2.1. Materials
The anticoagulant warfarin was the drug investigated in the present study. Warfarin sodium (WS) loaded into the orodispersible films (ODFs) was acquired from Sigma-Aldrich (St. Louis, MO, USA), and the WS present in the compounded oral powders in unit dose sachets (OPSs) was obtained from commercial Marevan forte 5 mg tablets (Orion Pharma, Espoo, Finland). Lactose monohydrate (parve granules, Oriola, Espoo, Finland) was used as a filler in the OPS together with the ground tablets. Hydroxypropylcellulose (HPC, Klucel™ EXF, MW 80,000), which was used as a film-forming agent for both the EXT and IJP ODFs, was kindly donated by Ashland (Schaffhausen, Switzerland). Quinoline yellow (Sigma-Aldrich, Bangalore, India) and propylene glycol (PG) ≥ 99.5% (Sigma-Aldrich, St. Louis, MO, USA) were added to the IJP ink due to their respective properties as a colorant and viscosity/surface tension modifier. Ethanol ≥ 94% (Etax A, Altia, Helsinki, Finland) and purified water (Milli-Q water, Millipore SA-67120, Millipore, Molsheim, France) were used for analytics and as solvents in the polymer and ink solutions.
2.2. Methods
2.2.1. Manufacturing of Personalized Doses
Target doses of 0.1, 0.5, 1, and 2 mg, were prepared by three different manufacturing methods. Two new innovative manufacturing methods in the pharmaceutical field, namely semi-solid extrusion 3D printing (EXT) and 2D inkjet printing (IJP), were compared to the established manufacturing method for oral powder unit dose sachets (OPSs) compounded at HUS Pharmacy, the hospital pharmacy of HUS Helsinki University Hospital (HUS) and subsequently used at New Children’s Hospital in Finland. The drug and the dose levels were selected based on an analysis of compounded OPSs at HUS Pharmacy in the year of 2018, which revealed that 1075 out of the 13,000 OPS manufactured at HUS Pharmacy contained the drug WS. The analysis additionally showed that the compounded WS doses were between 0.1 and 2.3 mg, where the most frequently compounded doses were 0.5 and 1 mg.
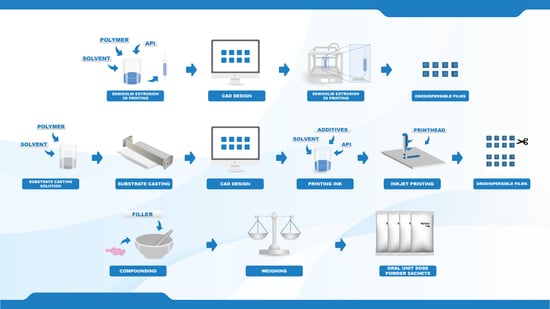
2.2.2. Film Designs
Squared films with four different aimed doses were designed (
Table 1) using a computer-aided design software (Inventor Professional software, version 2019, Autodesk, San Rafael, CA, USA) for the EXT ODFs and PowerPoint (version 2016, Microsoft Office, Microsoft Corporation, Redmond, WA, USA) for the IJP ODFs. EXT and IJP films were designed to have the same size, however, the printed area was designed to be slightly smaller for the IJP film to allow for manual cutting of the film after the printing step. A cutting template with the final size of the IJP film was also designed in PowerPoint. The EXT ODF designs were saved as .stl files and imported into the slicer software (RepertierHost v1.6.1, Hot-World GmbH and Co. KG, Willich, Germany) where the print settings were set, and the g-code was generated. The IJP designs were saved as .bmp files and imported into the printing software where the printing parameters were determined.
The film sizes were determined based on the assumption of what could be handled by a nurse at the hospital as well as physical considerations of pediatrics. The smallest film size was restricted by the size still manageable to handle, and the biggest film was limited by the size of a child’s mouth. The different sizes for the EXT ODFs were designed to increase in volume in the same ratio as the dose escalation in order to enable the use of the same printing solution for manufacturing of all sizes. The final sizes of the IJP ODFs were designed to be equal to the sizes designed for the EXT, as displayed in
Table 1.
2.2.3. Semisolid Extrusion 3D Printing
Printing Solution
The drug concentration in the HPC solution was determined by printing films (n = 6) of all designed sizes using a placebo HPC solution. The wet weight of the printed films was used to calculate the percent WS needed in the polymer solution to obtain the targeted doses. The average WS drug load for all of the different sizes based on the calculations was the selected drug load.
The placebo printing solution for the EXT was prepared by dissolving 15% (w/w) HPC in an ethanol and purified water mixture (ratio 1:1). The drug-loaded printing solution was prepared in a similar manner where 1.5% (w/w) WS and 15% (w/w) HPC were dissolved in a mixture of ethanol and purified water (ratio 1:1). The solutions were left on a magnetic stirrer overnight at room temperature to allow the polymer to fully dissolve.
Semisolid Extrusion 3D Printing
The prepared printing solutions were transferred into 10 mL disposable syringes attached to a single-use 25 G electro-polished tip (1/4″ Techcon TE Needle, Ellsworth adhesives, Norsborg, Sweden). The Biobots 1 (Biobot, Philadelphia, PA, USA) EXT equipped with an air compressor was used to print both placebo and drug-loaded ODFs. Films were printed on pieces of transparency sheets with a set pressure of 25 PSI and a printing layer height of 0.1 mm. One vertical shell was printed, and the outlines were subsequently filled in using a rectilinear fill pattern with a 45° fill angle, an infill density of 100% and an infill overlap of 15%. All printing steps were conducted with a speed of 8 mm/s. Within each batch, one film was printed at a time, and the films were let to dry overnight in room temperature.
EXT films were printed on three different days, referred to as batch 1, 2, and 3, to evaluate the day-to-day and batch variability of the manufacturing method. The same printing solution was used for printing of all batches in order to obtain information regarding the robustness of the technique rather than differences possibly originating from the preparation of the print solution. As the same solution was used for printing on three different days (day 1, 2, and 4), the stability of the drug-loaded printing solution stored in room temperature was determined by UV-spectrophotometry (Lambda 35, PerkinElmer, Singapore, Singapore) at 207 nm.
2.2.4. Inkjet Printing
Preparation of Solvent Cast Printing Substrates
The polymeric substrates used in the IJP process were prepared by solvent casting. A drug-free HPC solution was prepared in the same manner as described for the placebo EXT ODFs and subsequently cast into films with a wet thickness of 600 µm utilizing a film applicator (Multicator 411, Erichsen, Hemer, Germany). The films were cast on top of transparency sheets (clear transparent X-10.0, Folex, Germany) and allowed to dry in room temperature minimum overnight (some longer, due to the printing of batches on different days).
Inkjet Printing
Inkjet printing was performed with a PixDro LP50 piezoelectric printer (Roth and Rau, Eindhoven, Netherlands) equipped with a print head with 128 nozzles (SL-128 AA, Fujifilm, Tokyo, Japan) and a camera for visualization of the jetted droplets. The printing resolution was set to 720 dpi based on calculations of the target dose, estimated droplet volume, ink concentration, as well as the size of the printed area. Printing was conducted with a jetting frequency of 1400 Hz, a voltage of 80 V, an ink pressure of –18 mbar and a pulse shape of 3-16-5 µs. The prepared ink (drug or drug-free) was filtered (0.45 µm polypropylene membrane syringe filter, VWR International, Radnor, PA, USA) and used to imprint the prefabricated solvent cast HPC films according to the premade designs using 40–60 nozzles, a quality factor of 3 and bi-directional printing. One printing run resulted in 32 printed films of a certain size that were allowed to dry in ambient conditions overnight and subsequently cut with a scalpel according to a template in order to obtain the final size.
2.2.5. Compounding of Oral Powders in Unit Dose Sachets
The OPSs, each individual sachet weighing 200 mg, were compounded at the manufacturing unit at HUS Pharmacy in the same routinely manner as when OPSs are prepared and delivered for patients at the hospital. Three batches per dose were manufactured on three different days. The batch size was 30 OPSs except for the last batch of the 2 mg doses, where the batch size was 120 OPSs.
The OPSs were manufactured following the standard operating procedures of HUS Pharmacy for extemporaneously prepared OPSs. A pharmacist prepared the masses, and a technician or pharmacist weighed the individual sachets. Marevan forte 5 mg tablets were crushed in a mortar and ground with a pestle to a fine powder. Lactose monohydrate was added in geometric amounts to receive the final concentration and amount needed for each dose and batch size. The content of the individual sachets was weighed into waxed powder papers (Herra Järvisen Verstas Oy, Helsinki, Finland) using an analytical balance (MettlerToledo XP204, Greifensee, Switzerland). All sachets were labeled and packed in plastic ziplock bags.
2.2.6. Identification Labeling using Printed Quick Response (QR) Codes
ODFs prepared by IJP and EXT were imprinted with a quick response (QR) code containing vital information about the dosage form, such as type of dosage form, API, strength, manufacturing date, expiration date, as well as the batch number. The QR code was generated utilizing the free online QR generator (goQR.me, Foundata GmbH, Karlsruhe, Germany), saved as a .bmp file and imported into the printing software. A placebo ink containing 1% (w/w) brilliant blue G dissolved in the ink base consisting of 27% (w/w) PG, 5% (w/w) purified water, and 67% (w/w) ethanol was used for printing the QR code.
The same IJP and print head as utilized for the printing of the IJP ODFs was used to print the QR code. The ink was filtered through a 0.45 µm polypropylene membrane syringe filter (VWR International, Radnor, PA, USA) to remove any undissolved particles and bi-directional printing was conducted with a pulse shape of 3-16-5, a jetting frequency of 1700 Hz, a voltage of 80V, and an ink pressure of −18 mbar. The QR code was printed with one nozzle, a quality factor 1 and a resolution of 400 dpi. The readability of the imprinted QR code on the ODFs was evaluated using QR reader for iPhone (version 6.8, Tapmedia Ltd., UK) and QR Code Reader (version 1.0.7, Google Commerce Ltd., Dublin, Ireland) for android.
2.2.7. Weight, Thickness, and Appearance of Dosage Forms
The overall appearance of the prepared dosage forms was evaluated visually. The thickness of the ODFs was measured at 5 locations (all corners and the middle of the film) utilizing a caliper (CD-6”CX, Mitutoyo, Kawasaki, Japan) and the weight of the ODFs was determined using an analytical balance (AND GH-252, A and D Instruments Ltd., Tokyo, Japan). The weight and thickness of the ODFs used in the content analysis were chosen to represent the respective batches (average ± SD, n = 10).
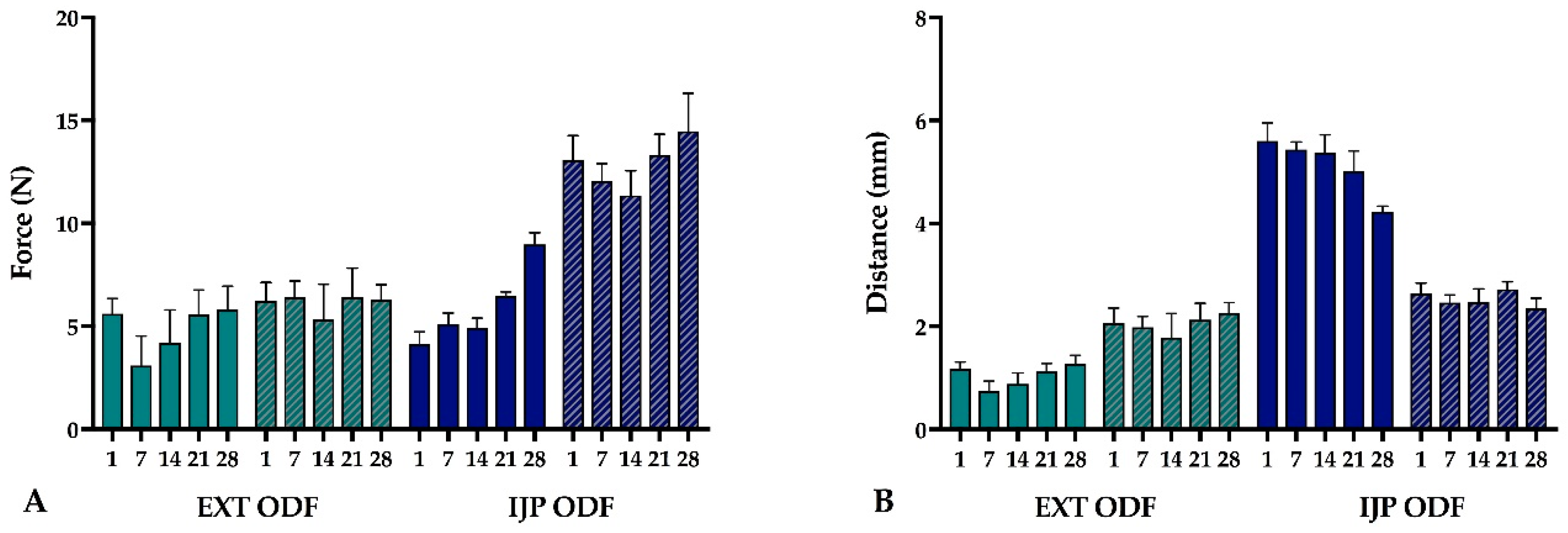
2.2.8. Mechanical Testing
The mechanical properties of the produced ODFs were investigated using a TA-XTplus (Stable Micro Systems, Godalming, UK) texture analyzer equipped with a 10 kg load cell. The largest ODFs (n = 5) were one at a time clamped between the Perspex film support platform and the aluminum circular top plate (Film support rig HDP/FSR, Stable Micro Systems). The spherical probe (ø 5 mm, SMS P/5S, Stable Micro Systems) was used to puncture the film with a constant speed of 1 mm/s until reaching the target distance of 5 mm (EXT films) or 15 mm (IJP films). The acquisition of data started when the trigger force of 0.049 N was reached, and the maximum applied force and penetration depth (mm) into the film before rupturing was recorded. Experiments were conducted at ambient conditions.
2.2.9. Surface pH
The surface pH of the prepared EXT and IJP ODFs (drug-loaded and placebo) as well as of the prepared OPSs was determined in room temperature by placing one 2 mg dosage form in a small glass vial and adding 1 mL of purified water. The electrode of the pH meter (Mettler Toledo FE20, Mettler Toledo AG, Greifensee, Switzerland) was lowered into the solution, and the surface pH was determined after being immersed for 1 min and 15 min, respectively. Measurements for each formulation were performed in triplicate.
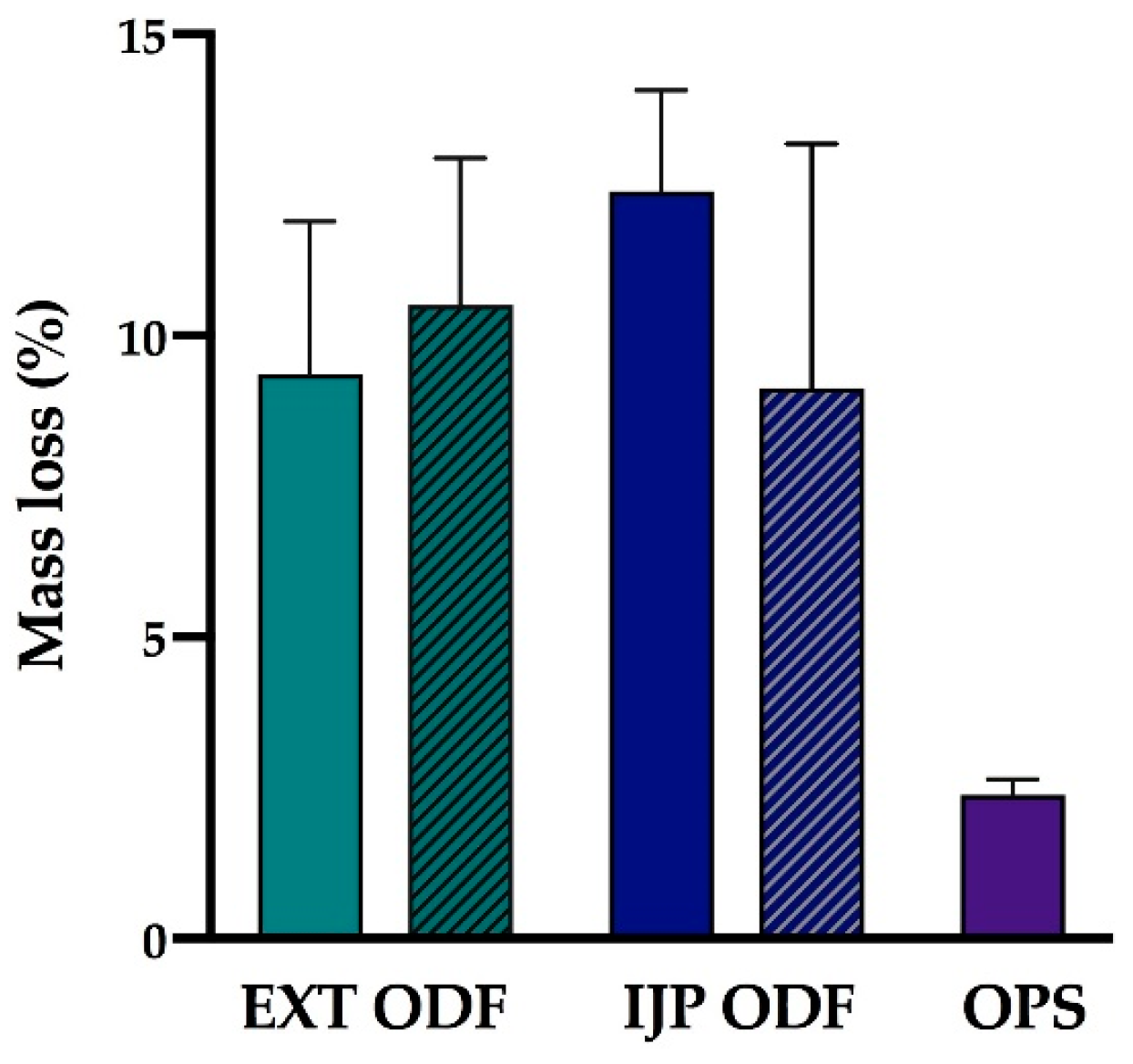
2.2.10. Moisture Content
The moisture content of the prepared dosage forms (n = 3) was investigated utilizing a moisture analyzer (Radwag Mac 50/NH, Radom, Poland). The sample with a target dose of 2 mg was placed on an aluminum pan, and the mass % weight loss corresponding to moisture evaporation was recorded as the sample was heated up to 120 °C. The end point of the measurement was set to when the change of mass had reached equilibrium and was less than 1 mg/min.
2.2.11. Disintegration
The disintegration time of the ODFs was investigated using the Petri dish method. The films were analyzed with regards to thickness and weight prior to the disintegration test. 10 mL of purified water was pipetted into a Petri dish, and the ODF was subsequently dropped on top of the liquid surface using tweezers. The time for the film to completely rupture in the middle into smaller film pieces utilizing this static method was recorded and reported as the time for the film to disintegrate. In other words, swelling (in any direction) of the film or small pieces wearing off at the edges was not defined as the endpoint.
2.2.12. Drug Content
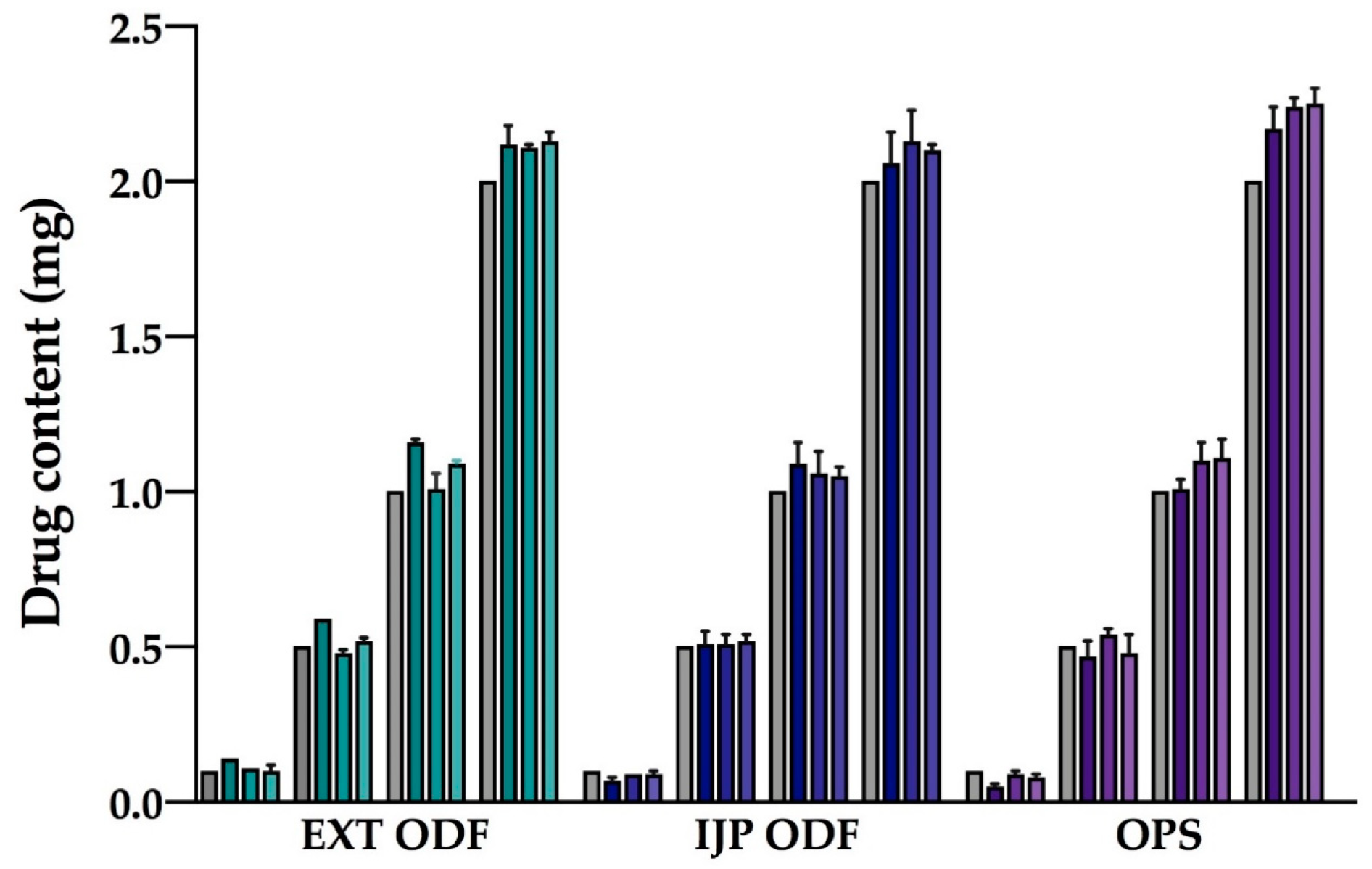
Drug content of the prepared doses was determined to evaluate the amount of drug obtained in the final dosage form utilizing the different manufacturing techniques. Briefly, one ODF or OPS was placed in 100 mL of purified water and shaken (Multi-shaker PSU 20, Biosan, Latvia) at 50 rpm for a minimum of 3 h. Samples were diluted when necessary, and the absorbance was subsequently spectrophotometrically (Lambda 35, PerkinElmer, Singapore, Singapore) analyzed at 207 nm. The absorption of the drug-free ODFs at 207 nm, consisting of either a HPC film or a HPC placebo-ink imprinted film, was used as a baseline for the measurements. For the filtered (0.2 µm cellulose acetate syringe filter) OPSs, the absorbance of purified water at 207 nm was considered as the baseline. Ten replicates of all prepared doses were analyzed for each batch and manufacturing method. For stability, ten replicates of the largest target dose were analyzed at each stability time point.
Uniformity of content of single-dose preparations (UC) was calculated according to the European Pharmacopeia (Ph. Eur. 9.0) 2.9.6, test B [
36]. The test complies with requirements if not more than one individual content is outside 85 and 115% of average content, and none is outside 75 and 125% of average content. If two or three individual dosage units are outside 85–115% of average content, a further 20 units should be tested. The test fails to comply with requirements if more than three individual contents are outside 85–115% of average content. Moreover, the prepared dosage forms were analyzed with regards to uniformity of dosage units as described in Ph. Eur. 2.9.40. The acceptability constant k = 2.4 (
n = 10) and T = 100% were used to calculate the acceptance values (AV). The AV (L1) should be ≤ 15.0 to meet the requirements. In this study, the acceptance UC and AV were based on ten replicates, an additional 20 dosage forms were not analyzed.
2.2.13. In Vitro Dissolution
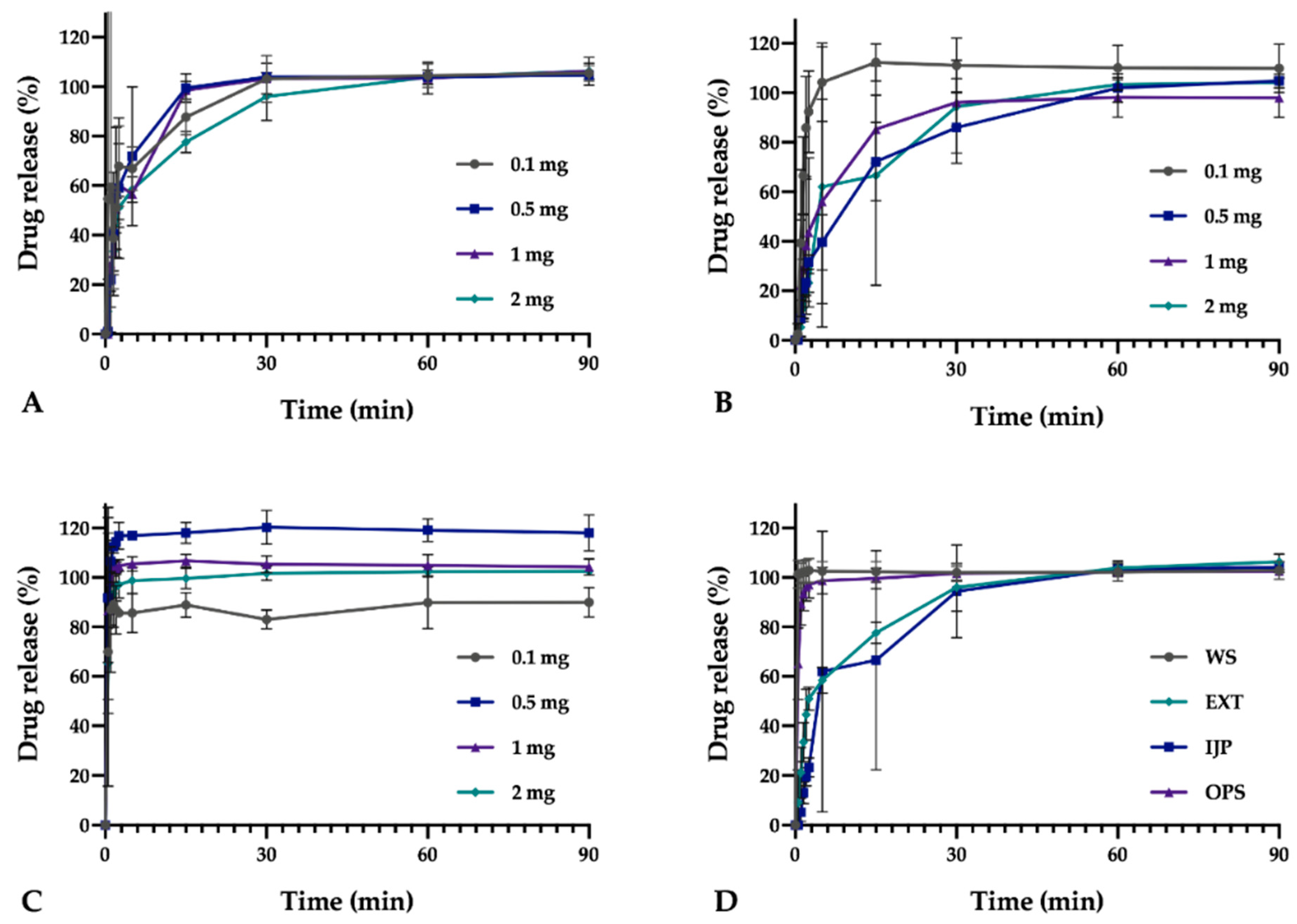
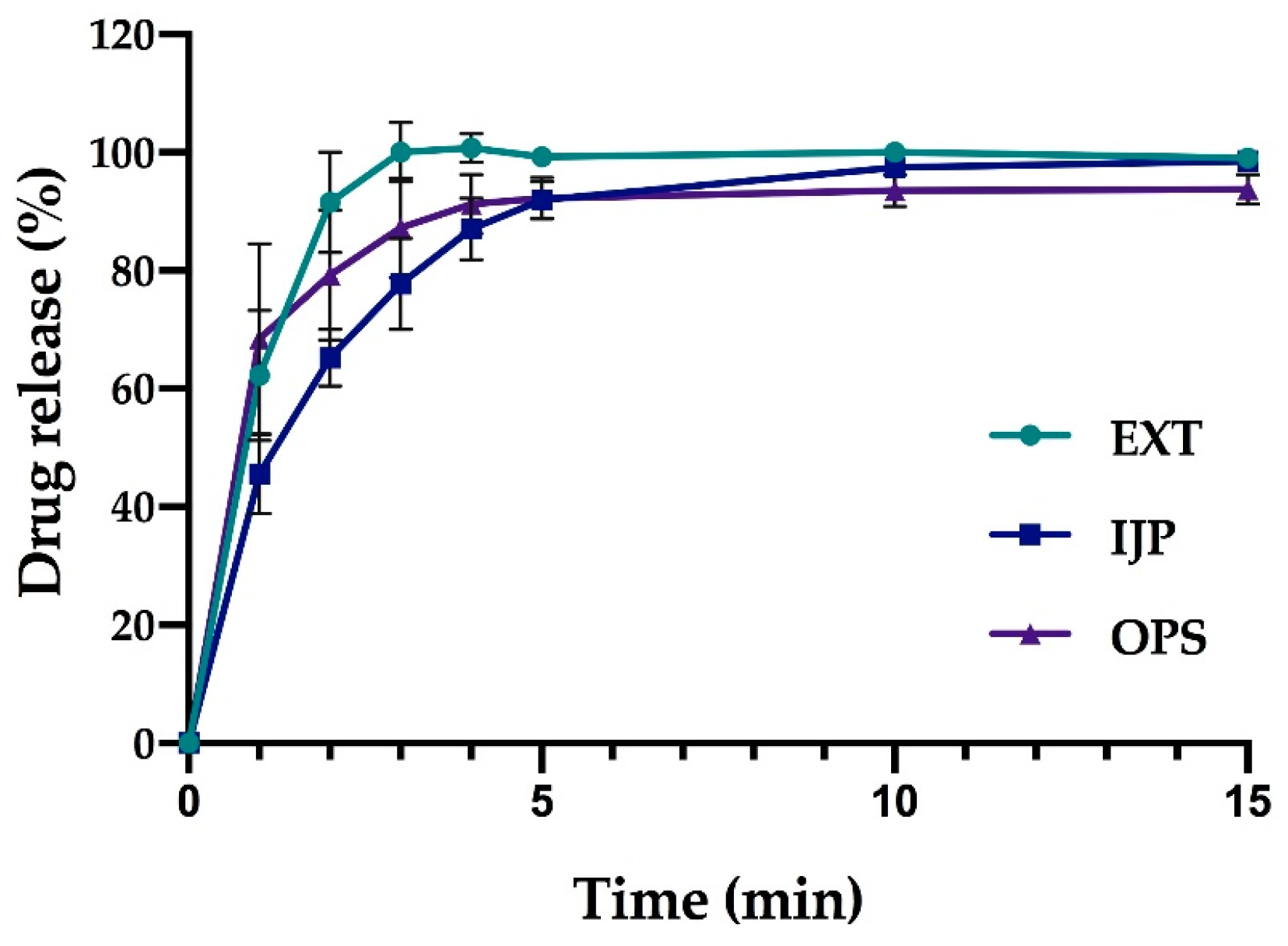
In vitro drug release studies were conducted for the pure drug as received, as well as for the prepared doses from the three different manufacturing techniques (EXT, IJP, and OPS) to study the drug release behavior of the dosage forms. The thickness and weight of the ODFs was documented prior to the dissolution study as well as the weight of the OPSs. The ODFs were placed in dissolution baskets and inserted in 250 mL glass bottles containing 100 mL of purified water, while the oral powder in the sachets were emptied from the sachets directly into the bottles. The bottles were kept on a shaking water bath at 37 °C and 50 rpm throughout the dissolution study. At each predetermined time point, 3 mL of media was manually withdrawn, and 3 mL of fresh media was added. The absorbance of the withdrawn solutions was measured at 207 nm using a UV–VIS spectrophotometer (Lambda 35, PerkinElmer, Singapore). The withdrawn solutions from the OPS samples were filtered through a 0.2 µm cellulose acetate syringe filter (rinsed with 30 mL of purified water prior to use) in order to remove undissolved particles. Samples were measured in triplicate, and the percent drug released was calculated based on the results obtained from the content measurements.
As a comparison to the manual dissolution, dosage forms with the highest drug load (target dose of 2 mg) were additionally studied utilizing an automated setup (Sotax AT 7smart, Basel, Switzerland). The ODFs were accurately weighed an inserted into baskets, while the oral powders from the OPSs were directly poured into the vessels filled with 500 mL of purified water at 37 ± 0.5 °C. The basket rotated with a speed of 50 rpm, and samples of the release media were automatically withdrawn at predefined time-points with the use of a pump (Sotax CY 6, Basel, Switzerland), filtered (glass microfiber filter GF/B, GE Healthcare Life Sciences, Little Chalfont, UK) and the absorbance measured at 207 nm utilizing an on-line UV–VIS spectrophotometer (Lambda 35, PerkinElmer, Singapore). The average percent drug release (n = 3) was once again calculated based on the results from the content measurements.
2.2.14. Evaluation of Drug Administration through a Naso-Gastric Tube
The administration of the produced dosage forms through a naso-gastric tube was mimicked to ensure that it would be possible to administer the prepared dosage forms to all patients at hospital wards. The amount of water used for administrating one OPS dose is not clearly standardized at HUS, but typically, the volume is as small as possible. To simulate the process used at the hospital ward, each dosage form was placed in a disposable plastic medicine cup and 2 mL purified water was added. The medicine cup was shaken for approximately 2 min whereafter the solution was administered into the naso-gastric tube (Nutricia Flocare pur tube, CH 6/60, inner diameter 1.1 mm, Nutricia Medical Devices BV, Zoetermeer, Netherlands) with the help of a disposable syringe and subsequently collected into a 100 mL volumetric flask. After administration of the dosage form, the naso-gastric tube was flushed with 2 mL of purified water, which likewise was collected in the volumetric flask. Purified water ad 100 mL was added, and the WS content was measured utilizing the same UV–VIS spectroscopy method as described in drug content measurement (
Section 2.2.12). Three replicates of the largest target dose for all three manufacturing methods were tested.
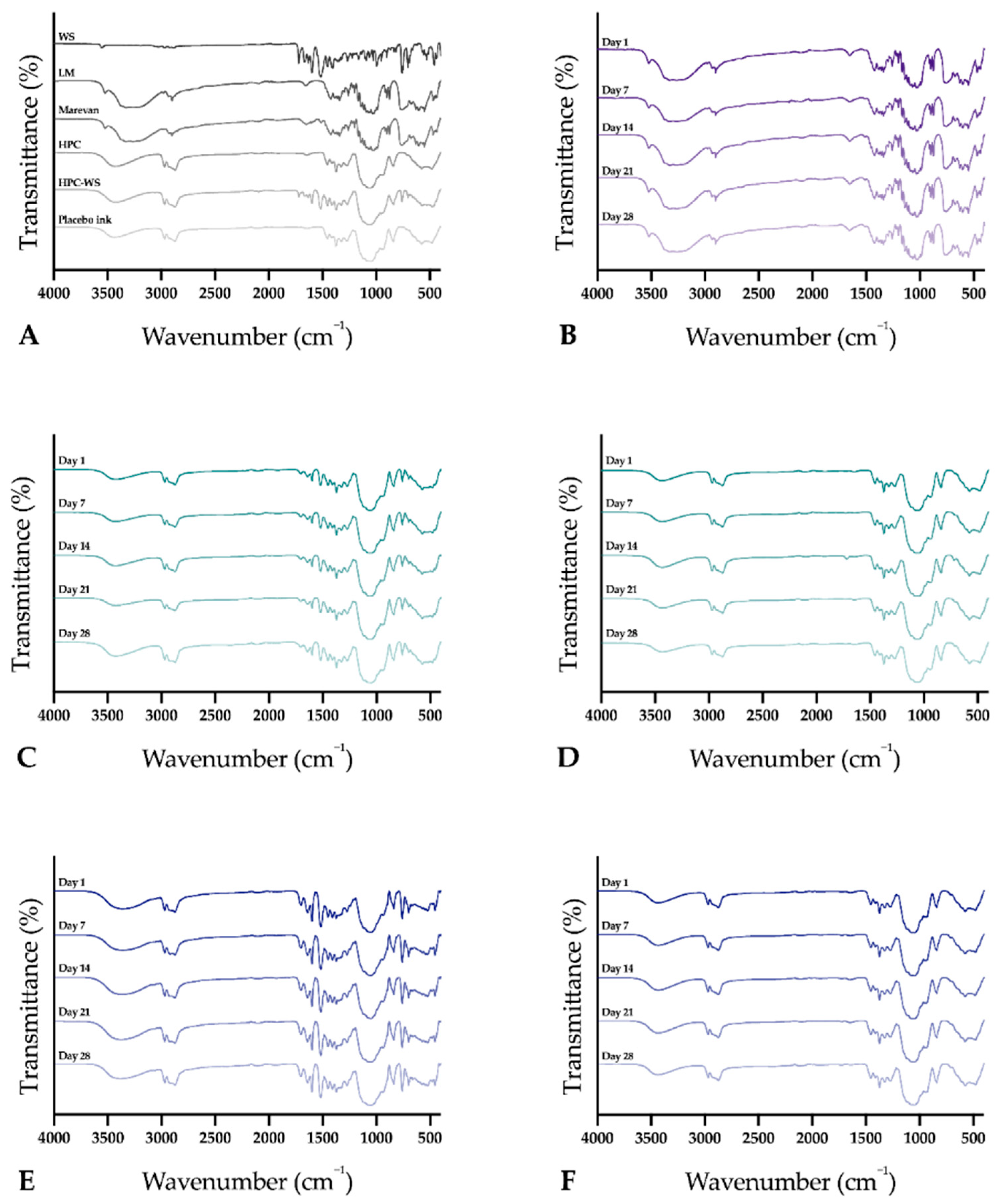
2.2.15. Attenuated Total Reflectance Fourier Transform Infrared (ATR-FTIR)
The infrared spectra of the raw materials, physical mixtures, and the prepared dosage forms were obtained using an Attenuated Total Reflectance Fourier Transform Infrared (ATR-FTIR) spectroscopy (Spectrum Two, PerkinElmer Inc., Beaconsfield, UK). The samples were placed on top of the diamond (IJP ODFs with the printed side facing the diamond), and a force of 75 N was applied during the measurement to attain a good signal. Samples were measured from 4000 to 400 cm–1 with four accumulations at a resolution of 4 cm−1. Spectra were obtained in duplicate, and a third measurement was performed in cases where differences were observed during the first two measurements. The software Spectrum (version 10.03.02, PerkinElmer) was used for acquisition of the spectra and for further data treatment utilizing baseline correction, normalization, and data tune-up.
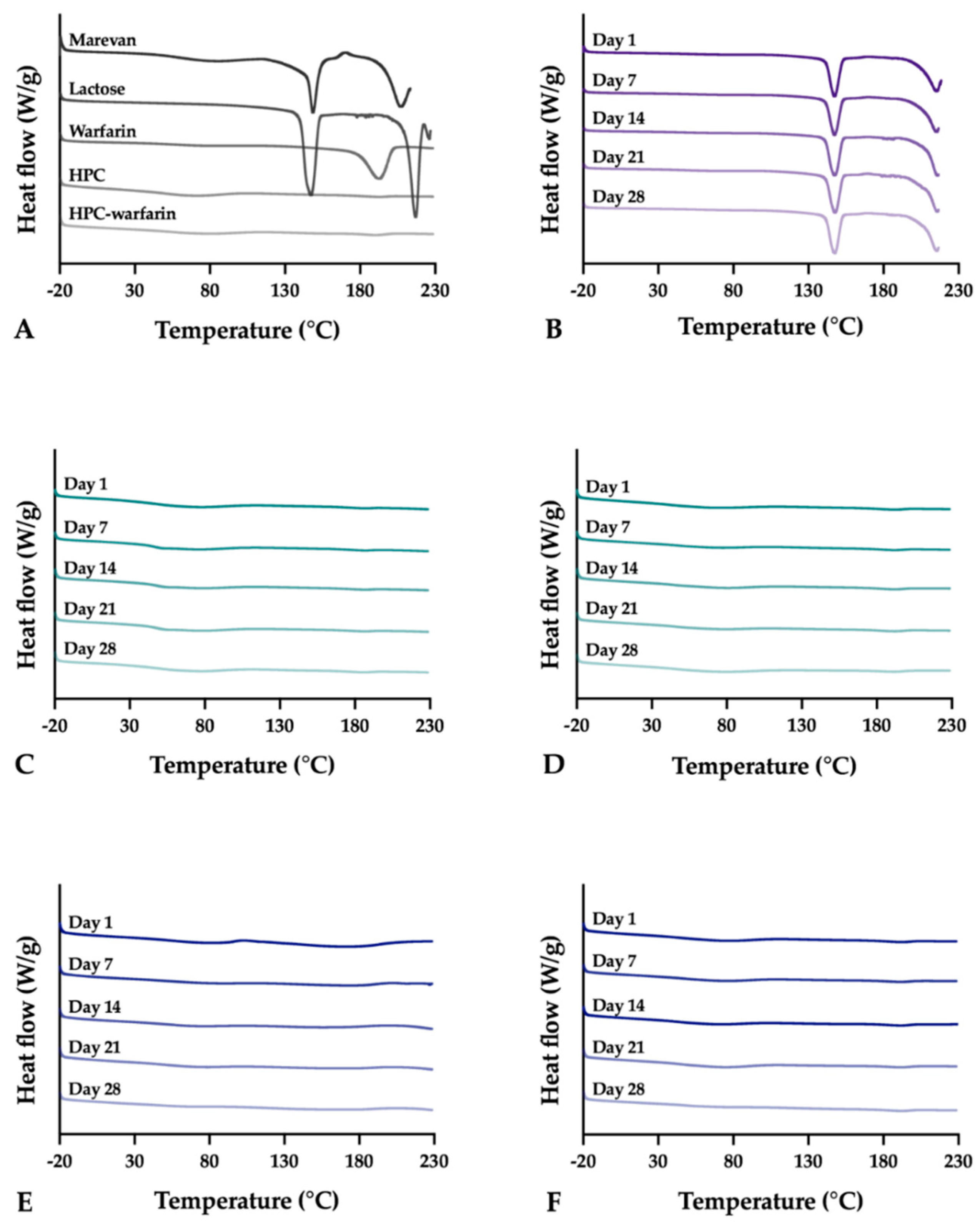
2.2.16. Differential scanning calorimetry (DSC)
Differential scanning calorimetry (DSC) was utilized to evaluate the thermal properties of the samples using the Q2000 (TA Instruments, New Castle, DE, USA). Samples weighing 3.0 ± 0.1 mg were analyzed in sealed Tzero aluminum pans from –20 to 230 °C with a heating rate of 10 °C/min. The OPSs were only heated up to 220 °C to avoid further degradation of the sample. Measurements were performed in duplicate and in triplicate if differences were observed during the first two runs. Nitrogen was used as purge gas with a flow rate of 50 mL/min during all measurements. The data was analyzed utilizing the TA Universal Analysis software (version 4.5A, TA Instruments).
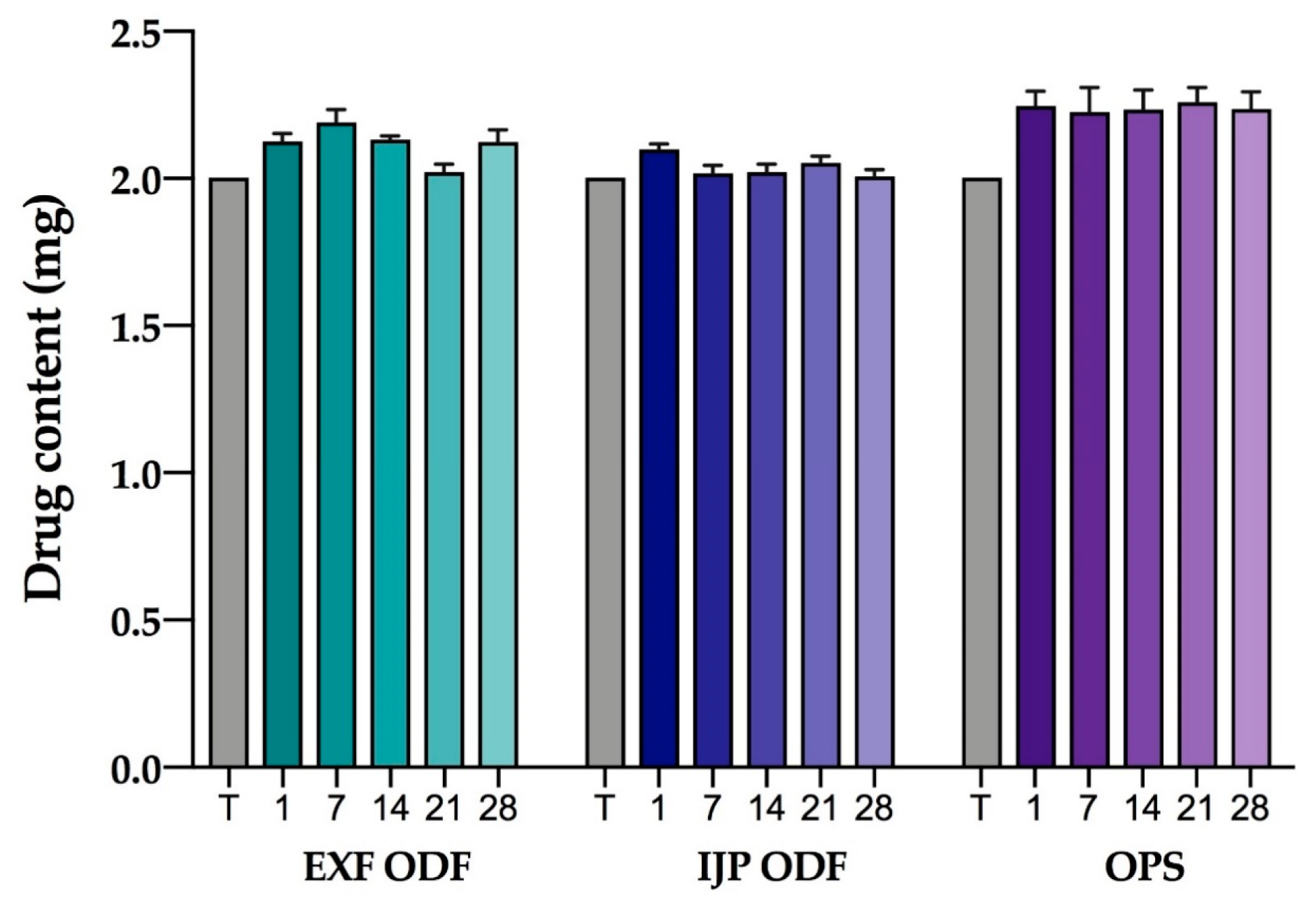
2.2.17. Stability
The stability of the EXT ODFs, IJP ODFs, and OPSs was investigated by visual inspection, mechanical analysis (of ODFs), UV–VIS spectroscopy (drug content), DSC, and ATR-FTIR. The EXT ODFs were stored in a Petri dish throughout the stability period and the IJP ODFs sheets were stacked on top of each other with a transparency sheet in between the printed samples and further covered with aluminum foil. The OPSs were stored in open ziplock bags as an external package. All samples were stored in ambient conditions in a cupboard protected from light. The temperature and relative humidity was tracked during the period using a humidity and temperature USB data logger (wk057, Wisemann Klein SL, Barcelona, Spain). Samples were analyzed at predefined time points, namely at day 1, 7, 14, 21, and 28.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










