Scaled-Up Production and Tableting of Grindable Electrospun Fibers Containing a Protein-Type Drug

 , , , ,
, , , ,  ,
,
Abstract
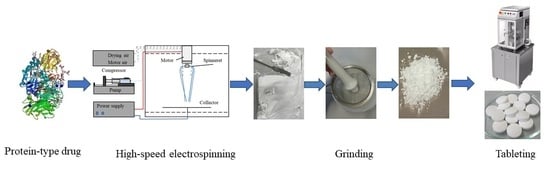
1. Introduction
2. Materials and Methods
2.1. Materials
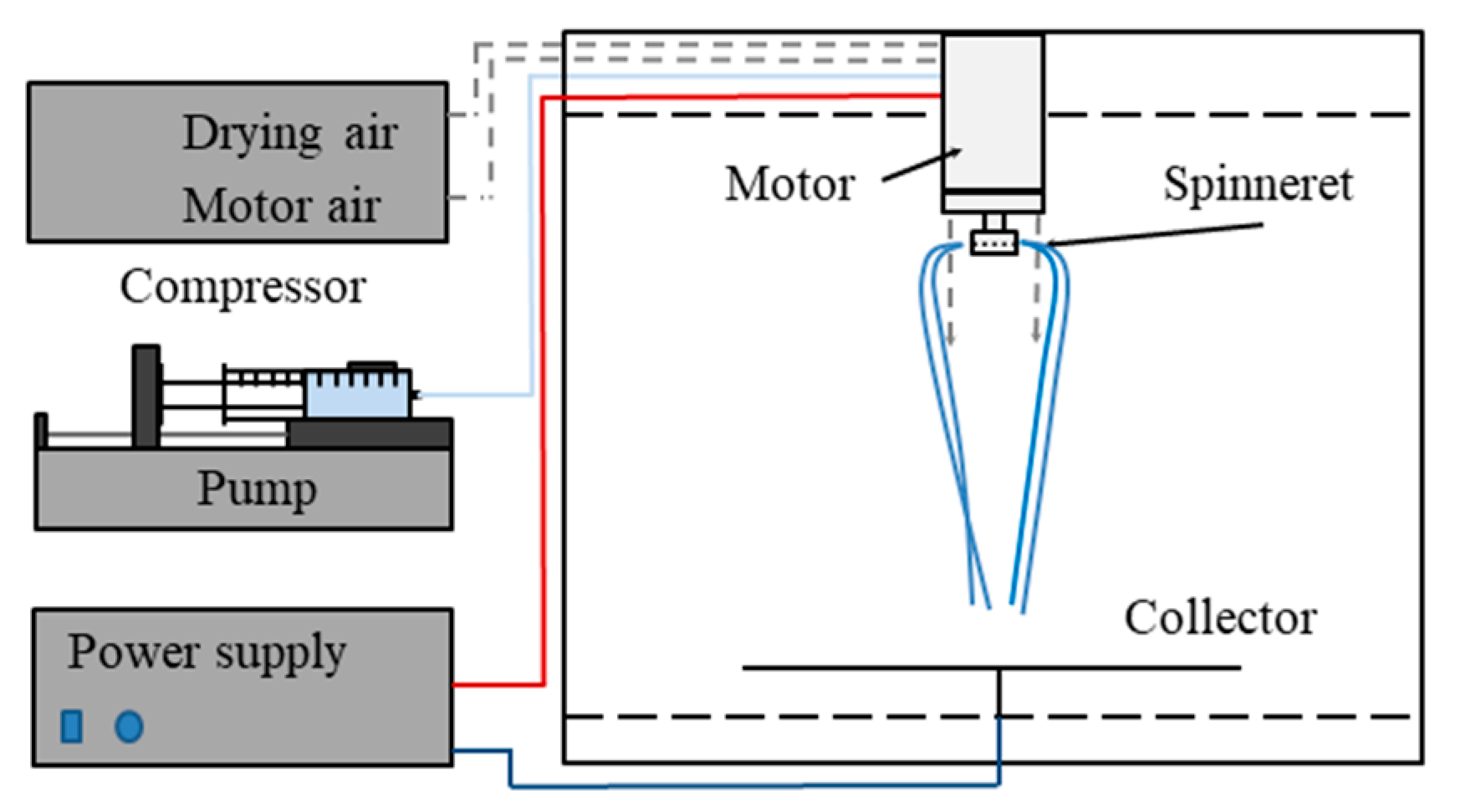
2.2. Scaled-Up Electrospinning of β-Galactosidase
2.3. Scanning Electron Microscopy
2.4. Determination of Residual Water Content
2.5. Grinding/Milling of the Electrospun Material
2.6. Modulated Differential Scanning Calorimetry (DSC)
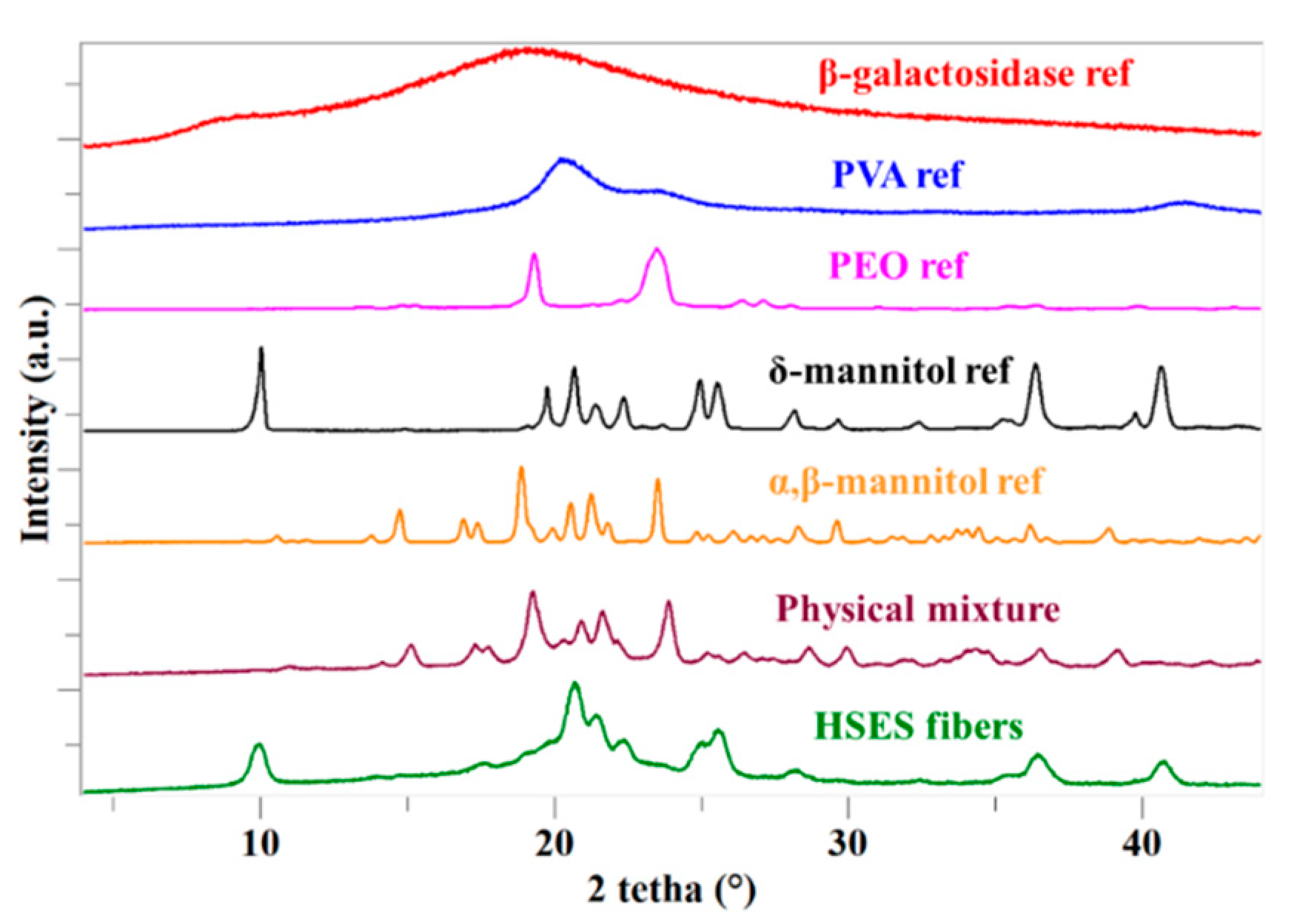
2.7. X-ray Powder Diffraction (XRPD)
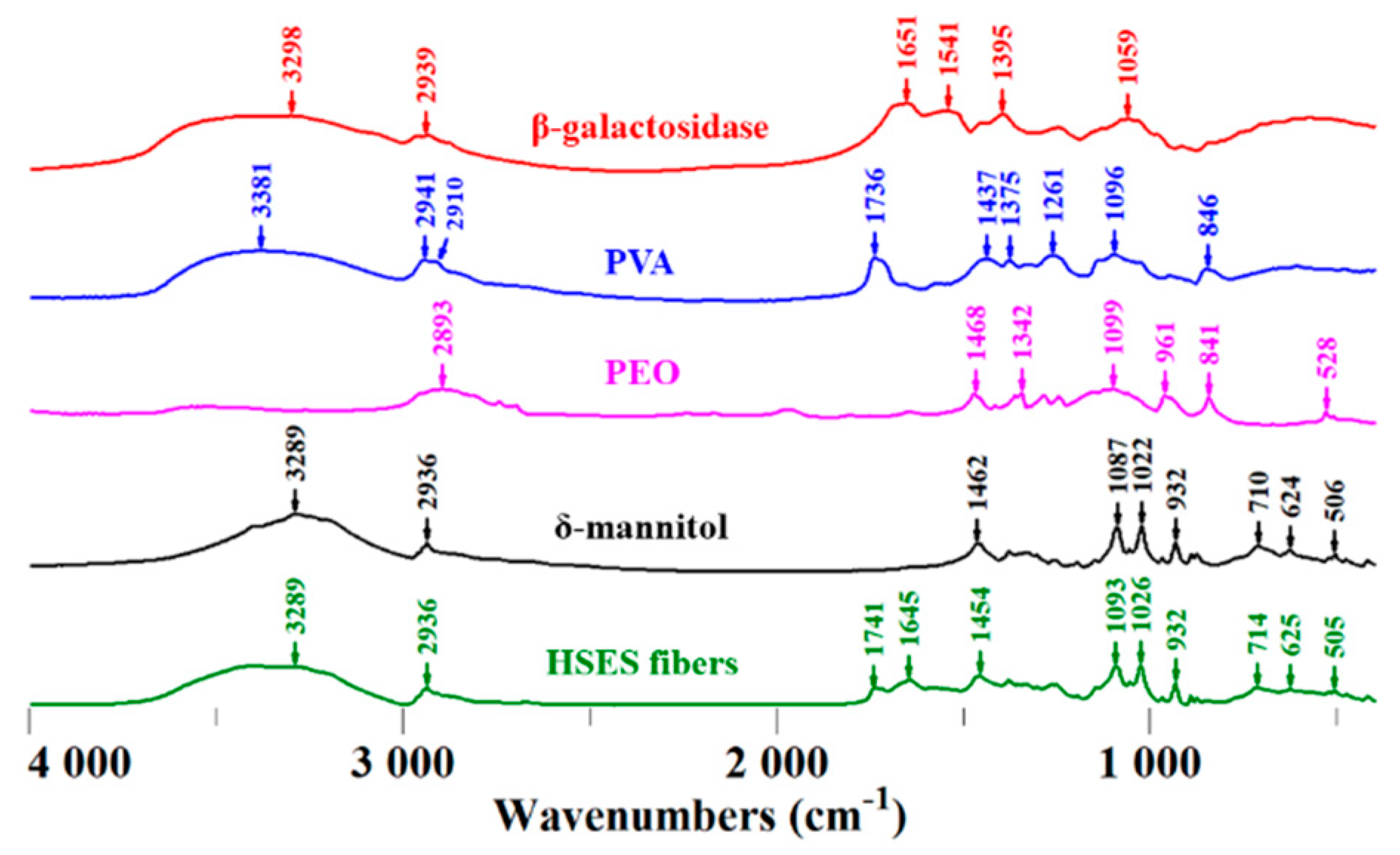
2.8. FTIR Measurement
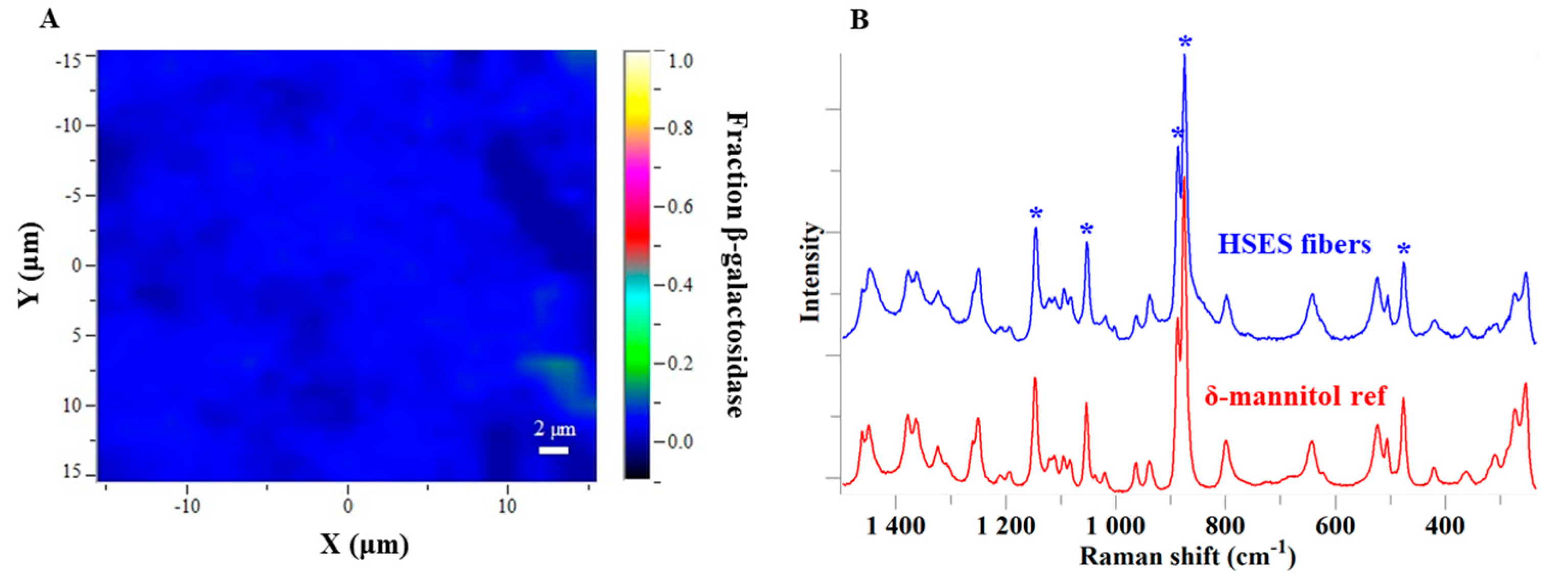
2.9. Raman Mapping
2.10. Tablet Preparation
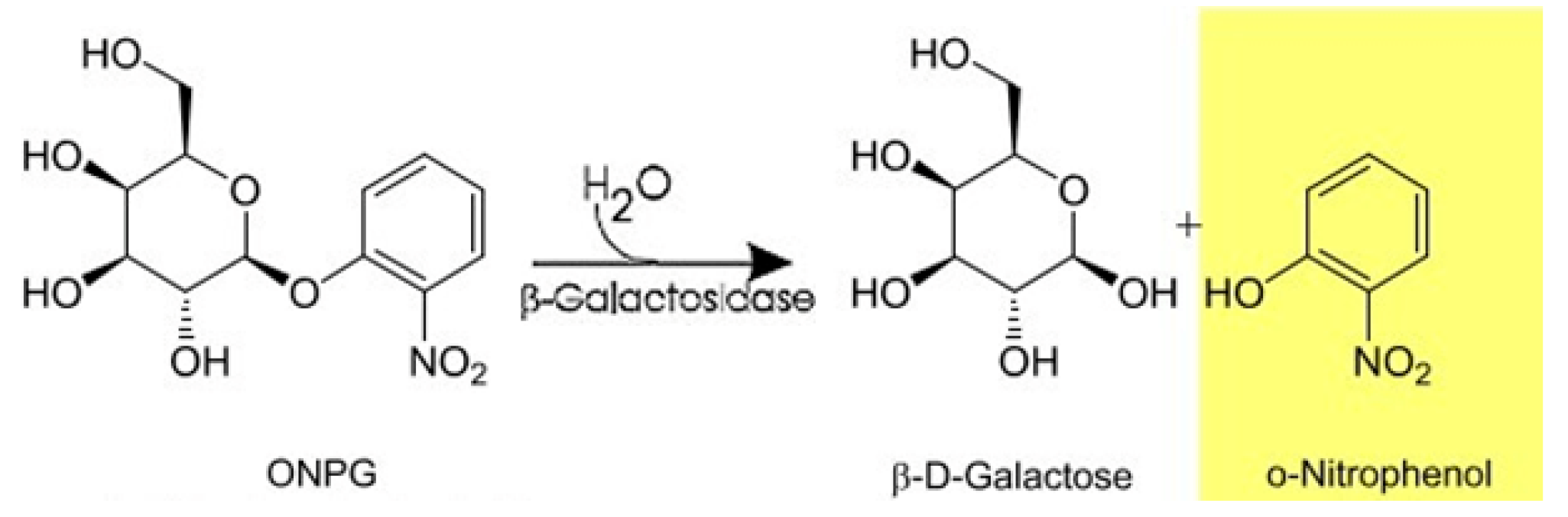
2.11. Determination of Enzyme Activity
2.12. Storage Stability Test
3. Results and Discussion
3.1. High-Speed Electrospinning of β-Galactosidase
3.2. Processing of the β-Galactosidase-Containing Fibers
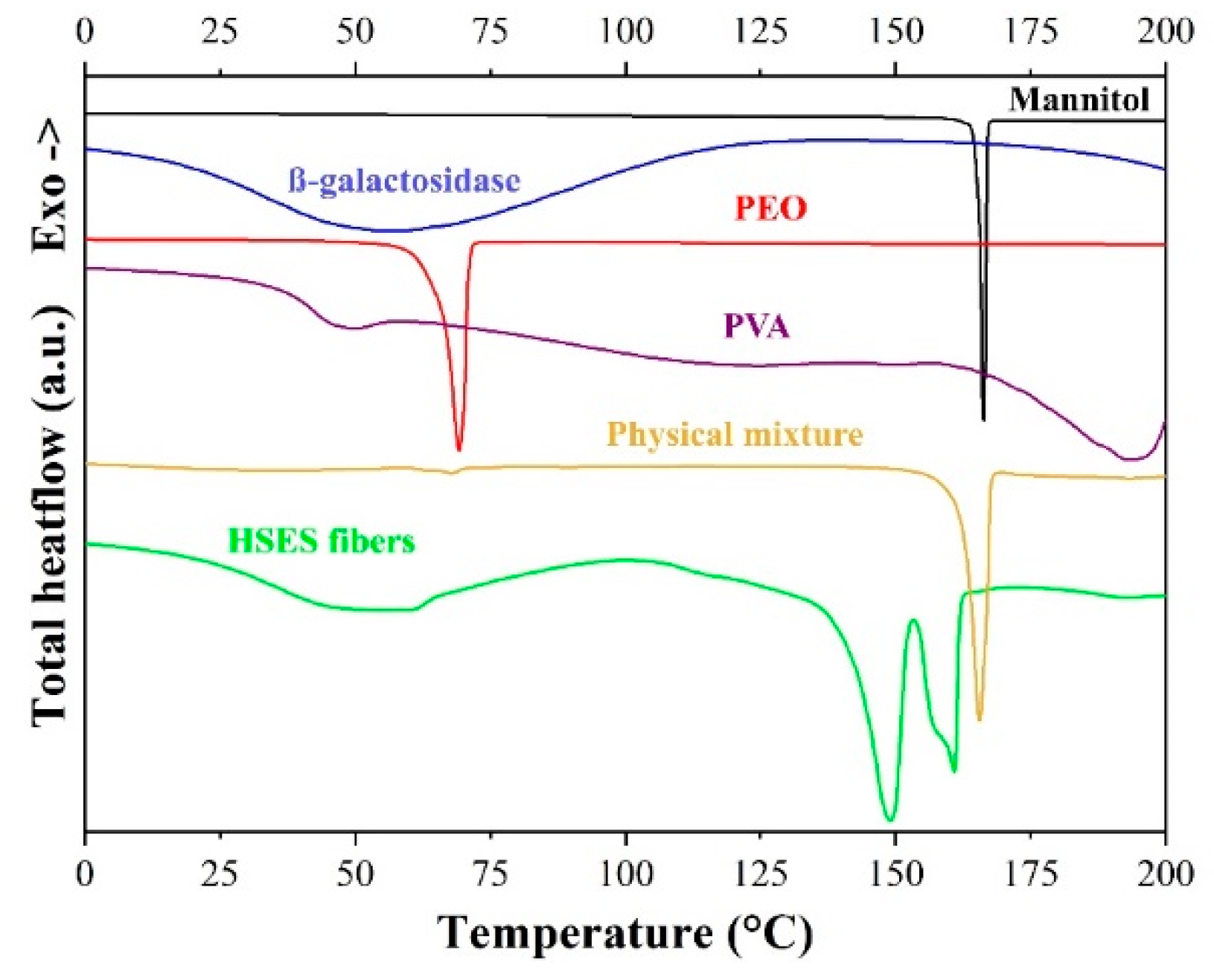
3.3. Characterization of the Fibers
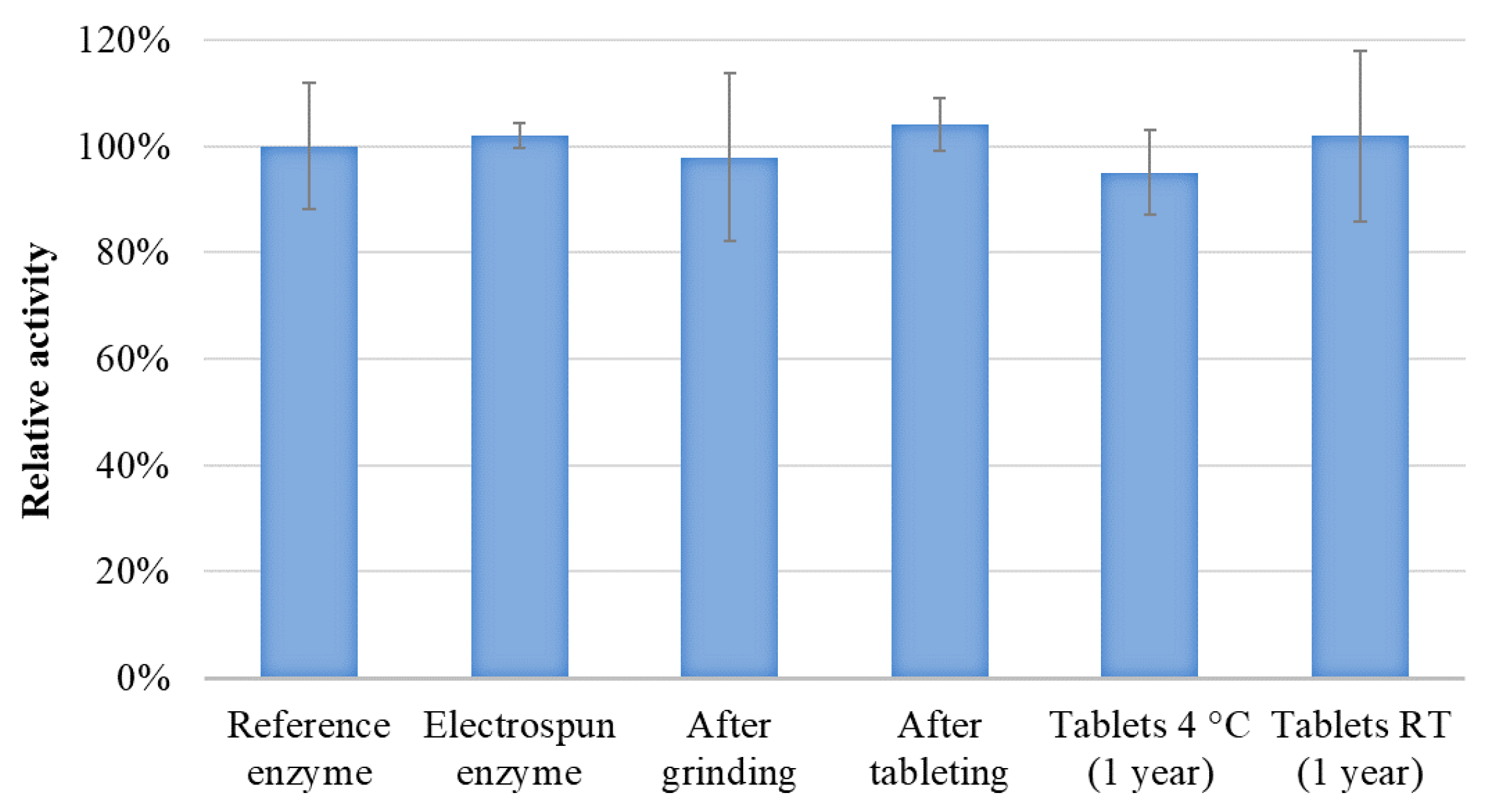
3.4. Tableting and Long-Term Stability Study of the Tablets
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Walsh, G. Biopharmaceutical benchmarks 2018. Nat. Biotechnol. 2018, 36, 1136–1145. [Google Scholar] [CrossRef] [PubMed]
- Jameel, F.; Hershenson, S. Formulation and Process Development Strategies for Manufacturing Biopharmaceuticals; John Wiley and Sons: Hoboken, NJ, USA, 2010. [Google Scholar]
- Langford, A.; Bhatnagar, B.; Walters, R.; Tchessalov, S.; Ohtake, S. Drying technologies for biopharmaceutical applications: Recent developments and future direction. Dry. Technol. 2018, 36, 677–684. [Google Scholar] [CrossRef]
- Ameri, M.; Maa, Y.F. Spray drying of biopharmaceuticals: Stability and process considerations. Dry. Technol. 2006, 24, 763–768. [Google Scholar] [CrossRef]
- Angkawinitwong, U.; Sharma, G.; Khaw, P.T.; Brocchini, S.; Williams, G.R. Solid-state protein formulations. Ther. Deliv. 2015, 6, 59–82. [Google Scholar] [CrossRef] [PubMed]
- Cramariuc, B.; Cramariuc, R.; Scarlet, R.; Manea, L.R.; Lupu, I.G.; Cramariuc, O. Fiber diameter in electrospinning process. J. Electrost. 2013, 71, 189–198. [Google Scholar] [CrossRef]
- Broeckx, G.; Vandenheuvel, D.; Claes, I.J.J.; Lebeer, S.; Kiekens, F. Drying techniques of probiotic bacteria as an important step towards the development of novel pharmabiotics. Int. J. Pharm. 2016, 505, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Wang, L.; Zhu, K. Coaxial electrospinning for encapsulation and controlled release of fragile water-soluble bioactive agents. J. Control. Release 2014, 193, 296–303. [Google Scholar] [CrossRef]
- Zussman, E. Encapsulation of cells within electrospun fibers. Polym. Adv. Technol. 2011, 22, 366–371. [Google Scholar] [CrossRef]
- Hu, X.; Liu, S.; Zhou, G.; Huang, Y.; Xie, Z.; Jing, X. Electrospinning of polymeric nanofibers for drug delivery applications. J. Control. Release 2014, 185, 12–21. [Google Scholar] [CrossRef]
- Vass, P.; Démuth, B.; Hirsch, E.; Nagy, B.; Andersen, S.K.; Vigh, T.; Verreck, G.; Csontos, I.; Nagy, Z.K.; Marosi, G. Drying technology strategies for colon-targeted oral delivery of biopharmaceuticals. J. Control. Release 2019, 296, 162–178. [Google Scholar] [CrossRef]
- Angkawinitwong, U.; Awwad, S.; Khaw, P.T.; Brocchini, S.; Williams, G.R. Electrospun formulations of bevacizumab for sustained release in the eye. Acta Biomater. 2017, 64, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Škrlec, K.; Zupančič, Š.; Prpar Mihevc, S.; Kocbek, P.; Kristl, J.; Berlec, A. Development of electrospun nanofibers that enable high loading and long-term viability of probiotics. Eur. J. Pharm. Biopharm. 2019, 136, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Nagy, Z.K.; Wagner, I.; Suhajda, A.; Tobak, T.; Harasztos, A.H.; Vigh, T.; Soti, P.L.; Pataki, H.; Molnar, K.; Marosi, G. Nanofibrous solid dosage form of living bacteria prepared by electrospinning. Express Polym. Lett. 2014, 8, 352–361. [Google Scholar] [CrossRef]
- Wagner, I.; Nagy, Z.K.; Vass, P.; Fehér, C.; Barta, Z.; Vigh, T.; Sóti, P.L.; Harasztos, A.H.; Pataki, H.; Balogh, A.; et al. Stable formulation of protein-type drug in electrospun polymeric fiber followed by tableting and scaling-up experiments. Polym. Adv. Technol. 2015, 26, 1461–1467. [Google Scholar] [CrossRef]
- Nagy, Z.K.; Balogh, A.; Demuth, B.; Pataki, H.; Vigh, T.; Szabo, B.; Molnar, K.; Schmidt, B.T.; Horak, P.; Marosi, G.; et al. High speed electrospinning for scaled-up production of amorphous solid dispersion of itraconazole. Int. J. Pharm. 2015, 480, 137–142. [Google Scholar] [CrossRef]
- Lukas, D.; Sarkar, A.; Pokorny, P. Self-organization of jets in electrospinning from free liquid surface: A generalized approach. J. Appl. Phys. 2008, 103, 084309. [Google Scholar] [CrossRef]
- Sebe, I.; Szabó, B.; Nagy, Z.K.; Szabó, D.; Zsidai, L.; Kocsis, B.; Zelkó, R. Polymer structure and antimicrobial activity of polyvinylpyrrolidone-based iodine nanofibers prepared with high-speed rotary spinning technique. Int. J. Pharm. 2013, 458, 99–103. [Google Scholar] [CrossRef]
- Démuth, B.; Farkas, A.; Szabó, B.; Balogh, A.; Nagy, B.; Vágó, E.; Vigh, T.; Tinke, A.P.; Kazsu, Z.; Demeter, Á.; et al. Development and tableting of directly compressible powder from electrospun nanofibrous amorphous solid dispersion. Adv. Powder Technol. 2017, 28, 1554–1563. [Google Scholar] [CrossRef]
- Szabo, E.; Demuth, B.; Nagy, B.; Molnar, K.; Farkas, A.; Szabo, B.; Balogh, A.; Hirsch, E.; Nagy, B.; Marosi, G.; et al. Scaled-up preparation of drug-loaded electrospun polymer fibres and investigation of their continuous processing to tablet form. Express Polym. Lett. 2018, 12, 436–451. [Google Scholar] [CrossRef]
- Hirsch, E.; Vass, P.; Démuth, B.; Pethő, Z.; Bitay, E.; Andersen, S.K.; Vigh, T.; Verreck, G.; Molnár, K.; Nagy, Z.K.; et al. Electrospinning scale-up and formulation development of PVA nanofibers aiming oral delivery of biopharmaceuticals. Express Polym. Lett. 2019, 13, 590–603. [Google Scholar] [CrossRef]
- Ugidos-Rodríguez, S.; Matallana-González, M.C.; Sánchez-Mata, M.C. Lactose malabsorption and intolerance: A review. Food Funct. 2018, 9, 4056–4068. [Google Scholar] [CrossRef] [PubMed]
- Branchu, S.; Forbes, R.T.; York, P.; Petrén, S.; Nyqvist, H.; Camber, O. Hydroxypropyl-β-cyclodextrin inhibits spray-drying-induced inactivation of β-galactosidase. J. Pharm. Sci. 1999, 88, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, S.; Aso, Y.; Izutsu, K.I.; Terao, T. Aggregates Formed During Storage of Galactosidase in Solution and in the Freeze-Dried State. Pharm. Res. 1993, 10, 687–691. [Google Scholar] [CrossRef] [PubMed]
- Burger, A.; Henck, J.-O.; Hetz, S.; Rollinger, J.M.; Weissnicht, A.A.; Stöttner, H. Energy/Temperature Diagram and Compression Behavior of the Polymorphs of d-Mannitol. J. Pharm. Sci. 2000, 89, 457–468. [Google Scholar] [CrossRef]
- Hao, H.; Su, W.; Barrett, M.; Caron, V.; Healy, A.-M.; Glennon, B. A Calibration-Free Application of Raman Spectroscopy to the Monitoring of Mannitol Crystallization and Its Polymorphic Transformation. Org. Process Res. Dev. 2010, 14, 1209–1214. [Google Scholar] [CrossRef]
- Sun, C.Q.; Wang, Y.; Tay, B.K.; Li, S.; Huang, H.; Zhang, Y.B. Correlation between the Melting Point of a Nanosolid and the Cohesive Energy of a Surface Atom. J. Phys. Chem. B 2002, 106, 10701–10705. [Google Scholar] [CrossRef]
- Cares-Pacheco, M.G.; Vaca-Medina, G.; Calvet, R.; Espitalier, F.; Letourneau, J.J.; Rouilly, A.; Rodier, E. Physicochemical characterization of d-mannitol polymorphs: The challenging surface energy determination by inverse gas chromatography in the infinite dilution region. Int. J. Pharm. 2014, 475, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.Y.; Wu, J.X.; Yang, M.; Young, P.M.; van den Berg, F.; Rantanen, J. Particle size dependence of polymorphism in spray-dried mannitol. Eur. J. Pharm. Sci. 2011, 44, 41–48. [Google Scholar] [CrossRef]
- Barth, A. Infrared spectroscopy of proteins. Biochim. Biophys. Acta 2007, 1767, 1073–1101. [Google Scholar] [CrossRef]
- Sun, X.-Z.; Williams, G.R.; Hou, X.-X.; Zhu, L.-M. Electrospun curcumin-loaded fibers with potential biomedical applications. Carbohydr. Polym. 2013, 94, 147–153. [Google Scholar] [CrossRef]
- Wen, S.J.; Richardson, T.J.; Ghantous, D.I.; Striebel, K.A.; Ross, P.N.; Cairns, E.J. FTIR characterization of PEO+LiN(CF3SO2)2 electrolytes. J. Electroanal. Chem. 1996, 408, 113–118. [Google Scholar] [CrossRef]
- Cornel, J.; Kidambi, P.; Mazzotti, M. Precipitation and Transformation of the Three Polymorphs ofd-Mannitol. Ind. Eng. Chem. Res. 2010, 49, 5854–5862. [Google Scholar] [CrossRef]
- De Beer, T.R.M.; Allesø, M.; Goethals, F.; Coppens, A.; Vander Heyden, Y.; Lopez De Diego, H.; Rantanen, J.; Verpoort, F.; Vervaet, C.; Remon, J.P.; et al. Implementation of a Process Analytical Technology System in a Freeze-Drying Process Using Raman Spectroscopy for In-Line Process Monitoring. Anal. Chem. 2007, 79, 7992–8003. [Google Scholar] [CrossRef] [PubMed]
- Hancock, B.C.; Shamblin, S.L. Water vapour sorption by pharmaceutical sugars. Pharm. Sci. Technol. Today 1998, 1, 345–351. [Google Scholar] [CrossRef]
- Burnett, D.J.; Thielmann, F.; Booth, J. Determining the critical relative humidity for moisture-induced phase transitions. Int. J. Pharm. 2004, 287, 123–133. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Amount (g) | Concentration (w/w%) | Ratio of Components in the Solid Product (%) | |||
|---|---|---|---|---|---|---|
| Original | Optimized | Original | Optimized | Original | Optimized | |
| PVA 130,000 | 1.000 | 1.000 | 7.2 | 7.6 | 26.0 | 32.5 |
| PEO 2M | 0.075 | 0.075 | 0.5 | 0.6 | 2.0 | 2.4 |
| Mannitol | 2.000 | 1.300 | 14.4 | 9.9 | 52.0 | 42.3 |
| β-galactosidase | 0.770 | 0.700 | 5.6 | 5.4 | 20.0 | 22.8 |
| Water | 10.00 | 10.00 | 72.2 | 76.5 | - | - |
| Ingredients | Amount (mg)/Tablet | Amount (%)/Tablet |
|---|---|---|
| MCC 200 | 150 | 30 |
| Mannitol | 150 | 30 |
| Crospovidone | 50 | 10 |
| Fibrous powder | 150 | 30 |
| ∑ | 500 | 100 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vass, P.; Hirsch, E.; Kóczián, R.; Démuth, B.; Farkas, A.; Fehér, C.; Szabó, E.; Németh, Á.; Andersen, S.K.; Vigh, T.; et al. Scaled-Up Production and Tableting of Grindable Electrospun Fibers Containing a Protein-Type Drug. Pharmaceutics 2019, 11, 329. https://doi.org/10.3390/pharmaceutics11070329
Vass P, Hirsch E, Kóczián R, Démuth B, Farkas A, Fehér C, Szabó E, Németh Á, Andersen SK, Vigh T, et al. Scaled-Up Production and Tableting of Grindable Electrospun Fibers Containing a Protein-Type Drug. Pharmaceutics. 2019; 11(7):329. https://doi.org/10.3390/pharmaceutics11070329
Chicago/Turabian StyleVass, Panna, Edit Hirsch, Rita Kóczián, Balázs Démuth, Attila Farkas, Csaba Fehér, Edina Szabó, Áron Németh, Sune K. Andersen, Tamás Vigh, and et al. 2019. "Scaled-Up Production and Tableting of Grindable Electrospun Fibers Containing a Protein-Type Drug" Pharmaceutics 11, no. 7: 329. https://doi.org/10.3390/pharmaceutics11070329
APA StyleVass, P., Hirsch, E., Kóczián, R., Démuth, B., Farkas, A., Fehér, C., Szabó, E., Németh, Á., Andersen, S. K., Vigh, T., Verreck, G., Csontos, I., Marosi, G., & Nagy, Z. K. (2019). Scaled-Up Production and Tableting of Grindable Electrospun Fibers Containing a Protein-Type Drug. Pharmaceutics, 11(7), 329. https://doi.org/10.3390/pharmaceutics11070329