Assessing Drug Interaction and Pharmacokinetics of Loxoprofen in Mice Treated with CYP3A Modulators

 ,
,
Abstract
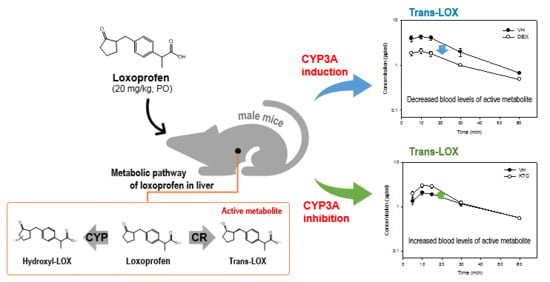
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animal Treatments and Sample Preparations
2.3. CYP Activities in the Mouse Liver
2.4. Instrument and Data Acquisition
2.5. Method Validation
2.6. Statistical Analysis
3. Results
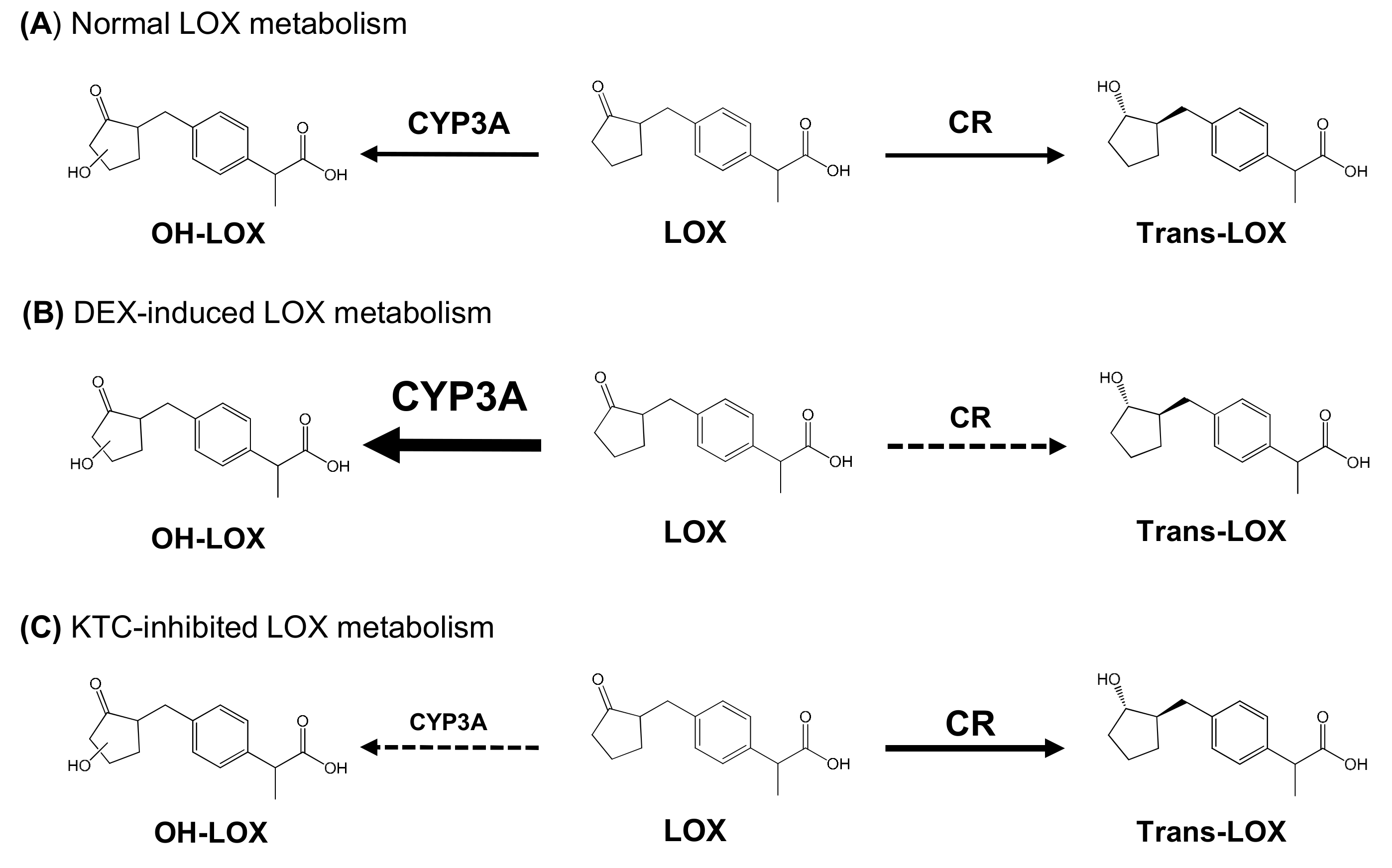
3.1. Identification of Loxoprofen and Its Metabolites
3.2. Evaluation Model for Determination of LOX–Drug Interaction
3.3. Pharmacokinetic Analysis
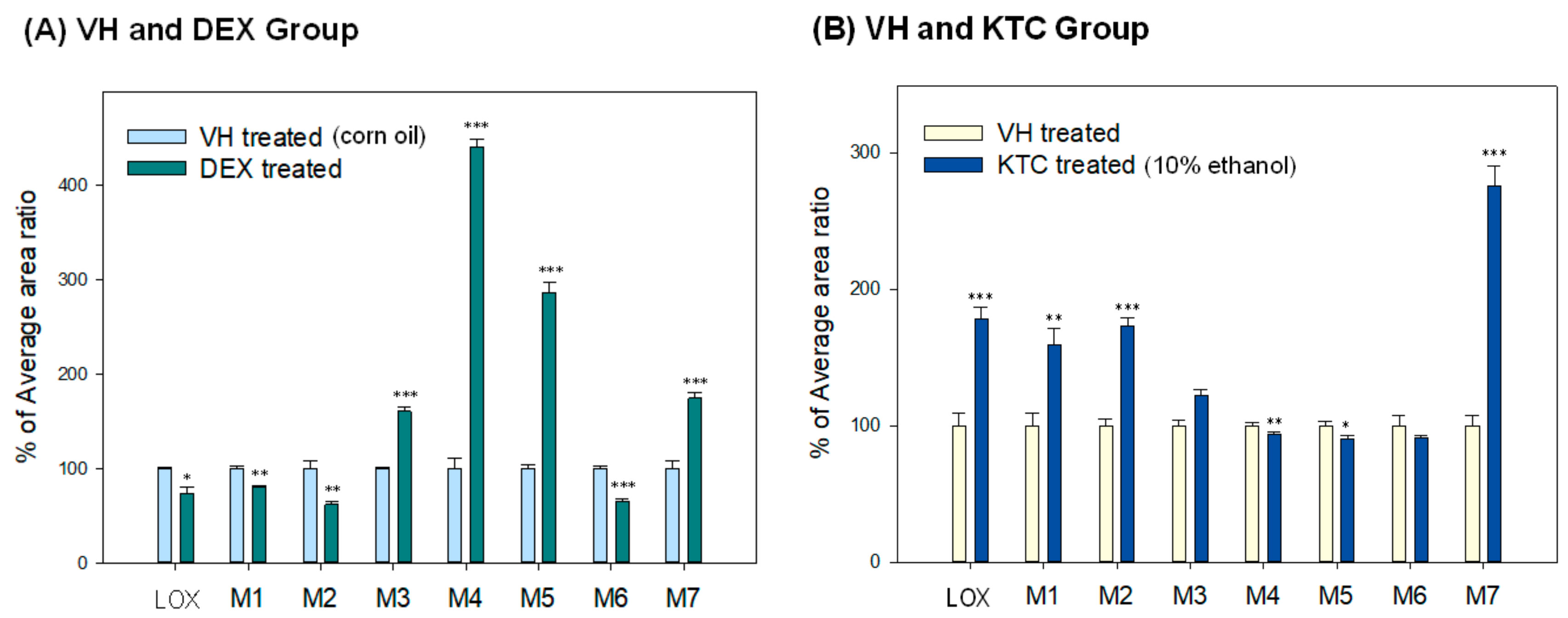
3.4. Metabolism and Metabolite Identification of LOX During DEX or KTC Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Greig, S.L.; Garnock-Jones, K.P. Loxoprofen: A review in pain and inflammation. Clin. Drug Investig. 2016, 36, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Wan, D.; Zhao, M.; Zhang, J.; Luan, L. Development and In Vitro-In Vivo Evaluation of a Novel Sustained-Release Loxoprofen Pellet with Double Coating Layer. Pharmaceutics 2019, 11, 260. [Google Scholar] [CrossRef] [PubMed]
- Helmy, S.A. Pharmacokinetics and Bioequivalence Evaluation of 2 Loxoprofen Tablets in Healthy Egyptian Male Volunteers. Clin. Pharmacol. Drug Dev. 2013, 2, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, N.; Suemasu, S.; Watanabe, H.; Tahara, K.; Tanaka, K.-I.; Okamoto, Y.; Ohtsuka, M.; Maruyama, T.; Mizushima, T. Comparison of pharmacokinetics between loxoprofen and its derivative with lower ulcerogenic activity, fluoro-loxoprofen. Drug Metab. Pharmacok. 2013, 28, 118–124. [Google Scholar] [CrossRef]
- Kang, H.-A.; Cho, H.-Y.; Lee, Y.-B. Bioequivalence of hana loxoprofen sodium tablet to dongwha Loxonin® tablet (Loxoprofen Sodium 60 mg). J. Pharm. Investig. 2011, 41, 117–123. [Google Scholar] [CrossRef]
- Moore, N.; Pollack, C.; Butkerait, P. Adverse drug reactions and drug–drug interactions with over-the-counter NSAIDs. Ther. Clin. Risk. Manag. 2015, 11, 1061–1075. [Google Scholar] [PubMed]
- Sawamura, R.; Kazui, M.; Kurihara, A.; Izumi, T. Absorption, distribution, metabolism and excretion of loxoprofen after dermal application of loxoprofen gel to rats. Xenobiotica 2014, 44, 1026–1038. [Google Scholar] [CrossRef]
- Takeda, M.; Khamdang, S.; Narikawa, S.; Kimura, H.; Hosoyamada, M.; Cha, S.H.; Sekine, T.; Endou, H. Characterization of methotrexate transport and its drug interactions with human organic anion transporters. J. Pharmacol. Exp. Ther. 2002, 302, 666–671. [Google Scholar] [CrossRef]
- Yue, Z.; Shi, J.; Jiang, P.; Sun, H. Acute kidney injury during concomitant use of valacyclovir and loxoprofen: Detecting drug-drug interactions in a spontaneous reporting system. Pharmacoepidemiol. Drug Saf. 2014, 23, 1154–1159. [Google Scholar] [CrossRef]
- Takahashi, H.; Kashima, T.; Kimura, S.; Murata, N.; Takaba, T.; Iwade, K.; Abe, T.; Tainaka, H.; Yasumori, T.; Echizen, H. Pharmacokinetic interaction between warfarin and a uricosuric agent, bucolome: Application of in vitro approaches to predicting in vivo reduction of (S)-warfarin clearance. Drug Metab. Dispos. 1999, 27, 1179–1186. [Google Scholar]
- Dinger, J.; Meyer, M.R.; Maurer, H.H. Development of an in vitro cytochrome P450 cocktail inhibition assay for assessing the inhibition risk of drugs of abuse. Toxicol. Lett. 2014, 230, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Ab Rahman, N.S.; Abd Majid, F.A.; Wahid, A.; Effendy, M.; Zainudin, A.N.; Zainol, S.N.; Ismail, H.F.; Wong, T.S.; Tiwari, N.K.; Giri, S. Evaluation of Herb-Drug Interaction of Synacinn™ and Individual Biomarker through Cytochrome 450 Inhibition Assay. Drug Metab. Lett. 2018, 12, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Nishikawa, Y.; Hayashi, R. Species differences in metabolism of sodium 2-[4-(2-oxocyclopentylmethyl)-phenyl] propionate dihydrate (loxoprofen sodium), a new anti-inflammatory agent. Chem. Pharm. Bull. 1983, 31, 3656–3664. [Google Scholar] [CrossRef] [PubMed]
- Jhee, O.H.; Lee, M.H.; Shaw, L.M.; Lee, S.E.; Park, J.H.; Kang, J.S. Pharmacokinetics and bioequivalence study of two brands of loxoprofen tablets in healthy volunteers. Arzneimittelforschung 2007, 57, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, R.; Cho, P.; Paudel, S.; Shrestha, A.; Kang, M.; Jeong, T.; Lee, E.-S.; Lee, S. Exploring the Metabolism of Loxoprofen in Liver Microsomes: The Role of Cytochrome P450 and UDP-Glucuronosyltransferase in Its Biotransformation. Pharmaceutics 2018, 10, 112. [Google Scholar] [CrossRef]
- Zhou, S.-F. Drugs behave as substrates, inhibitors and inducers of human cytochrome P450 3A4. Curr. Drug Metab. 2008, 9, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Basheer, L.; Kerem, Z. Interactions between CYP3A4 and dietary polyphenols. Oxid. Med. Cell. Longev. 2015, 2015. [Google Scholar] [CrossRef]
- Naruto, S.; Terada, A. Synthesis of the eight possible optically active isomers of 2-[4-(2-hydroxycyclopentylmethyl) phenyl] propionic acid. Chem. Pharm. Bull. 1983, 31, 4319–4323. [Google Scholar] [CrossRef]
- Seervi, M.; Lotankar, S.; Barbar, S.; Sathaye, S. Assessment of cytochrome P450 inhibition and induction potential of lupeol and betulin in rat liver microsomes. Drug Metab. Pers. Ther. 2016, 31, 115–122. [Google Scholar] [CrossRef]
- Seneca, N.; Zoghbi, S.S.; Shetty, H.U.; Tuan, E.; Kannan, P.; Taku, A.; Innis, R.B.; Pike, V.W. Effects of ketoconazole on the biodistribution and metabolism of [11C] loperamide and [11C] N-desmethyl-loperamide in wild-type and P-gp knockout mice. Nucl. Med. Biol. 2010, 37, 335–345. [Google Scholar] [CrossRef]
- Shrestha, R.; Kim, J.-H.; Nam, W.; Lee, H.S.; Lee, J.-M.; Lee, S. Selective inhibition of CYP2C8 by fisetin and its methylated metabolite, geraldol, in human liver microsomes. Drug Metab. Pharmacok. 2018, 33, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Sim, J.; Nam, W.; Lee, D.; Lee, S.; Hungchan, O.; Joo, J.; Liu, K.-H.; Han, J.Y.; Ki, S.H.; Jeong, T.C. Selective induction of hepatic cytochrome P450 2B activity by leelamine in vivo, as a potent novel inducer. Arch. Pharmacal. Res. 2015, 38, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Hamaguchi, M.; Seno, T.; Yamamoto, A.; Kohno, M.; Kadoya, M.; Ishino, H.; Ashihara, E.; Kimura, S.; Tsubakimoto, Y.; Takata, H. Loxoprofen sodium, a non-selective NSAID, reduces atherosclerosis in mice by reducing inflammation. J. Clin. Biochem. Nutr. 2010, 47, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, H.; Inoue, G.; Nakazawa, T.; Imura, T.; Saito, W.; Uchida, K.; Miyagi, M.; Takahira, N.; Takaso, M. Loxoprofen sodium and celecoxib for postoperative pain in patients after spinal surgery: A randomized comparative study. J. Orthop. Sci. 2015, 20, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, H.; Tanaka, Y.; Watanabe, H.; Hayashi, R.; Kawada, K. Optical inversion of (2R)-to (2S)-isomers of 2-[4-(2-oxocyclopentylmethyl)-phenyl] propionic acid (loxoprofen), a new anti-inflammatory agent, and its monohydroxy metabolites in the rat. Chem. Pharm. Bull. 1984, 32, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Foti, R.S.; Dalvie, D.K. Cytochrome P450 and non–cytochrome P450 oxidative metabolism: Contributions to the pharmacokinetics, safety, and efficacy of xenobiotics. Drug Metab. Dispos. 2016, 44, 1229–1245. [Google Scholar] [CrossRef]
- Tanaka, Y.; Nishikawa, Y.; Matsuda, K.; Yamazaki, M.; Hayashi, R. Purification and some properties of ketone reductase forming an active metabolite of sodium 2-[4-(2-oxocyclopentylmethyl)-phenyl] propionate dihydrate (loxoprofen sodium), a new anti-inflammatory agent, in rabbit liver cytosol. Chem. Pharm. Bull. 1984, 32, 1040–1048. [Google Scholar] [CrossRef]
- Sawamura, R.; Kazui, M.; Kurihara, A.; Izumi, T. Pharmacokinetics of loxoprofen and its active metabolite after dermal application of loxoprofen gel to rats. Pharm. 2015, 70, 74–80. [Google Scholar]
- Verhoef, C.M.; van Roon, J.A.; Vianen, M.E.; Lafeber, F.P.; Bijlsma, J.W. The immune suppressive effect of dexamethasone in rheumatoid arthritis is accompanied by upregulation of interleukin 10 and by differential changes in interferon γ and interleukin 4 production. Ann. Rheum. Dis. 1999, 58, 49–54. [Google Scholar] [CrossRef]
- Liu, D.; Ahmet, A.; Ward, L.; Krishnamoorthy, P.; Mandelcorn, E.D.; Leigh, R.; Brown, J.P.; Cohen, A.; Kim, H. A practical guide to the monitoring and management of the complications of systemic corticosteroid therapy. Allergy Asthma Clin. Immunol. 2013, 9, 30. [Google Scholar] [CrossRef] [PubMed]
- Cook, A.M.; McDonnell, A.M.; Lake, R.A.; Nowak, A.K. Dexamethasone co-medication in cancer patients undergoing chemotherapy causes substantial immunomodulatory effects with implications for chemo-immunotherapy strategies. Oncoimmunology 2016, 5, e1066062. [Google Scholar] [CrossRef] [PubMed]
- Pufall, M.A. Glucocorticoids and Cancer. Adv. Exp. Med. Biol. 2015, 872, 315–333. [Google Scholar] [CrossRef] [PubMed]
- Al Katheeri, N.; Wasfi, I.; Lambert, M.; Albo, A.G.; Nebbia, C. In vivo and in vitro metabolism of dexamethasone in the camel. Vet. J. 2006, 172, 532–543. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, E.; Maggs, J.; Park, B.; Back, D. Dexamethasone metabolism in vitro: Species differences. J. Steroid. Biochem. 1997, 62, 345–352. [Google Scholar] [CrossRef]
- Tomlinson, E.; Lewis, D.; Maggs, J.; Kroemer, H.; Park, B.; Back, D. In vitro metabolism of dexamethasone (DEX) in human liver and kidney: The involvement of CYP3A4 and CYP17 (17, 20 LYASE) and molecular modelling studies. Biochem. Pharmacol. 1997, 54, 605–611. [Google Scholar] [CrossRef]
- Iwanaga, K.; Honjo, T.; Miyazaki, M.; Kakemi, M. Time-dependent changes in hepatic and intestinal induction of cytochrome P450 3A after administration of dexamethasone to rats. Xenobiotica 2013, 43, 765–773. [Google Scholar] [CrossRef]
- Li, J.; Chen, R.; Yao, Q.-Y.; Liu, S.-J.; Tian, X.-Y.; Hao, C.-Y.; Lu, W.; Zhou, T.-Y. Time-dependent pharmacokinetics of dexamethasone and its efficacy in human breast cancer xenograft mice: A semi-mechanism-based pharmacokinetic/pharmacodynamic model. Acta Pharmacol. Sin. 2018, 39, 472–481. [Google Scholar] [CrossRef]
- Sohn, C.A. Evaluation of ketoconazole. Clin. Pharm. 1982, 1, 217–224. [Google Scholar]
- Jones, H.E. Ketoconazole. Arch. Dermatol. 1982, 118, 217–219. [Google Scholar] [CrossRef]
- Rodriguez, R.J.; Acosta, D. Metabolism of ketoconazole and deacetylated ketoconazole by rat hepatic microsomes and flavin-containing monooxygenases. Drug Metab. Dispos. 1997, 25, 772–777. [Google Scholar] [PubMed]
- Kim, J.-H.; Choi, W.-G.; Lee, S.; Lee, H. Revisiting the metabolism and bioactivation of ketoconazole in human and mouse using liquid chromatography-mass spectrometry-based metabolomics. Int. J. Mol. Sci. 2017, 18, 621. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Chan, S.Y.; Goh, B.C.; Chan, E.; Duan, W.; Huang, M.; McLeod, H.L. Mechanism-based inhibition of cytochrome P450 3A4 by therapeutic drugs. Clin. Pharmacok. 2005, 44, 279–304. [Google Scholar] [CrossRef] [PubMed]
- Thummel, K.; Wilkinson, G. In vitro and in vivo drug interactions involving human CYP3A. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 389–430. [Google Scholar] [CrossRef] [PubMed]
- Bourcier, K.; Hyland, R.; Kempshall, S.; Jones, R.; Maximilien, J.; Irvine, N.; Jones, B. Investigation into UDP-glucuronosyltransferase (UGT) enzyme kinetics of imidazole-and triazole-containing antifungal drugs in human liver microsomes and recombinant UGT enzymes. Drug Metab. Dispos. 2010, 38, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Colaizzi, J.; Bierman, R.; Woestenborghs, R.; Heykants, J. Pharmacokinetics and dose proportionality of ketoconazole in normal volunteers. Antimicrob. Agents Chemother. 1986, 30, 206–210. [Google Scholar] [CrossRef]
- Hukkanen, J. Induction of cytochrome P450 enzymes: A view on human in vivo findings. Expert Rev. Clin. Pharmacol. 2012, 5, 569–585. [Google Scholar] [CrossRef]
- Gentile, D.M.; Tomlinson, E.S.; Maggs, J.L.; Park, B.K.; Back, D.J. Dexamethasone metabolism by human liver in vitro. Metabolite identification and inhibition of 6-hydroxylation. J. Pharmacol. Exp. Ther. 1996, 277, 105–112. [Google Scholar]
- Kaeser, B.; Zandt, H.; Bour, F.; Zwanziger, E.; Schmitt, C.; Zhang, X. Drug-drug interaction study of ketoconazole and ritonavir-boosted saquinavir. Antimicrob. Agents Chemother. 2009, 53, 609–614. [Google Scholar] [CrossRef]
- Fujimori, S.; Hanada, R.; Hayashida, M.; Sakurai, T.; Ikushima, I.; Sakamoto, C. Celecoxib Monotherapy Maintained Small Intestinal Mucosa Better Compared with Loxoprofen Plus Lansoprazole Treatment. J. Clin. Gastroenterol. 2016, 50, 218–226. [Google Scholar] [CrossRef]
- Mizukami, K.; Murakami, K.; Yamauchi, M.; Matsunari, O.; Ogawa, R.; Nakagawa, Y.; Okimoto, T.; Kodama, M.; Fujioka, T. Evaluation of selective cyclooxygenase-2 inhibitor-induced small bowel injury: Randomized cross-over study compared with loxoprofen in healthy subjects. Dig. Endosc. 2013, 25, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Cascorbi, I. Drug interactions-principles, examples and clinical consequences. Dtsch. Arztebl. Int. 2012, 109, 546–556. [Google Scholar] [PubMed]
- Li, X.-Q.; Andersson, T.B.; Ahlström, M.; Weidolf, L. Comparison of inhibitory effects of the proton pump-inhibiting drugs omeprazole, esomeprazole, lansoprazole, pantoprazole, and rabeprazole on human cytochrome P450 activities. Drug Metab. Dispos. 2004, 32, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, K. Metabolism of tacrolimus (FK506) and recent topics in clinical pharmacokinetics. Drug Metab. Pharmacok. 2007, 22, 328–335. [Google Scholar] [CrossRef]
- Milanov, I.; Georgiev, D. Mechanisms of tizanidine action on spasticity. Acta Neurol. Scand. 1994, 89, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jin, Y. Irreversible profound symptomatic bradycardia requiring pacemaker after tizanidine/loxoprofen combination therapy: A case report. J. Int. Med.Res. 2018, 46, 2466–2469. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analytes | Parameters | VH (Corn Oil) | DEX |
|---|---|---|---|
| LOX | Cmax (µg/mL) | 4.8 ± 0.9 | 2.5 ± 0.2 ** |
| Tmax (min) | 5.0 ± 0.0 | 5.0 ± 0.0 | |
| AUC(0–60) (µg·min/mL) | 95.7 ± 14.5 | 53.5 ± 6.1 ** | |
| T1/2 (min) | 14.9 ± 0.6 | 12.0 ± 0.7 * | |
| AUC(0–∞) (µg·min/mL) | 102.2 ± 15.0 | 56.2 ± 6.9 *** | |
| cis-LOX | Cmax (µg/mL) | 2.4 ± 0.3 | 1.1 ± 0.2 ** |
| Tmax (min) | 10.4 ± 0.7 | 10.0 ± 1.0 | |
| AUC(0–60) (µg·min/mL) | 72.7 ± 12.4 | 29.9 ± 4.4 ** | |
| T1/2 (min) | 12.3 ± 0.3 | 13.9 ± 0.6 * | |
| AUC(0–∞) (µg·min/mL) | 75.8 ± 12.8 | 31.5 ± 4.4 ** | |
| trans-LOX | Cmax (µg/mL) | 4.4 ± 0.5 | 2.1 ± 0.2 ** |
| Tmax (min) | 9.1 ± 1.2 | 10.4 ± 0.9 | |
| AUC(0–60) (µg·min/mL) | 137.4 ± 19.0 | 67.6 ± 5.7 ** | |
| T1/2 (min) | 18.2 ± 0.6 | 26.4 ± 1.6 ** | |
| AUC(0–∞) (µg·min/mL) | 154.8 ± 19.2 | 85.8 ± 5.0 ** |
| Analytes | Parameters | VH (10% Ethanol) | KTC |
|---|---|---|---|
| LOX | Cmax (µg/mL) | 2.7 ± 0.3 | 3.5 ± 0.5 |
| Tmax (min) | 5.0 ± 0.0 | 5.0 ± 0.0 | |
| AUC(0–60) (µg·min/mL) | 54.7 ± 4.6 | 66.6 ± 6.6 | |
| T1/2 (min) | 14.6 ± 1.1 | 12.2 ± 0.9 | |
| AUC(0–∞) (µg·min/mL) | 59.1 ± 5.6 | 68.9 ± 6.5 | |
| cis-LOX | Cmax (µg/mL) | 1.2 ± 0.1 | 1.6 ± 0.1 * |
| Tmax (min) | 13.3 ± 0.8 | 11.6 ± 0.8 | |
| AUC(0–60) (µg·min/mL) | 36.0 ± 3.9 | 49.0 ± 5.9 * | |
| T1/2 (min) | 13.3 ± 0.9 | 13.4 ± 0.6 | |
| AUC(0–∞) (µg·min/mL) | 38.4 ± 4.4 | 51.9 ± 6.1 * | |
| trans-LOX | Cmax (µg/mL) | 2.1 ± 0.2 | 3.1 ± 0.3 ** |
| Tmax (min) | 11.6 ± 0.8 | 11.7 ± 0.8 | |
| AUC(0–60) (µg·min/mL) | 69.9 ± 6.0 | 80.4 ± 9.6 * | |
| T1/2 (min) | 26.0 ± 0.5 | 19.8 ± 0.7 ** | |
| AUC(0–∞) (µg·min/mL) | 90.9 ± 7.1 | 94.7 ± 11.0 |
| Compounds | Parent Ions (m/z) | Elemental Composition | Error (ppm) | Product Ions (m/z) | Description |
|---|---|---|---|---|---|
| Lox | 245.1179 | C15H17O3 | 0.4 | 83.0492 | LOX |
| M1 | 247.1339 | C15H19O3 | 2.0 | 233.1181, 217.1230, 201.1279, 191.1071 | Trans-LOX |
| M2 | 247.1336 | C15H19O3 | 0.5 | 217.1230, 191.1071 | Cis-LOX |
| M3 | 261.1138 | C15H17O4 | 4.2 | 99.0441, 81.0335 | OH-LOX |
| M4 | 261.1133 | C15H17O4 | 2.3 | 99.0441 | OH-LOX |
| M5 | 263.1288 | C15H19O4 | 1.9 | 233.1181, 207.1022, 133.0650, 99.0442 | OH-trans-LOX |
| M6 | 354.1382 | C17H24O5NS | 2.0 | 149.9859, 124.0065, 79.9563 | Taurine conjugate |
| M7 | 421.1514 | C21H25O9 | 3.6 | 245.1182, 193.0348, 175.0242, 83.0492 | Glucuronide conjugate |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paudel, S.; Shrestha, A.; Cho, P.; Shrestha, R.; Kim, Y.; Lee, T.; Kim, J.-H.; Jeong, T.C.; Lee, E.-S.; Lee, S. Assessing Drug Interaction and Pharmacokinetics of Loxoprofen in Mice Treated with CYP3A Modulators. Pharmaceutics 2019, 11, 479. https://doi.org/10.3390/pharmaceutics11090479
Paudel S, Shrestha A, Cho P, Shrestha R, Kim Y, Lee T, Kim J-H, Jeong TC, Lee E-S, Lee S. Assessing Drug Interaction and Pharmacokinetics of Loxoprofen in Mice Treated with CYP3A Modulators. Pharmaceutics. 2019; 11(9):479. https://doi.org/10.3390/pharmaceutics11090479
Chicago/Turabian StylePaudel, Sanjita, Aarajana Shrestha, Piljoung Cho, Riya Shrestha, Younah Kim, Taeho Lee, Ju-Hyun Kim, Tae Cheon Jeong, Eung-Seok Lee, and Sangkyu Lee. 2019. "Assessing Drug Interaction and Pharmacokinetics of Loxoprofen in Mice Treated with CYP3A Modulators" Pharmaceutics 11, no. 9: 479. https://doi.org/10.3390/pharmaceutics11090479
APA StylePaudel, S., Shrestha, A., Cho, P., Shrestha, R., Kim, Y., Lee, T., Kim, J.-H., Jeong, T. C., Lee, E.-S., & Lee, S. (2019). Assessing Drug Interaction and Pharmacokinetics of Loxoprofen in Mice Treated with CYP3A Modulators. Pharmaceutics, 11(9), 479. https://doi.org/10.3390/pharmaceutics11090479