- Sieving, P.A.; Caruso, R.C.; Tao, W.; Coleman, H.R.; Thompson, D.J.S.; Fullmer, K.R.; Bush, R.A. Ciliary neurotrophic factor (CNTF) for human retinal degeneration: Phase I trial of CNTF delivered by encapsulated cell intraocular implants. Proc. Natl. Acad. Sci. USA 2006, 103, 3896–3901. [Google Scholar] [CrossRef]
- Kontturi, L.S.; Collin, E.C.; Murtomäki, L.; Pandit, A.S.; Yliperttula, M.; Urtti, A. Encapsulated cells for long-term secretion of soluble VEGF receptor 1: Material optimization and simulation of ocular drug response. Eur. J. Pharm. Biopharm. 2015, 95, 387–397. [Google Scholar] [CrossRef]
- Miao, H.; Wu, B.D.; Tao, Y.; Li, X.X. Diffusion of macromolecules through sclera. Acta Ophthalmol. 2013, 91, 1–6. [Google Scholar] [CrossRef]
- Angkawinitwong, U.; Awwad, S.; Khaw, P.T.; Brocchini, S.; Williams, G.R. Electrospun formulations of bevacizumab for sustained release in the eye. Acta Biomater. 2017, 64, 126–136. [Google Scholar] [CrossRef]
- de Souza, S.O.L.; Guerra, M.C.A.; Heneine, L.G.D.; de Oliveira, C.R.; Junior, A.D.S.C.; Fialho, S.L.; Oréfice, R.L. Biodegradable core-shell electrospun nanofibers containing bevacizumab to treat age-related macular degeneration. J. Mater. Sci. Mater. Med. 2018, 29, 173. [Google Scholar] [CrossRef] [PubMed]
- Pandit, J.; Sultana, Y.; Aqil, M. Chitosan-coated PLGA nanoparticles of bevacizumab as novel drug delivery to target retina: Optimization, characterization, and in vitro toxicity evaluation. Artif. Cells Nanomed. Biotechnol. 2017, 45, 1397–1407. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.S.; Green, C.R.; Wang, K.; Danesh-Meyer, H.V.; Rupenthal, I.D. Sustained intravitreal delivery of connexin43 mimetic peptide by poly(d,l-lactide-co-glycolide) acid micro- and nanoparticles-Closing the gap in retinal ischaemia. Eur. J. Pharm. Biopharm. 2015, 95, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Awwad, S.; Abubakre, A.; Angkawinitwong, U.; Khaw, P.T.; Brocchini, S. In situ antibody-loaded hydrogel for intravitreal delivery. Eur. J. Pharm. Sci. 2019, 137, 104993. [Google Scholar] [CrossRef]
- Liu, W.; Borrell, M.A.; Venerus, D.C.; Mieler, W.F.; Kang-Mieler, J.J. Characterization of biodegradable microsphere-hydrogel ocular drug delivery system for controlled and extended release of ranibizumab. Transl. Vis. Sci. Technol. 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Osswald, C.R.; Guthrie, M.J.; Avila, A.; Valio, J.A.; Mieler, W.F.; Kang-Mieler, J.J. In Vivo Efficacy of an Injectable Microsphere-Hydrogel Ocular Drug Delivery System. Curr. Eye Res. 2017, 2, 1293–1301. [Google Scholar] [CrossRef]
- Drapala, P.W.; Brey, E.M.; Mieler, W.F.; Venerus, D.C.; Kang Derwent, J.J.; Pérez-Luna, V.H. Role of Thermo-responsiveness and Poly(ethylene glycol) Diacrylate Cross-link Density on Protein Release from Poly(N-isopropylacrylamide) Hydrogels. J. Biomater. Sci. Polym. Ed. 2011, 22, 59–75. [Google Scholar] [CrossRef]
- Kim, S.; Kang-Mieler, J.J.; Liu, W.; Wang, Z.; Yiu, G.; Teixeira, L.B.C.; Mieler, W.F.; Thomasy, S.M. Safety and biocompatibility of aflibercept-loaded microsphere thermo-responsive hydrogel drug delivery system in a nonhuman primate model. Transl. Vis. Sci. Technol. 2020, 9, 30. [Google Scholar] [CrossRef]
- Xue, K.; Zhao, X.; Zhang, Z.; Qiu, B.; Tan, Q.S.W.; Ong, K.H.; Liu, Z.; Parikh, B.H.; Barathi, V.A.; Yu, W.; et al. Sustained delivery of anti-VEGFs from thermogel depots inhibits angiogenesis without the need for multiple injections. Biomater. Sci. 2019, 7, 4603–4614. [Google Scholar] [CrossRef]
- Yu, Y.; Lau, L.C.M.; Lo, A.C.-Y.; Chau, Y. Injectable Chemically Crosslinked Hydrogel for the Controlled Release of Bevacizumab in Vitreous: A 6-Month in Vivo Study. Transl. Vis. Sci. Technol. 2015, 4, 5. [Google Scholar] [CrossRef]
- Tyagi, P.; Barros, M.; Stansbury, J.W.; Kompella, U.B. Light-activated, in situ forming gel for sustained suprachoroidal delivery of bevacizumab. Mol. Pharm. 2013, 10, 2858–2867. [Google Scholar] [CrossRef]
- Awwad, S.; Al-Shohani, A.; Khaw, P.T.; Brocchini, S. Comparative Study of In Situ Loaded Antibody and PEG-Fab NIPAAM Gels. Macromol. Biosci. 2018, 18, 1700255. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Weng, Y.; Xu, L.; Chen, H. Sustained release of avastin® from polysaccharides cross-linked hydrogels for ocular drug delivery. Int. J. Biol. Macromol. 2013, 60, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Xu, X.; Yao, F.; Jin, L.; Xie, B.B.; Shi, S.; Ma, H.; Li, X.Y.; Chen, H. In situ covalently cross-linked PEG hydrogel for ocular drug delivery applications. Int. J. Pharm. 2014, 470, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Jalalvandi, E.; Shavandi, A. Shear thinning/self-healing hydrogel based on natural polymers with secondary photocrosslinking for biomedical applications. J. Mech. Behav. Biomed. Mater. 2019, 90, 191–201. [Google Scholar] [CrossRef]
- Guvendiren, M.; Lu, H.D.; Burdick, J.A. Shear-thinning hydrogels for biomedical applications. Soft Matter. 2012, 8, 260–272. [Google Scholar] [CrossRef]
- Purcell, B.P.; Lobb, D.; Charati, M.B.; Dorsey, S.M.; Wade, R.J.; Zellars, K.N.; Doviak, H.; Pettaway, S.; Logdon, C.B.; Shuman, J.A.; et al. Injectable and bioresponsive hydrogels for on-demand matrix metalloproteinase inhibition. Nat. Mater. 2014, 13, 653–661. [Google Scholar] [CrossRef]
- Mulyasasmita, W.; Lee, J.S.; Heilshorn, S.C. Molecular-level engineering of protein physical hydrogels for predictive sol-gel phase behavior. Biomacromolecules 2011, 12, 3406–3411. [Google Scholar] [CrossRef]
- Uman, S.; Dhand, A.; Burdick, J.A. Recent advances in shear-thinning and self-healing hydrogels for biomedical applications. J. Appl. Polym. Sci. 2020, 137, 48668. [Google Scholar] [CrossRef]
- Bastings, M.M.C.; Koudstaal, S.; Kieltyka, R.E.; Nakano, Y.; Pape, A.C.H.; Feyen, D.A.M.; van Slochteren, F.J.; Doevendans, P.A.; Sluijter, J.P.G.; Meijer, E.W.; et al. A fast pH-switchable and self-healing supramolecular hydrogel carrier for guided, local catheter injection in the infarcted myocardium. Adv. Healthc. Mater. 2014, 3, 70–78. [Google Scholar] [CrossRef]
- Chen, M.H.; Wang, L.L.; Chung, J.J.; Kim, Y.H.; Atluri, P.; Burdick, J.A. Methods to Assess Shear-Thinning Hydrogels for Application as Injectable Biomaterials. ACS Biomater. Sci. Eng. 2017, 3, 3146–3160. [Google Scholar] [CrossRef]
- Wang, L.L.; Sloand, J.N.; Gaffey, A.C.; Venkataraman, C.M.; Wang, Z.; Trubelja, A.; Hammer, D.A.; Atluri, P.; Burdick, J.A. Injectable, Guest-Host Assembled Polyethylenimine Hydrogel for siRNA Delivery. Biomacromolecules 2017, 18, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Alarcon, C.D.; Pennadam, S.; Alexander, C. Stimuli responsive polymers for biomedical applications. Chem. Soc. Rev. 2005, 34, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Liu, Y.; Li, X.; Dereje, K.; Zhang, B.; Ren, J.; Lu, J.; Li, J.; Du, S.; Liu, Z. Research progress of in-situ gelling ophthalmic drug delivery system. Asian J. Pharm. Sci. 2019, 14, 1–15. [Google Scholar] [CrossRef]
- Bisht, R.; Jaiswal, J.K.; Rupenthal, I.D. Nanoparticle-loaded biodegradable light-responsive in situ forming injectable implants for effective peptide delivery to the posterior segment of the eye. Med. Hypotheses 2017, 103, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Lohani, A.; Singh, G.; Bhattacharya, S.S.; Verma, A. Interpenetrating Polymer Networks as Innovative Drug Delivery Systems. J. Drug Deliv. 2014, 2014, 583612. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Lee, B.S.; Mieler, W.F.; Kang-Mieler, J.J. Biodegradable Microsphere-Hydrogel Ocular Drug Delivery System for Controlled and Extended Release of Bioactive Aflibercept in Vitro. Curr. Eye Res. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Awwad, S.S.H. The Vitreous as a Depot to Prolong the Action of Ocular Therapies. 2016. Available online: https://discovery.ucl.ac.uk/id/eprint/1530892 (accessed on 14 October 2020).
- Huelsmann, P.M.; Kopetzki, E. Fusion Proteins for Ophthalmology with Increased Eye Retention. Patent Application No. US20190225676A1, 25 July 2019. [Google Scholar]
- Rimpelä, A.K.; Reinisalo, M.; Hellinen, L.; Grazhdankin, E.; Kidron, H.; Urtti, A.; del Amo, E.A. Implications of melanin binding in ocular drug delivery. Adv. Drug Deliv. Rev. 2018, 126, 23–43. [Google Scholar] [CrossRef]
- Jakubiak, P.; Reutlinger, M.; Mattei, P.; Schuler, F.; Urtti, A.; Alvarez-Sánchez, R. Understanding Molecular Drivers of Melanin Binding to Support Rational Design of Small Molecule Ophthalmic Drugs. J. Med. Chem. 2018, 61, 10106–10115. [Google Scholar] [CrossRef]
- Michael, I.P.; Westenskow, P.D.; Hacibekiroglu, S.; Greenwald, A.C.; Ballios, B.G.; Kurihara, T.; Li, Z.; Warren, C.M.; Zhang, P.; Aguilar, E.; et al. Local acting Sticky-trap inhibits vascular endothelial growth factor dependent pathological angiogenesis in the eye. EMBO Mol. Med. 2014, 6, 604–623. [Google Scholar] [CrossRef]
- Shatz, W.; Aaronson, J.; Yohe, S.; Kelley, R.F.; Kalia, Y.N. Strategies for modifying drug residence time and ocular bioavailability to decrease treatment frequency for back of the eye diseases. Expert Opin. Drug Deliv. 2019, 16, 43–57. [Google Scholar] [CrossRef]
- Ghosh, J.; Roguska, M.; Nguyen, A.A.; Pietzonka, T.; Machacek, M.; Golosov, A. Compositions and Methods for Long Acting Molecules. Patent Application No. US20160297854A1, 13 October 2016. [Google Scholar]
- Dong, X. Current strategies for brain drug delivery. Theranostics 2018, 8, 1481–1493. [Google Scholar] [CrossRef]
- Yi, X.; Manickam, D.S.; Brynskikh, A.; Kabanov, A.V. Agile delivery of protein therapeutics to CNS. J. Control. Release 2014, 190, 637–663. [Google Scholar] [CrossRef]
- Bors, L.A.; Erdö, F. Overcoming the blood-brain barrier. Challenges and tricks for CNS drug delivery. Sci. Pharm. 2019, 87, 6. [Google Scholar] [CrossRef]
- Pakulska, M.M.; Ballios, B.G.; Shoichet, M.S. Injectable hydrogels for central nervous system therapy. Biomed. Mater. 2012, 7. [Google Scholar] [CrossRef]
- Cooke, M.J.; Wang, Y.; Morshead, C.M.; Shoichet, M.S. Controlled epi-cortical delivery of epidermal growth factor for the stimulation of endogenous neural stem cell proliferation in stroke-injured brain. Biomaterials 2011, 32, 5688–5697. [Google Scholar] [CrossRef]
- Brasnjevic, I.; Steinbusch, H.W.M.; Schmitz, C.; Martinez-Martinez, P. Delivery of peptide and protein drugs over the blood-brain barrier. Prog. Neurobiol. 2009, 87, 212–251. [Google Scholar] [CrossRef]
- Spencer, B.J.; Verma, I.M. Targeted delivery of proteins across the blood-brain barrier. Proc. Natl. Acad. Sci. USA 2007, 104, 7594–7599. [Google Scholar] [CrossRef] [PubMed]
- Ulapane, K.R.; Kopec, B.M.; Siahaan, T.J. Improving in vivo brain delivery of monoclonal antibody using novel cyclic peptides. Pharmaceutics 2019, 11, 568. [Google Scholar] [CrossRef] [PubMed]
- Oller-Salvia, B.; Sánchez-Navarro, M.; Giralt, E.; Teixidó, M. Blood-brain barrier shuttle peptides: An emerging paradigm for brain delivery. Chem. Soc. Rev. 2016, 45, 4690–4707. [Google Scholar] [CrossRef] [PubMed]
- Sjöstedt, N.; Kortejärvi, H.; Kidron, H.; Vellonen, K.S.; Urtti, A.; Yliperttula, M. Challenges of using in vitro data for modeling P-glycoprotein efflux in the blood-brain barrier. Pharm. Res. 2014, 31, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Arvanitis, C.D.; Ferraro, G.B.; Jain, R.K. The blood–brain barrier and blood–tumour barrier in brain tumours and metastases. Nat. Rev. Cancer 2020, 20, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R.; Junnuthula, V.R.; Gowd, G.S.; Ashokan, A.; Thomas, J.; Peethambaran, R.; Thomas, A.; Unni, A.K.K.; Panikar, D.; Nair, S.V.; et al. Theranostic 3-Dimensional nano brain-implant for prolonged and localized treatment of recurrent glioma. Sci. Rep. 2017, 7, 43271. [Google Scholar] [CrossRef] [PubMed]
- Shapira-Furman, T.; Serra, R.; Gorelick, N.; Doglioli, M.; Tagliaferri, V.; Cecia, A.; Peters, M.; Kumar, A.; Rottenberg, Y.; Langer, R.; et al. Biodegradable wafers releasing Temozolomide and Carmustine for the treatment of brain cancer. J. Control. Release 2019, 295, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Bota, D.A.; Desjardins, A.; Quinn, J.A.; Affronti, M.L.; Friedman, H.S. Interstitial chemotherapy with biodegradable BCNU (Gliadel®) wafers in the treatment of malignant gliomas. Ther. Clin. Risk Manag. 2007, 3, 707–715. [Google Scholar] [PubMed]
- He, Y.; Munn, D.; Falo, L.D. Recombinant lentivector as a genetic immunization vehicle for antitumor immunity. Expert. Rev. Vaccines 2007, 6, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Tuladhar, A.; Obermeyer, J.M.; Payne, S.L.; Siu, R.C.W.; Zand, S.; Morshead, C.M.; Shoichet, M.S. Injectable hydrogel enables local and sustained co-delivery to the brain: Two clinically approved biomolecules, cyclosporine and erythropoietin, accelerate functional recovery in rat model of stroke. Biomaterials 2020, 235, 119794. [Google Scholar] [CrossRef]
- Kang, C.E.; Tator, C.H.; Shoichet, M.S. Poly(ethylene glycol) modification enhances penetration of fibroblast growth factor 2 to injured spinal cord tissue from an intrathecal delivery system. J. Control. Release 2010, 144, 25–31. [Google Scholar] [CrossRef]
- Wang, Y.; Cooke, M.J.; Lapitsky, Y.; Wylie, R.G.; Sachewsky, N.; Corbett, D.; Morshead, C.M.; Shoichet, M.S. Transport of epidermal growth factor in the stroke-injured brain. J. Control. Release 2011, 149, 225–235. [Google Scholar] [CrossRef]
- Sousa, F.; Dhaliwal, H.K.; Gattacceca, F.; Sarmento, B.; Amiji, M.M. Enhanced anti-angiogenic effects of bevacizumab in glioblastoma treatment upon intranasal administration in polymeric nanoparticles. J. Control. Release 2019, 309, 37–47. [Google Scholar] [CrossRef]
- Ozdemir-Kaynak, E.; Qutub, A.A.; Yesil-Celiktas, O. Advances in glioblastoma multiforme treatment: New models for nanoparticle therapy. Front. Physiol. 2018, 9, 170. [Google Scholar] [CrossRef] [PubMed]
- Sousa, F.; Cruz, A.; Fonte, P.; Pinto, I.M.; Neves-Petersen, M.T.; Sarmento, B. A new paradigm for antiangiogenic therapy through controlled release of bevacizumab from PLGA nanoparticles. Sci. Rep. 2017, 7, 3736. [Google Scholar] [CrossRef]
- Lampe, K.J.; Kern, D.S.; Mahoney, M.J.; Bjugstad, K.B. The administration of BDNF and GDNF to the brain via PLGA microparticles patterned within a degradable PEG-based hydrogel: Protein distribution and the glial response. J. Biomed. Mater. Res. Part A 2011, 96, 595–607. [Google Scholar] [CrossRef]
- Huang, R.; Ke, W.; Liu, Y.; Wu, D.; Feng, L.; Jiang, C.; Pei, Y. Gene therapy using lactoferrin-modified nanoparticles in a rotenone-induced chronic Parkinson model. J. Neurol. Sci. 2010, 290, 123–130. [Google Scholar] [CrossRef]
- Herrán, E.; Ruiz-Ortega, J.Á.; Aristieta, A.; Igartua, M.; Requejo, C.; Lafuente, J.V.; Ugedo, L.; Pedraz, J.L.; Hernández, R.M. In vivo administration of VEGF- and GDNF-releasing biodegradable polymeric microspheres in a severe lesion model of Parkinson’s disease. Eur. J. Pharm. Biopharm. 2013, 85, 1183–1190. [Google Scholar] [CrossRef]
- Garbayo, E.; Montero-Menei, C.N.; Ansorena, E.; Lanciego, J.L.; Aymerich, M.S.; Blanco-Prieto, M.J. Effective GDNF brain delivery using microspheres—A promising strategy for Parkinson’s disease. J. Control. Release 2009, 135, 119–126. [Google Scholar] [CrossRef]
- Jollivet, C.; Aubert-Pouessel, A.; Clavreul, A.; Venier-Julienne, M.-C.; Montero-Menei, C.N.; Benoit, J.-P.; Menei, P. Long-term effect of intra-striatal glial cell line-derived neurotrophic factor-releasing microspheres in a partial rat model of Parkinson’s disease. Neurosci. Lett. 2004, 356, 207–210. [Google Scholar] [CrossRef]
- Gouhier, C.; Chalon, S.; Aubert-Pouessel, A.; Venier-Julienne, M.-C.; Jollivet, C.; Benoit, J.-P.; Guilloteau, D. Protection of dopaminergic nigrostriatal afferents by GDNF delivered by microspheres in a rodent model of Parkinson’s disease. Synapse 2002, 44, 124–131. [Google Scholar] [CrossRef]
- Aubert-Pouëssel, A.; Venier-Julienne, M.C.; Clavreul, A.; Sergent, M.; Jollivet, C.; Montero-Menei, C.N.; Garcion, E.; Bibby, D.C.; Menei, P.; Benoit, J.-P. In vitro study of GDNF release from biodegradable PLGA microspheres. J. Control. Release 2004, 95, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Gujral, C.; Minagawa, Y.; Fujimoto, K.; Kitano, H.; Nakaji-Hirabayashi, T. Biodegradable microparticles for strictly regulating the release of neurotrophic factors. J. Control. Release 2013, 168, 307–316. [Google Scholar] [CrossRef]
- Kurakhmaeva, K.B.; Djindjikhashvili, I.A.; Petrov, V.E.; Balabanyan, V.U.; Voronina, T.A.; Trofimov, S.S.; Kreuter, J.; Gelperina, S.; Begley, D.; Alyautdin, R.N. Brain targeting of nerve growth factor using poly(butyl cyanoacrylate) nanoparticles. J. Drug Target. 2009, 17, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chen, J.; Feng, C.; Shao, X.; Liu, Q.; Zhang, Q.; Pang, Z.; Jiang, X. Intranasal nanoparticles of basic fibroblast growth factor for brain delivery to treat Alzheimer’s disease. Int. J. Pharm. 2014, 461, 192–202. [Google Scholar] [CrossRef] [PubMed]






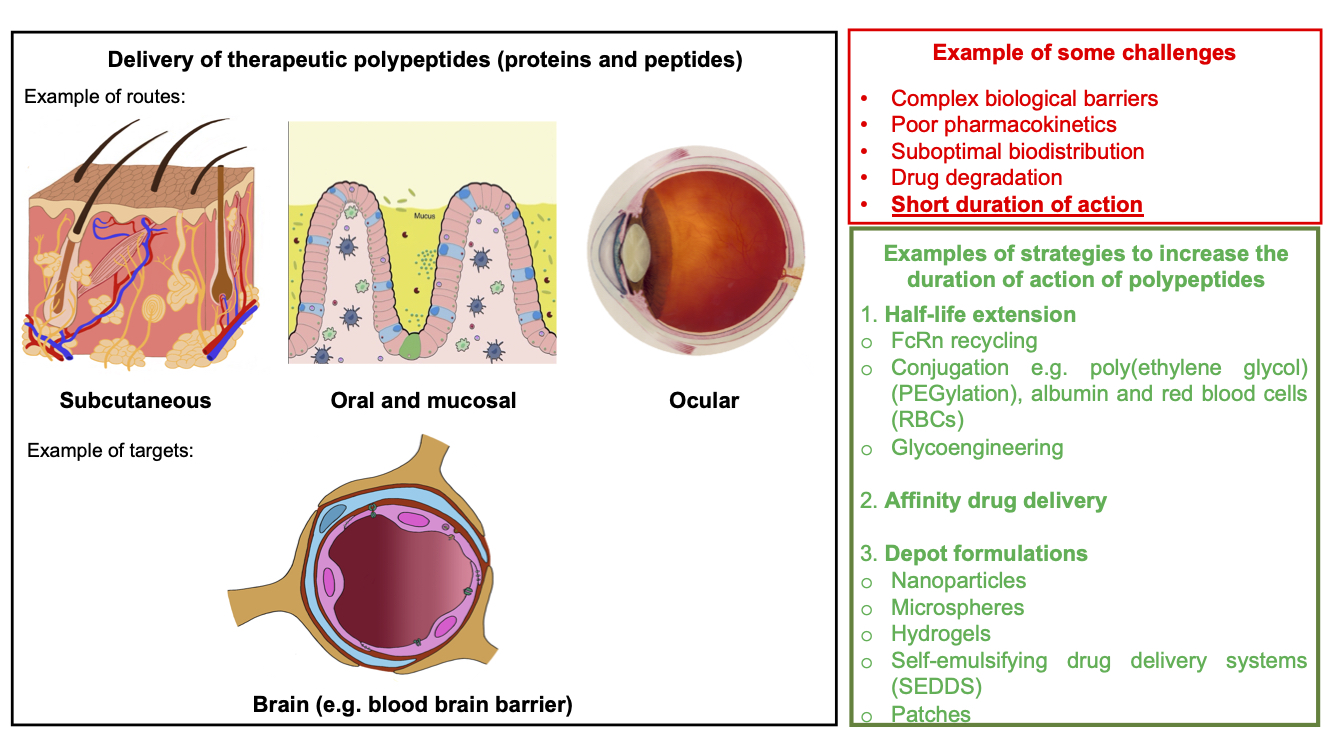{kind=link}
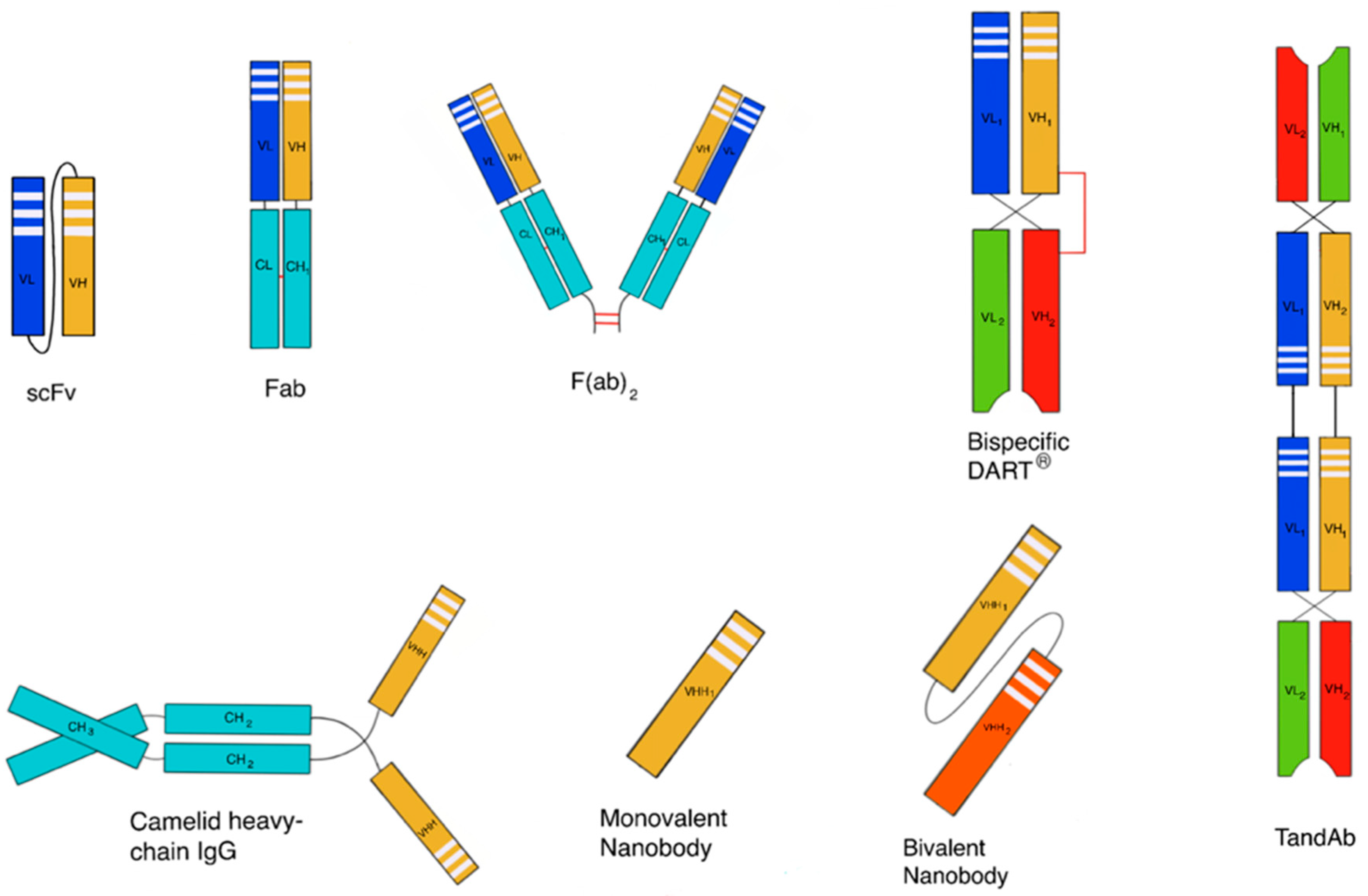{kind=link}
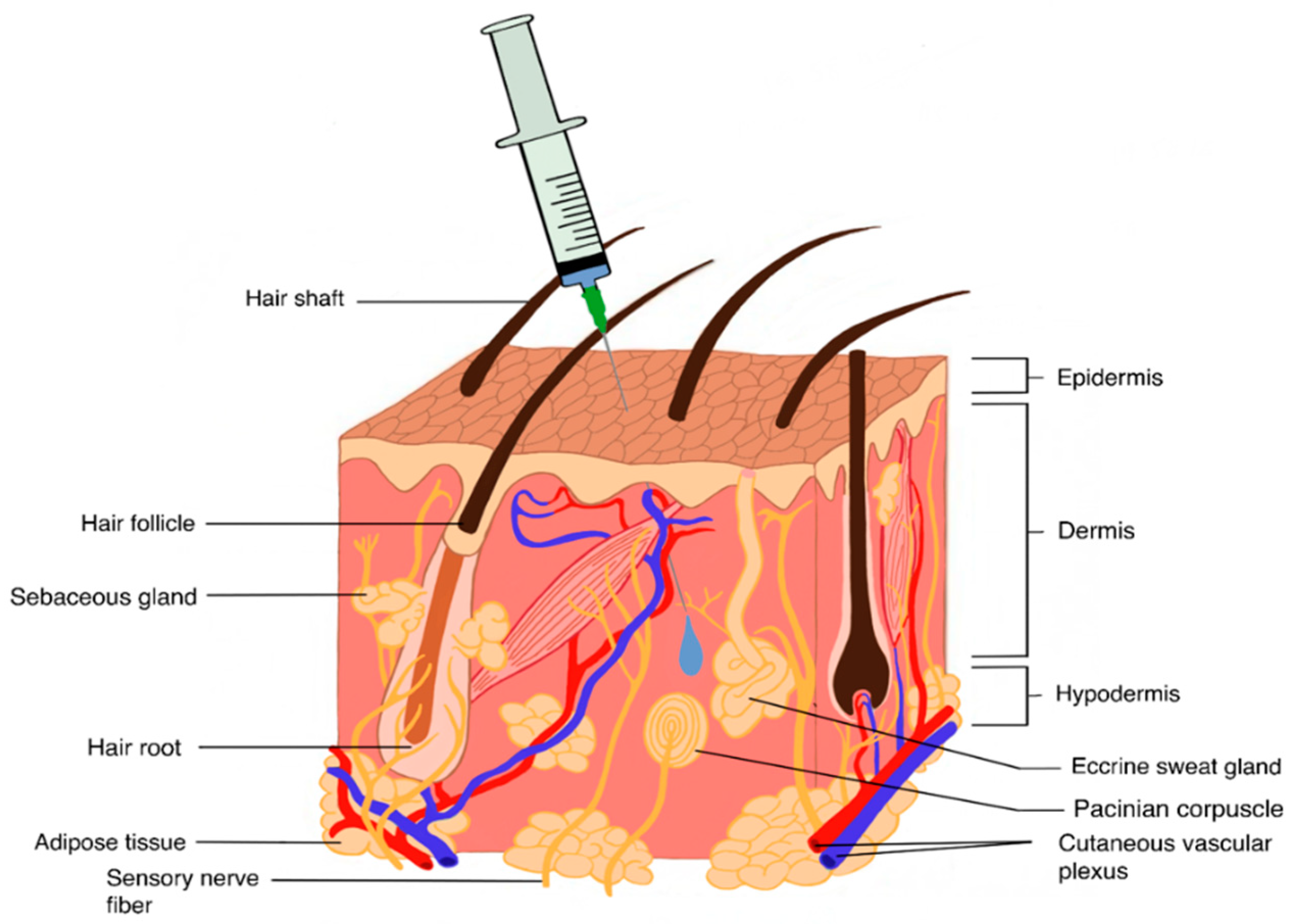{kind=link}
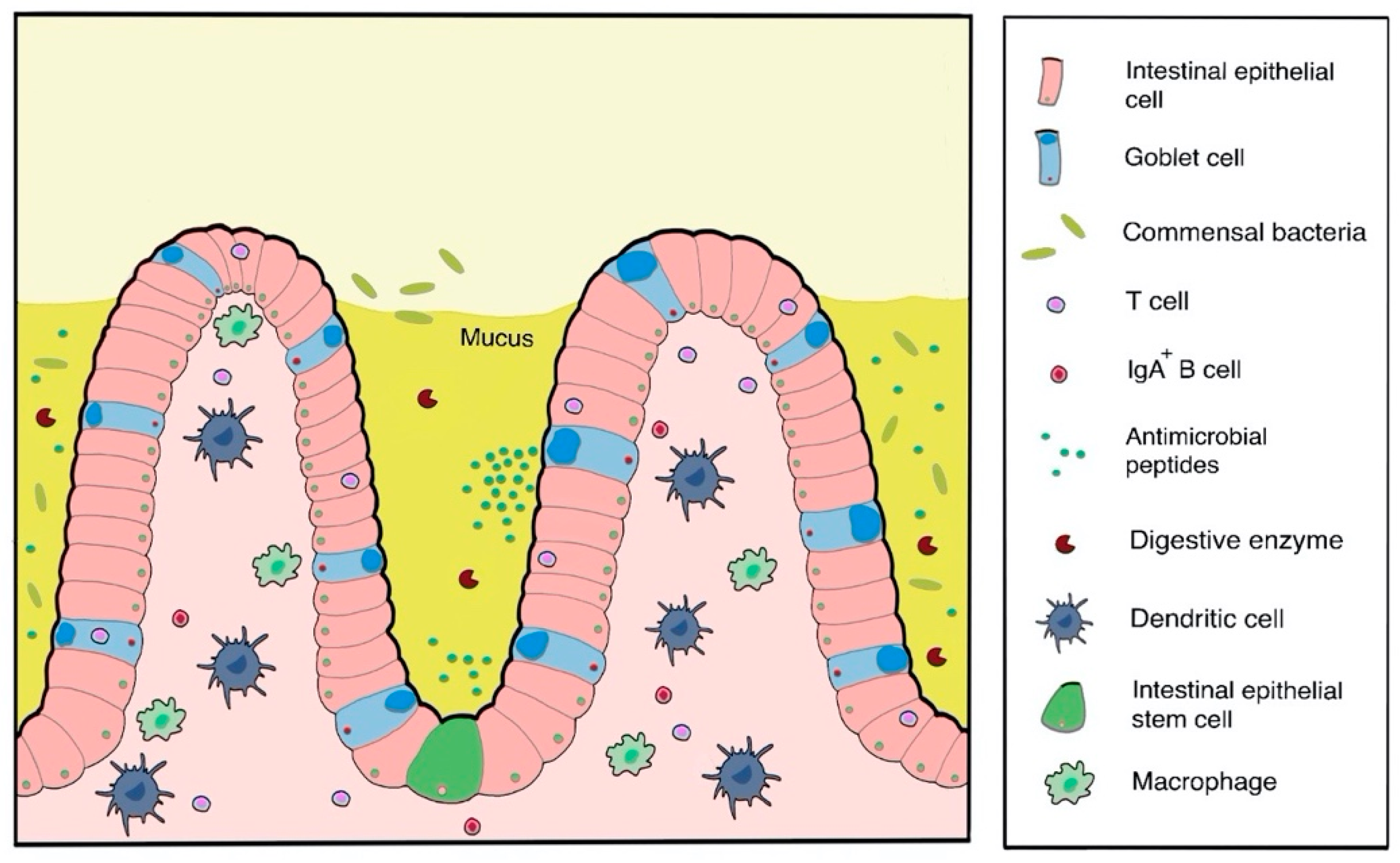{kind=link}
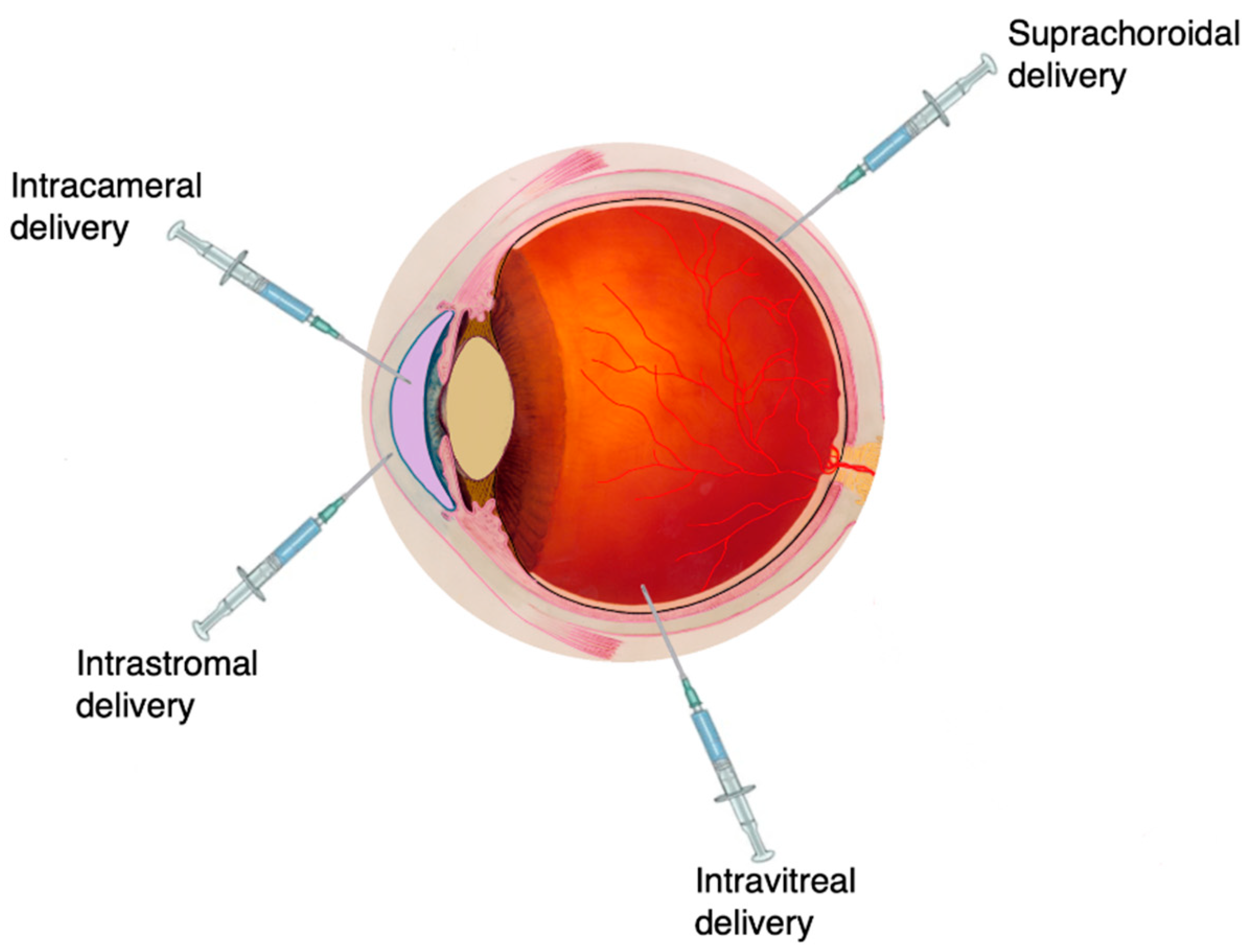{kind=link}
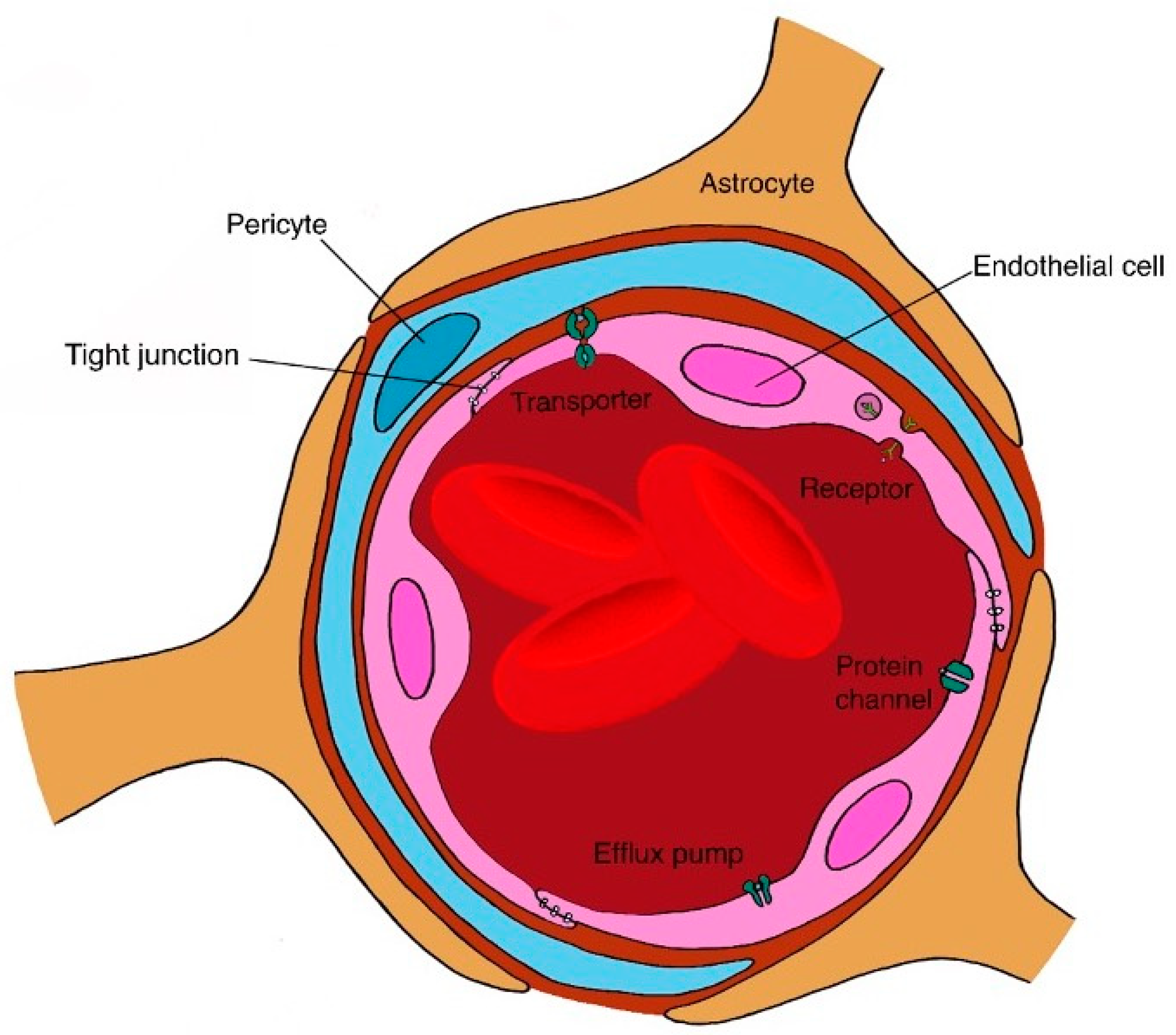{kind=link}
| Route/Target | Examples of Targeted Biomolecules | Barriers to Delivery | Challenges | Examples of Formulation Strategies |
|---|---|---|---|---|
| Routes of delivery | ||||
| SC | Insulin, exenatide, insulin growth factor-1, canakinumab somatotropin, mecasermin, omalizumab, etanercept, and IFN-α2b | Injection site catabolism and limited volume in SC space | Reduced bioavailability, drug degradation | Microspheres, thermoresponsive hydrogels, microparticles, conjugation (e.g., polymers) and affinity DDS |
| Oral | Insulin, octreotide, exenatide, salmon calcitonin, parathyroid hormone, desmopressin, GCSF, erythropoietin, leuprolide and semaglutide | Intestinal mucosa, cellular tight junctions, efflux pumps, pH and enzymes | Drug degradation, decreased absorption and loss of biological activity | Protease inhibitors, absorption enhancers, mucoadhesive systems and polymeric carriers |
| Ocular | Bevacizumab, aflibercept, brolucizumab, pegaptanib, insulin and ranibizumab | Corneal tight junctions, the sclera, efflux pumps, BRB and ILM | Rapid elimination, short half-life and degradation | Nanoparticles, microparticles, liposomes, microneedles, hydrogels, dendrimers, conjugation (e.g., PEG/other polymers), affinity DDS and micelles |
| Target of delivery | ||||
| Brain | Human EGF, FGF-2 and bevacizumab | BBB, CNS, vascular barriers, cellular tight junctions | Short half-life, decreased drug concentration and poor drug distribution | Microparticles, 11 functionalised nanocarriers, conjugation (e.g., PEG) and liposomes |
| Type | Name | Indication | Strategy | Release Mechanism | Dosing |
|---|---|---|---|---|---|
| Leuprolide | Lupron Depot | Prostate cancer | PLGA microparticles | Gradual release from the particulates | Monthly or every 3, 4 or 6 months |
| Eligard | In-situ forming PLGA depot | Gradual release from a depot | Monthly | ||
| Octreotide | Sandostatin LAR | Acromegaly cancer | PLGA microparticles | Gradual release from the particulates | Monthly |
| Lanreotide | Somatuline Autogel | Acromegaly cancer | Self-assembly into nanotubes | Release over a long period | Monthly |
| Goserelin | Zoladex | Prostate cancer | PLGA implant | Gradual release from the implant | Up to 12 weeks administration |
| Insulin | NPH | T1 diabetes | Formulated with Zn[2]+ and protamine | Slowly released from crystalline suspension | 18–24 h |
| Glargine | T1 and T2 diabetes | Formulation in acidic pH (initially, substitution of α Asn21 with Gly addition of 2 Arg groups to β chain) | Initially soluble formulation precipitates upon injection | 20–24 h | |
| GLP-1 agonist | Exenatide (Bydureon®) | T2 diabetes | Encapsulation in PLGA microspheres | Release follows polymer degradation | Weekly |
| Depot System | Therapeutic Biomolecule | Delivery System Design | Example Studies |
|---|---|---|---|
| Patches | Salmon calcitonin, insulin, exenatide | Polymeric matrix system of sodium CMC, carbopol and pectin coated with ethyl cellulose | 3-, 13- and 80-fold increase in half-life of calcitonin, insulin and exenatide, respectively [244] |
| Insulin | Surfactant-coated chitosan-ethyl cellulose patch system | Increase in local insulin concentration and rapid insulin permeation across intestinal epithelium [255] | |
| Insulin | Patch-permeation enhancer system in pH-responsive enteric coated capsule | Unidirectional insulin over 3–4 h demonstrated in release studies in rats [243] | |
| Hydrogels | Calcitonin, human growth hormone | Poly(methacrylic acid-grafted-PEG) and poly(methacrylic acid-co-N-vinyl pyrrolidone) hydrogels | Slow drug release at low pH [256] |
| Anti-TNF antibody | Poly(methacrylic acid-grafted-PEG) and poly(methacrylic acid-co-N-vinyl pyrrolidone) hydrogel microspheres | Serum antibody levels detectable after 4 h [257] | |
| SEDDs | Leuprolide | PEG-35 castor oil, glycerol monocaprylocaprate, propylene glycol and triglyceride-containing SMEDDs | Sustained drug release (50%) within 30 h [258] |
| Octreotide | Oily phase ion-paired complex of octreotide and deoxycholate in SNEDDs | Sustained drug release for at least 24 h [259] | |
| Polymeric and mucoadhesive particulate carriers | Protamine insulin | Mucoadhesive nanoparticles | Increased gut retention time and sustained hypoglycaemic effect in rat models [260] |
| Insulin | Chitosan/dextran sulphate nanoparticles | Sustained release over 10 h [257] | |
| Insulin | EDTA-poly(glutamic acid)-chitosan degradable nanoparticles | Prolonged hypoglycaemic effect by tight junction permeation [261] | |
| Insulin | HA pH-responsive nanoparticles | Hypoglycaemic effect lasting up to 8 h and drug protection against enzymatic degradation [262] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibeanu, N.; Egbu, R.; Onyekuru, L.; Javaheri, H.; Tee Khaw, P.; R. Williams, G.; Brocchini, S.; Awwad, S. Injectables and Depots to Prolong Drug Action of Proteins and Peptides. Pharmaceutics 2020, 12, 999. https://doi.org/10.3390/pharmaceutics12100999
Ibeanu N, Egbu R, Onyekuru L, Javaheri H, Tee Khaw P, R. Williams G, Brocchini S, Awwad S. Injectables and Depots to Prolong Drug Action of Proteins and Peptides. Pharmaceutics. 2020; 12(10):999. https://doi.org/10.3390/pharmaceutics12100999
Chicago/Turabian StyleIbeanu, Nkiruka, Raphael Egbu, Lesley Onyekuru, Hoda Javaheri, Peng Tee Khaw, Gareth R. Williams, Steve Brocchini, and Sahar Awwad. 2020. "Injectables and Depots to Prolong Drug Action of Proteins and Peptides" Pharmaceutics 12, no. 10: 999. https://doi.org/10.3390/pharmaceutics12100999
APA StyleIbeanu, N., Egbu, R., Onyekuru, L., Javaheri, H., Tee Khaw, P., R. Williams, G., Brocchini, S., & Awwad, S. (2020). Injectables and Depots to Prolong Drug Action of Proteins and Peptides. Pharmaceutics, 12(10), 999. https://doi.org/10.3390/pharmaceutics12100999