Effects of Hydrophilic Carriers on Structural Transitions and In Vitro Properties of Solid Self-Microemulsifying Drug Delivery Systems

Abstract
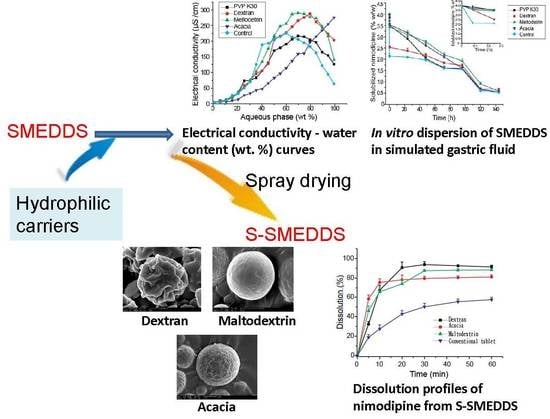
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Preparation of SMEDDS and Droplet Size Determination
2.3. Effects of Carriers on Microstructure of SMEDDS
2.4. Effects of Carriers on Drug Loading of SMEDDS
2.5. Effects of Carriers on Dispersion and Precipitation of SMEDDS in Simulated Gastric Fluid
2.6. Preparation of S-SMEDDS
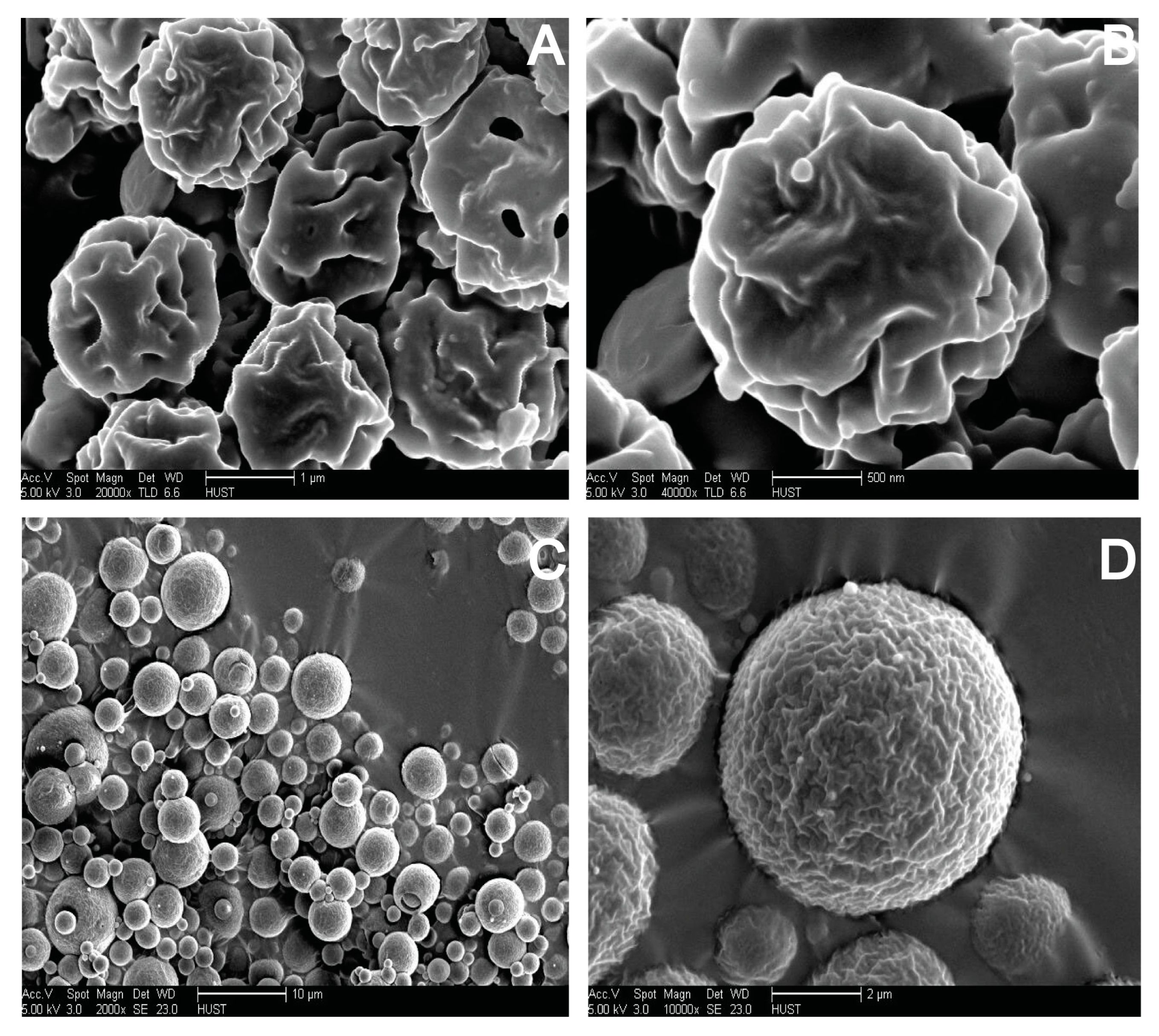
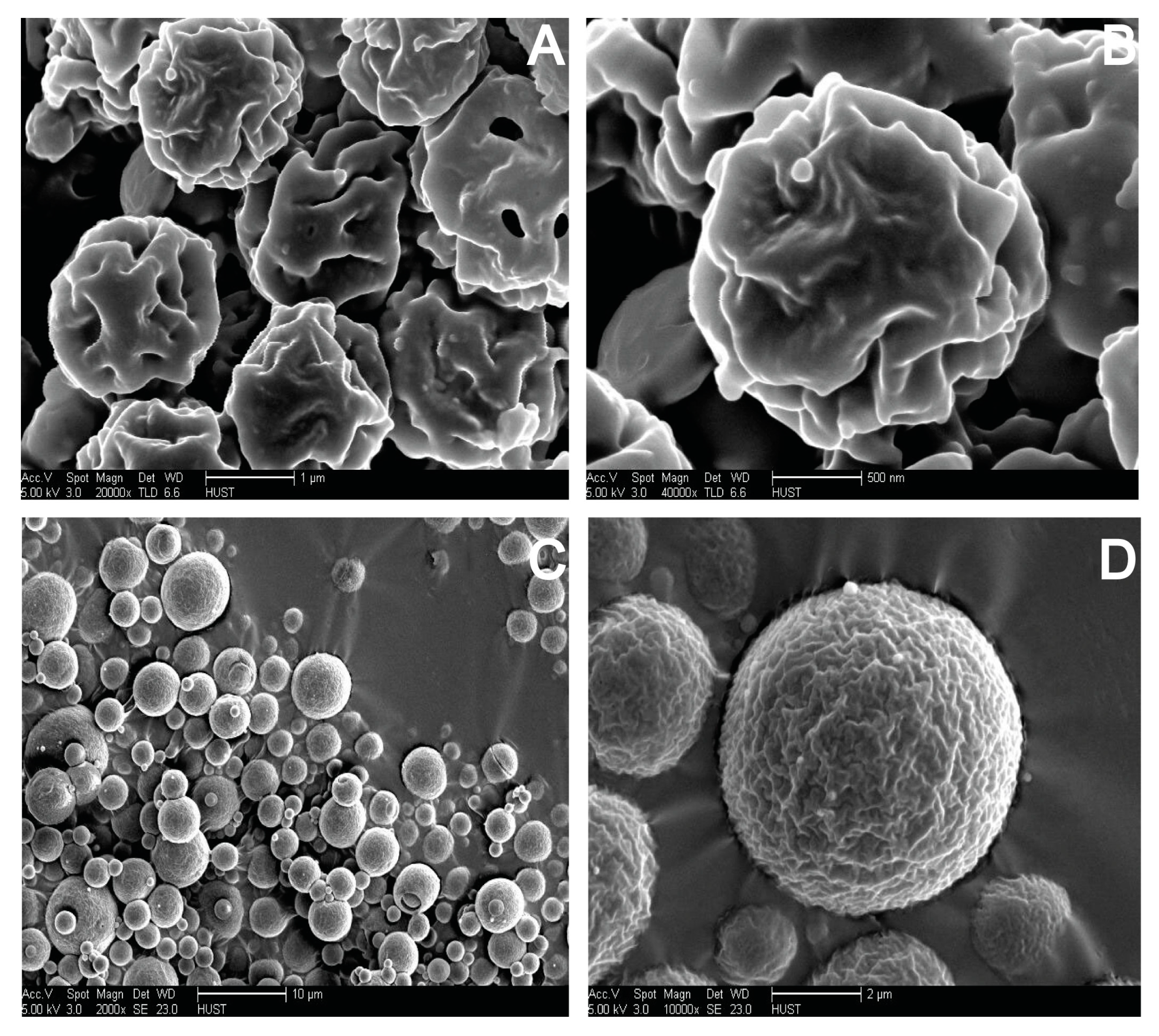
2.7. Morphological Analysis of S-SMEDDS
2.8. Reconstitution Properties of S-SMEDDS
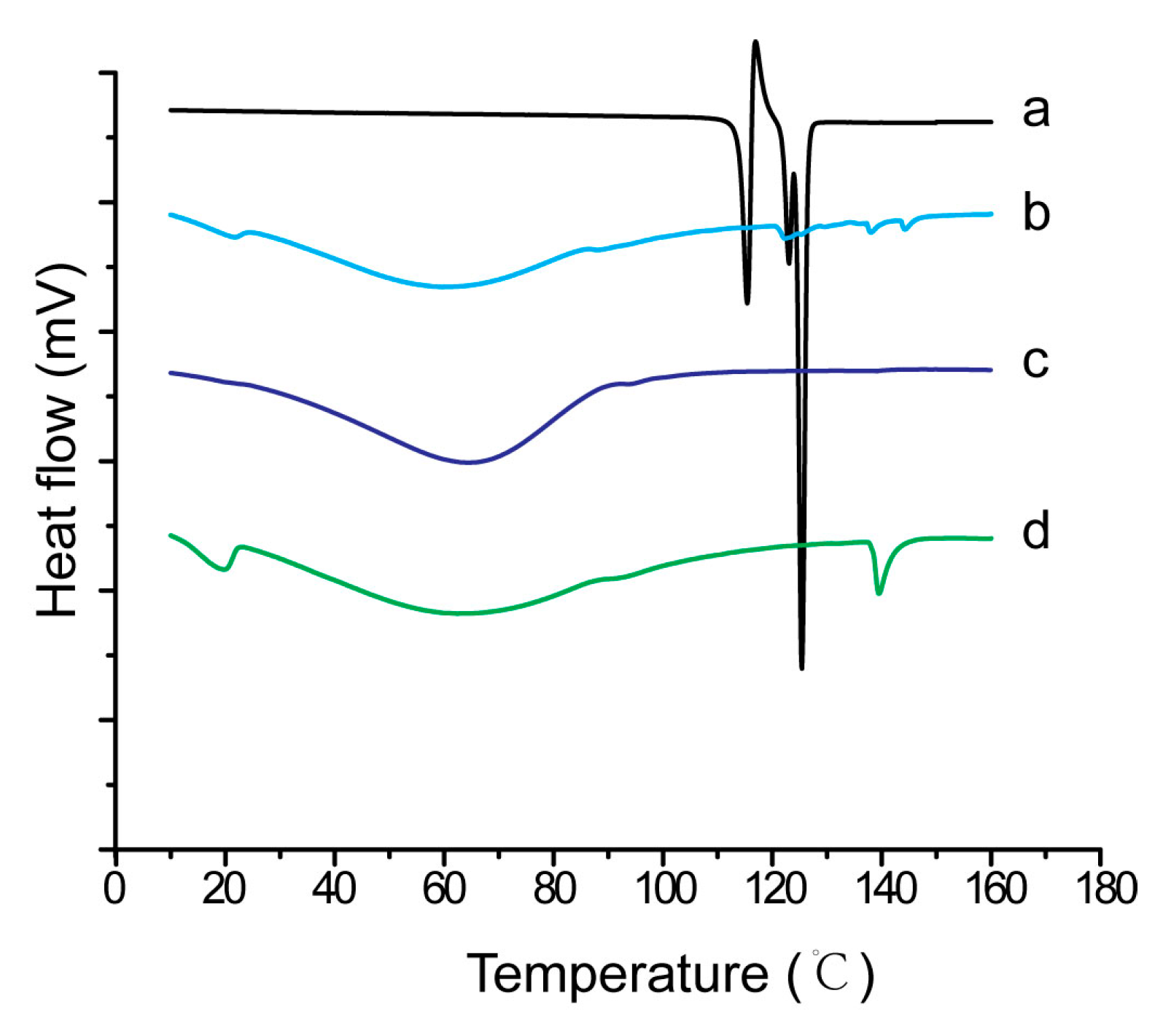
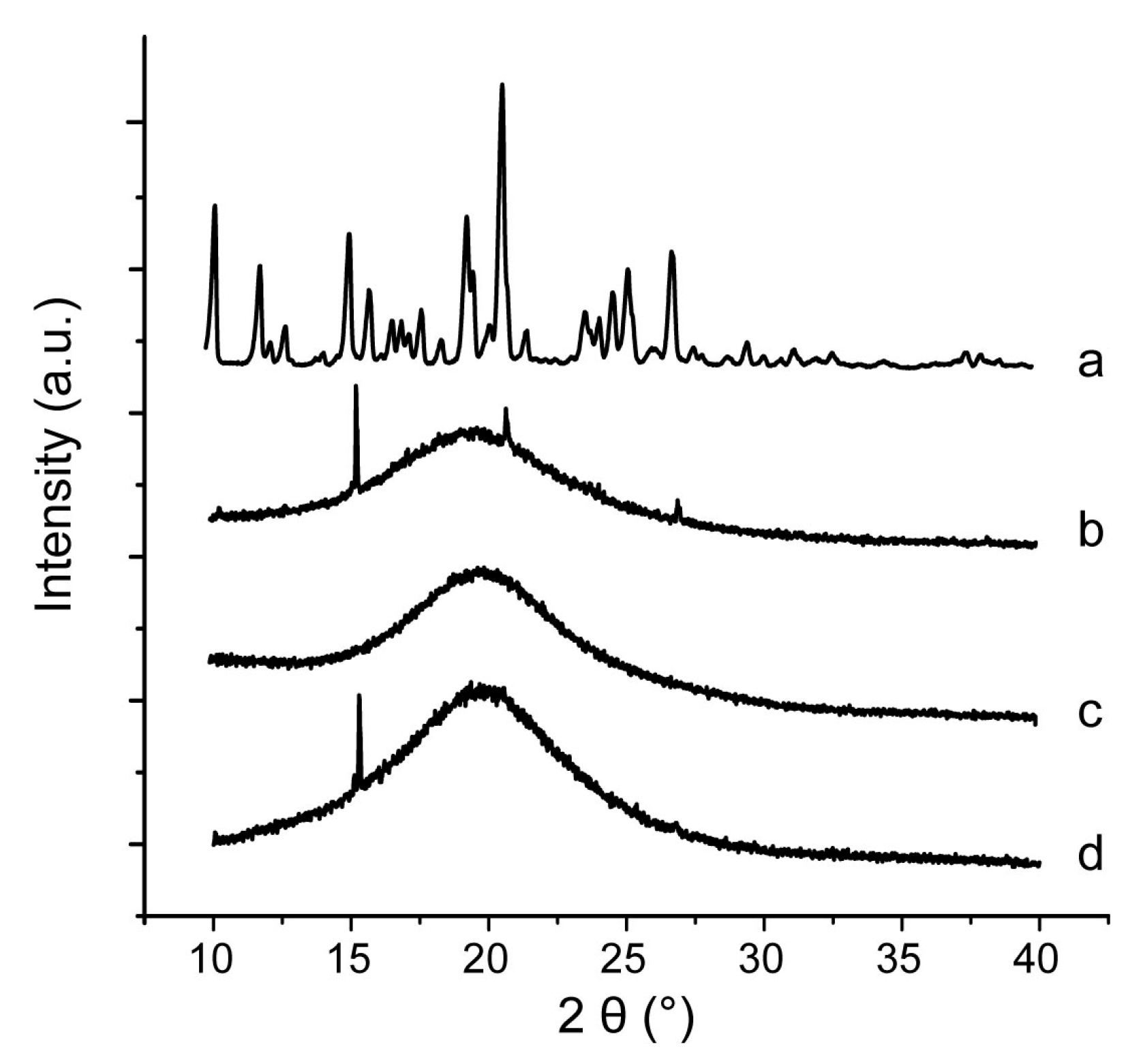
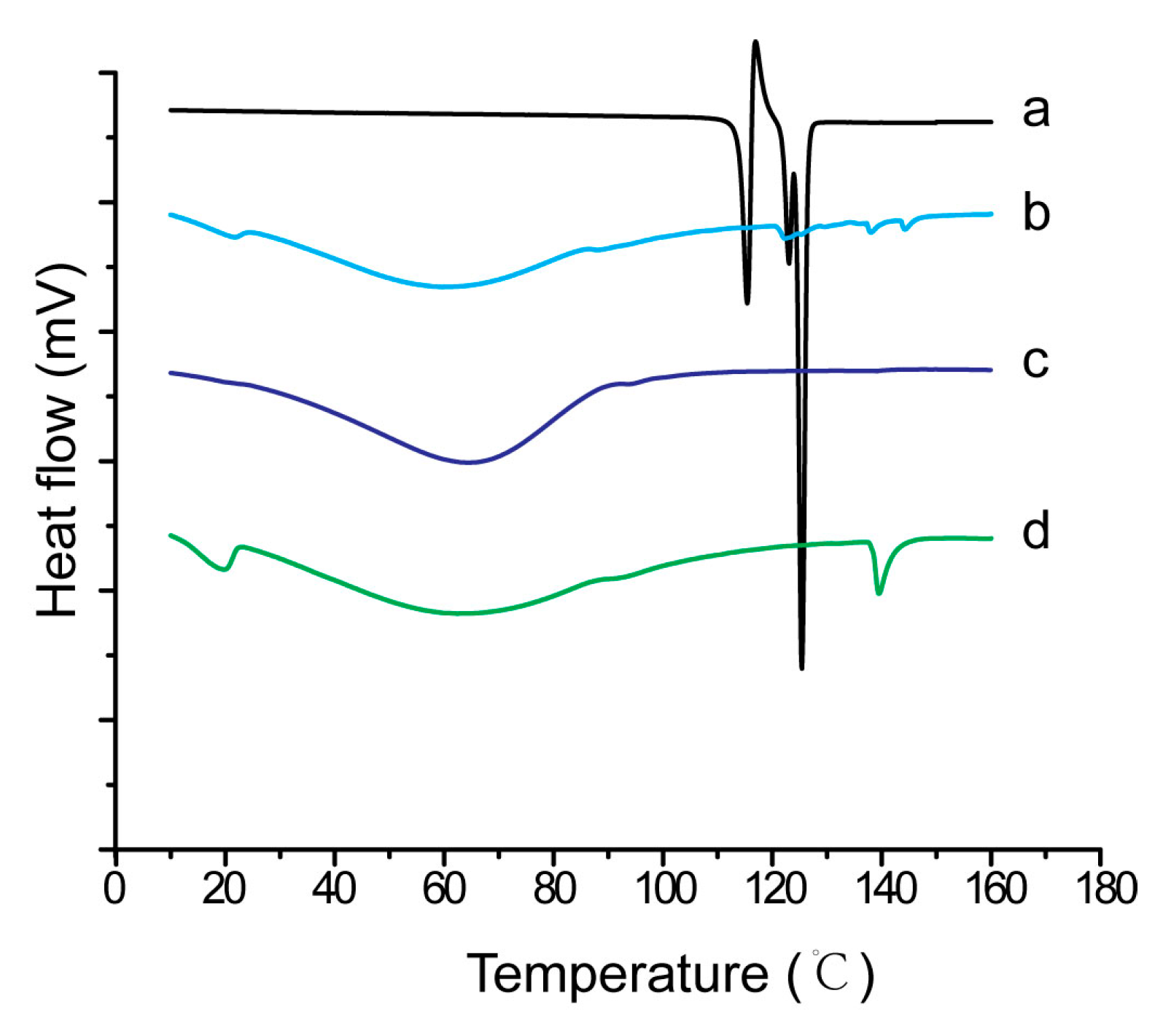
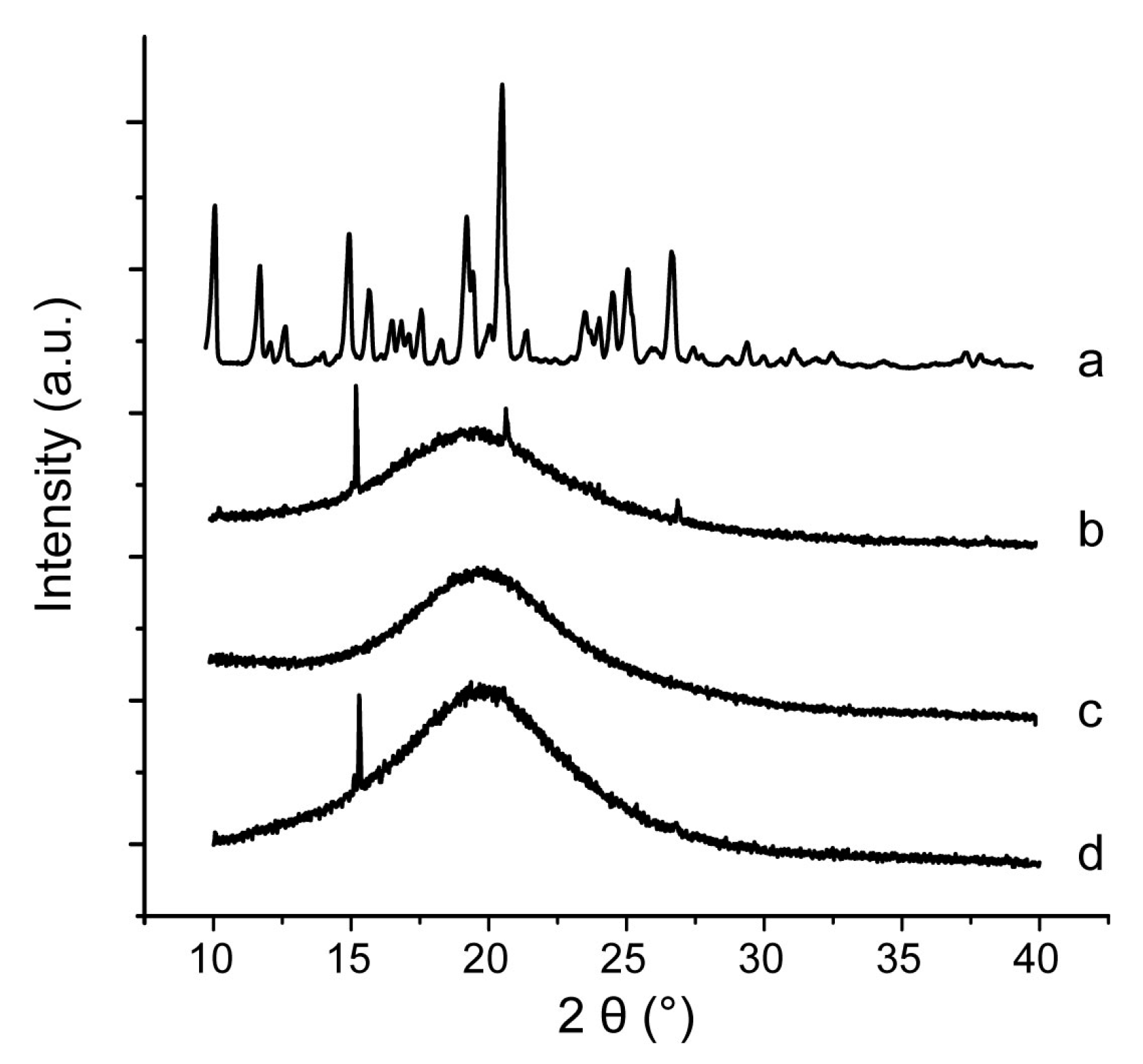
2.9. Characterization of Inner Physical Structure of S-SMEDDS
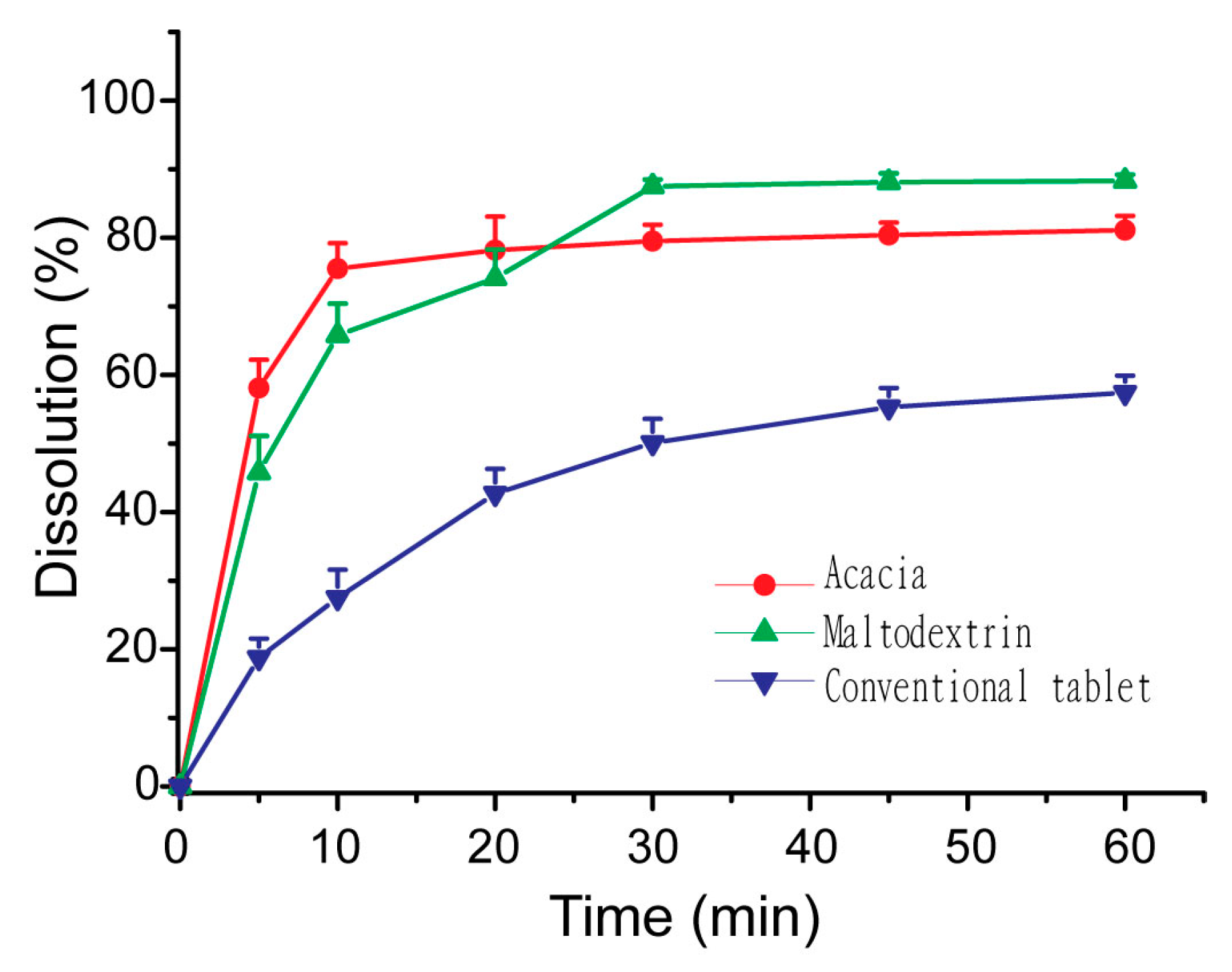
2.10. In Vitro Dissolution Studies of S-SMEDDS
2.11. Statistical Analyses
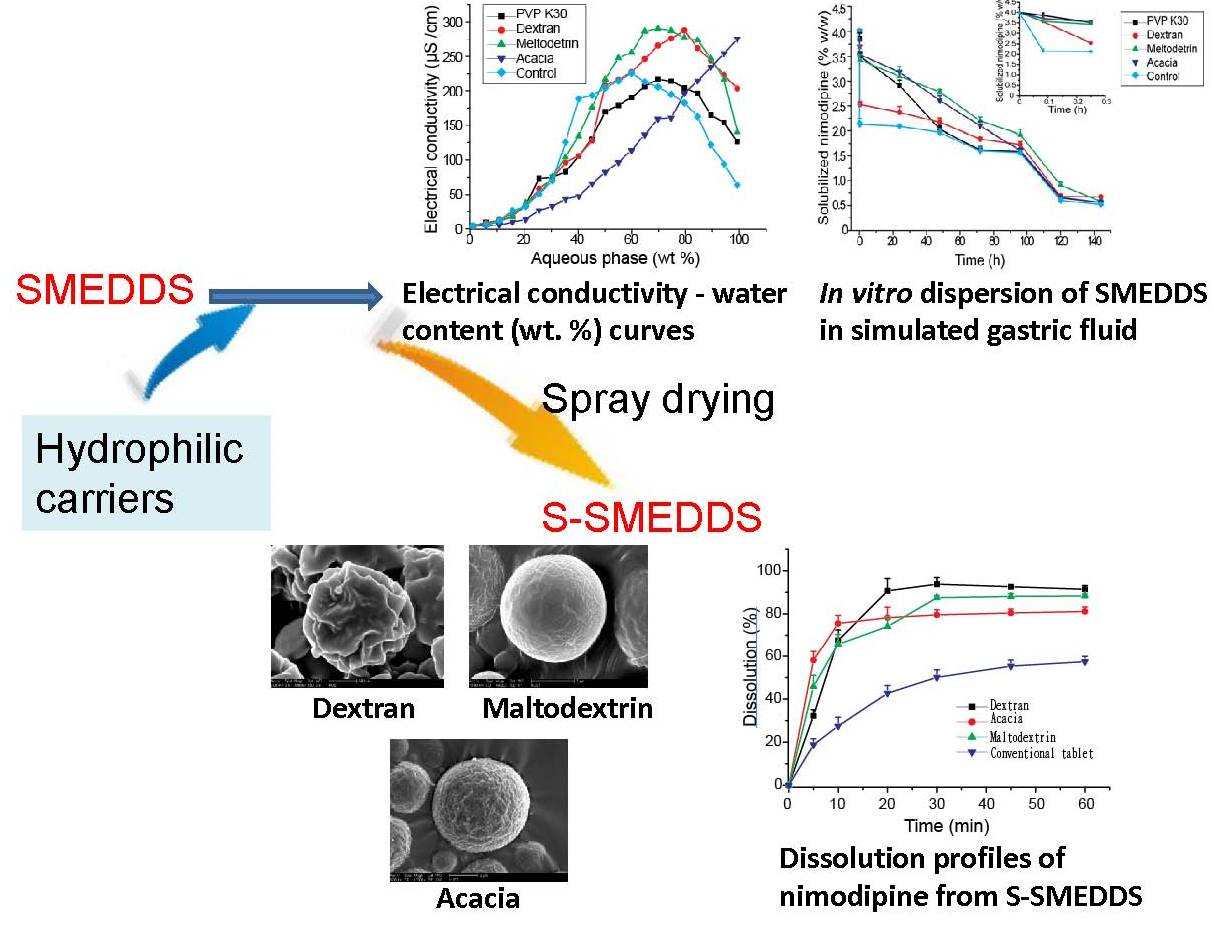
3. Results and Discussions
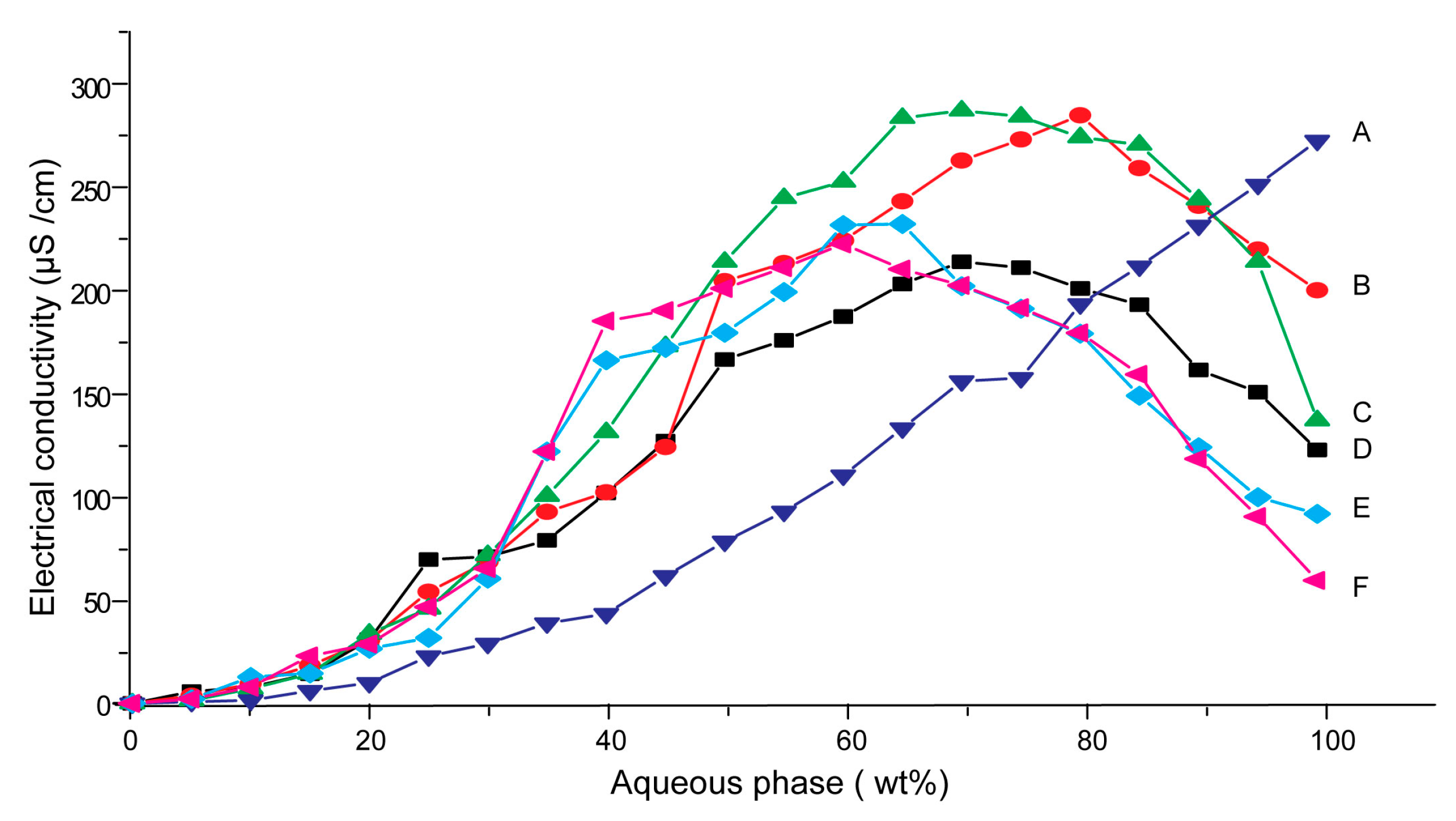
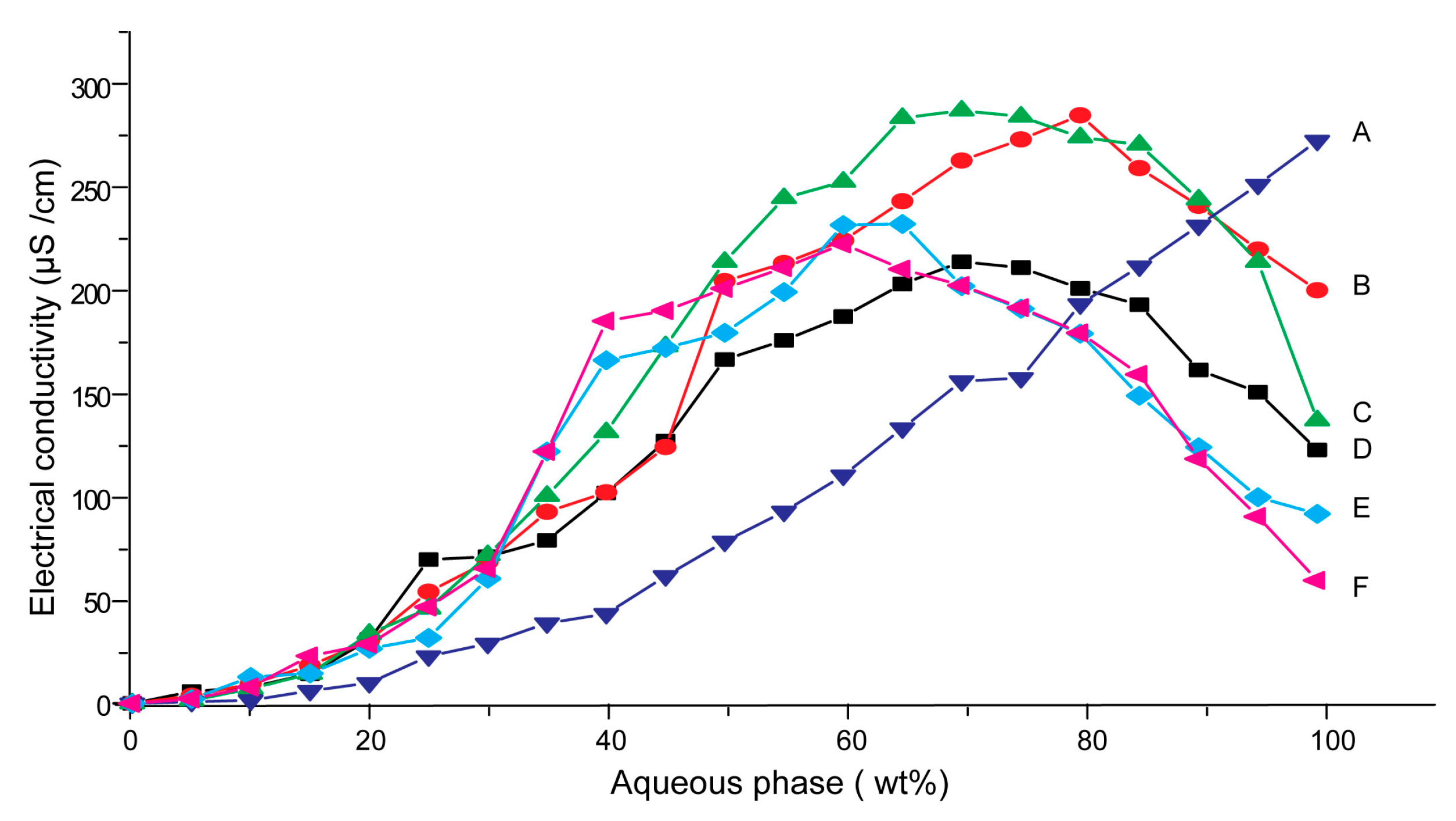
3.1. Effects of Carriers on Microstructure of SMEDDS
3.2. Influences of Carriers on Drug Loading of SMEDDS
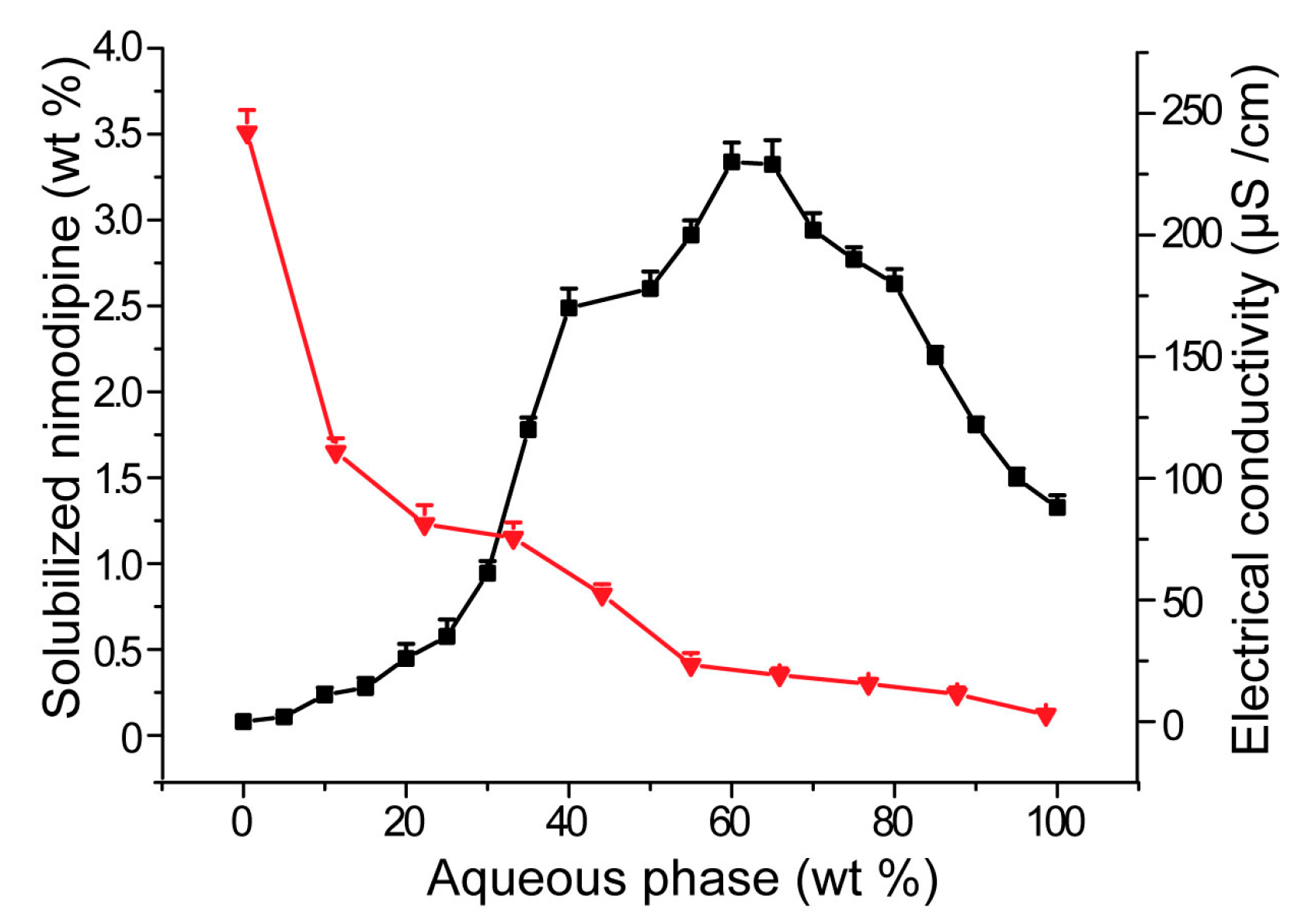
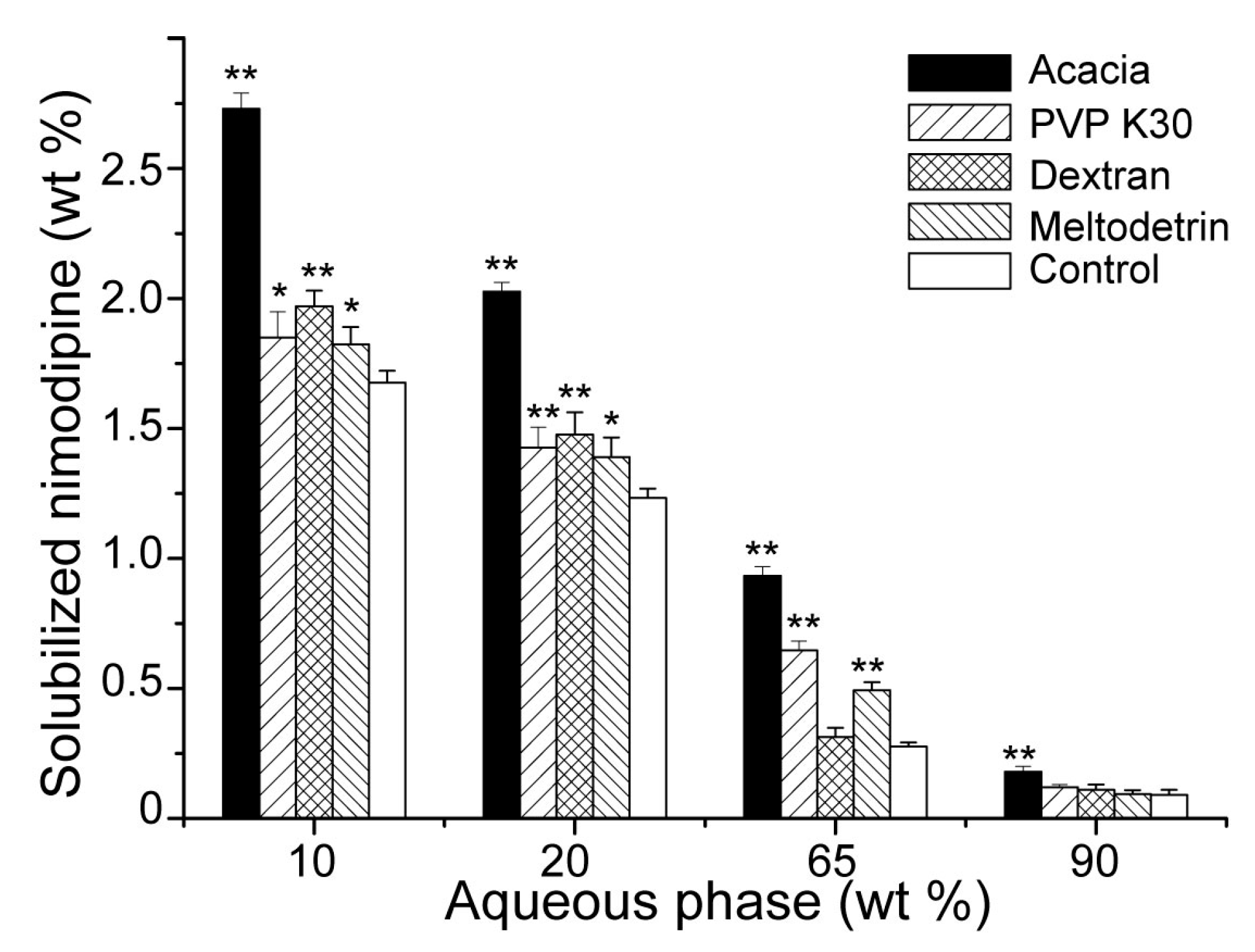
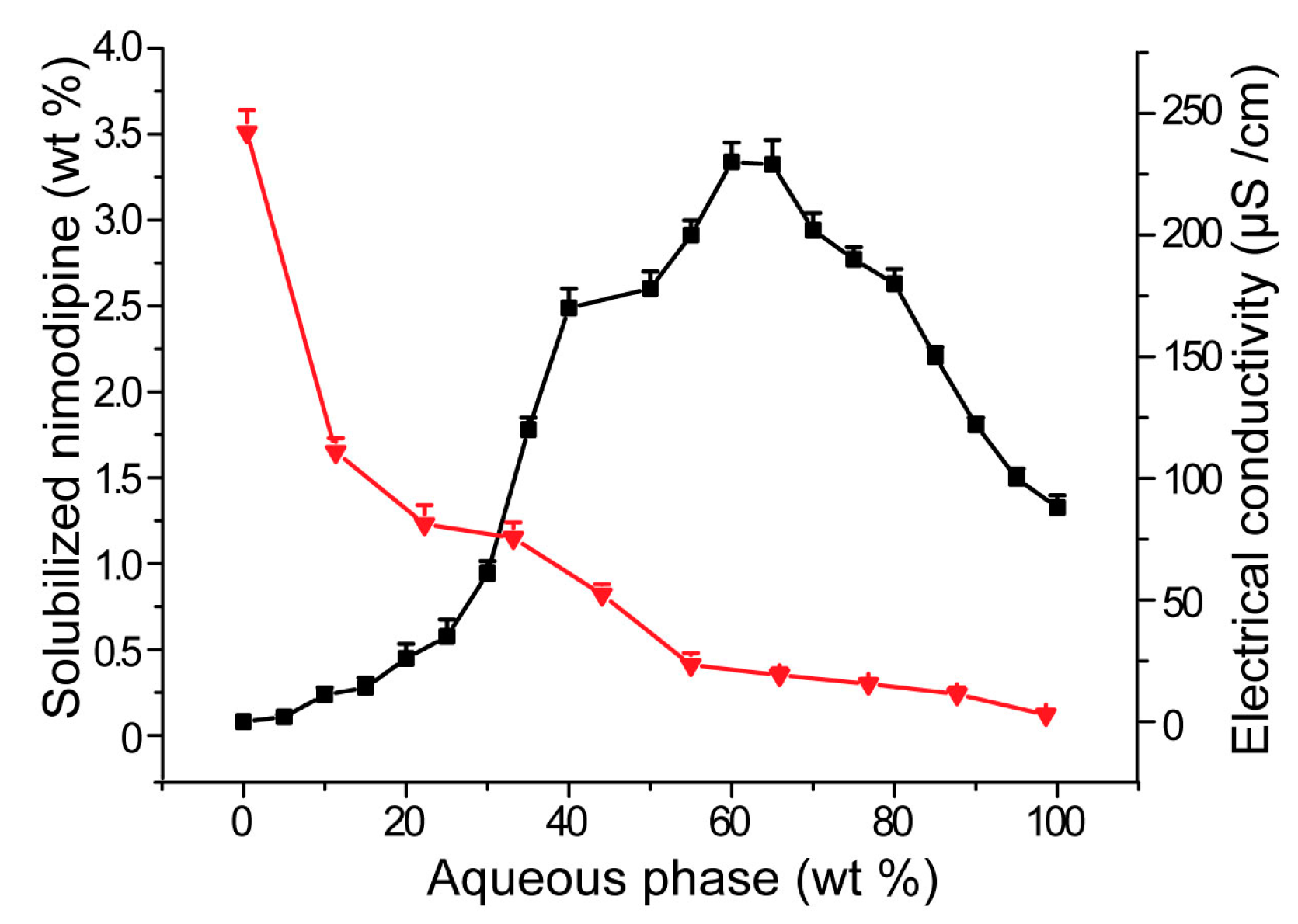
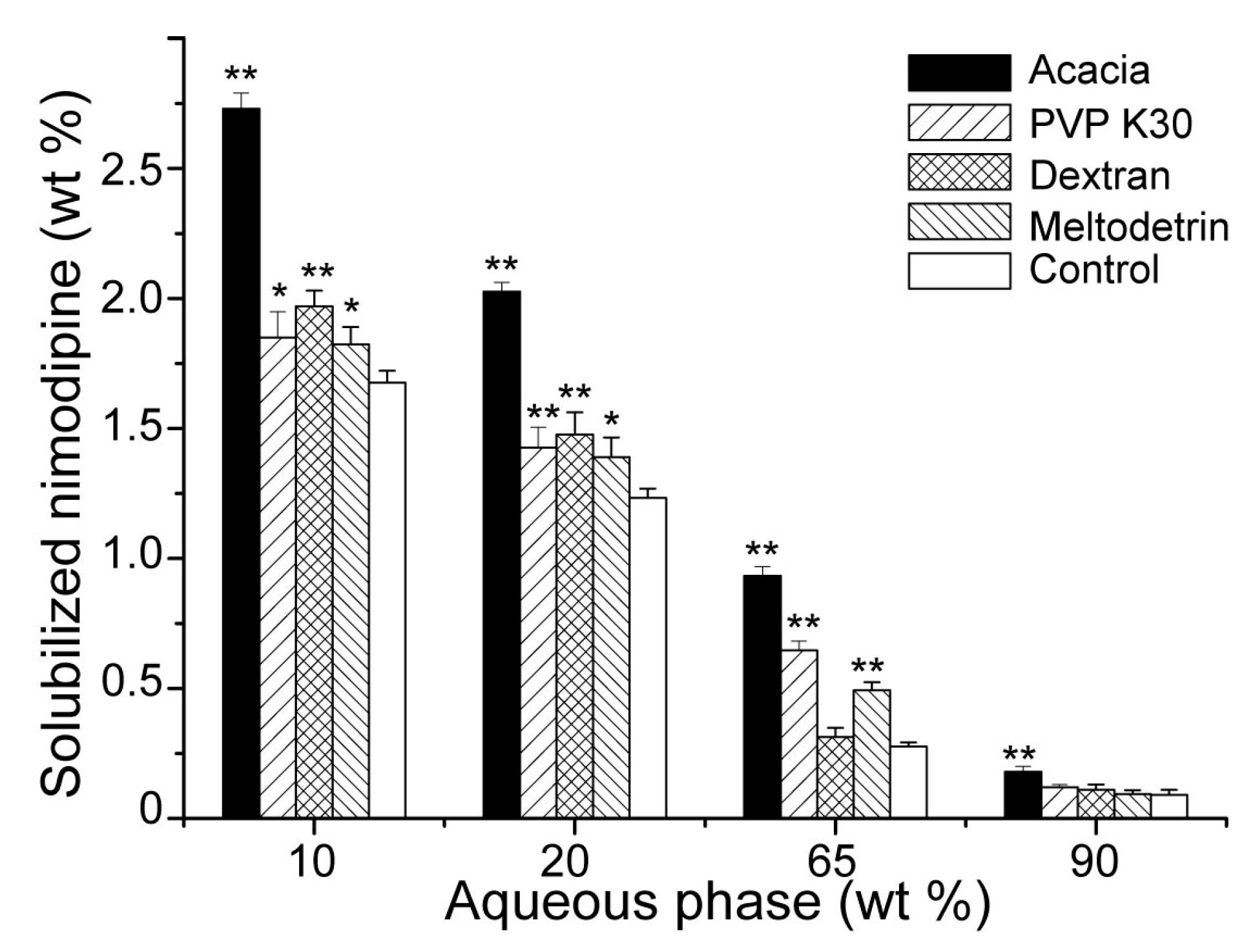
3.3. Influences of Carriers on Dispersion and Precipitation of SMEDDS in Simulated Gastric Fluid
3.4. Influences of Carriers on Characterization of S-SMEDDS
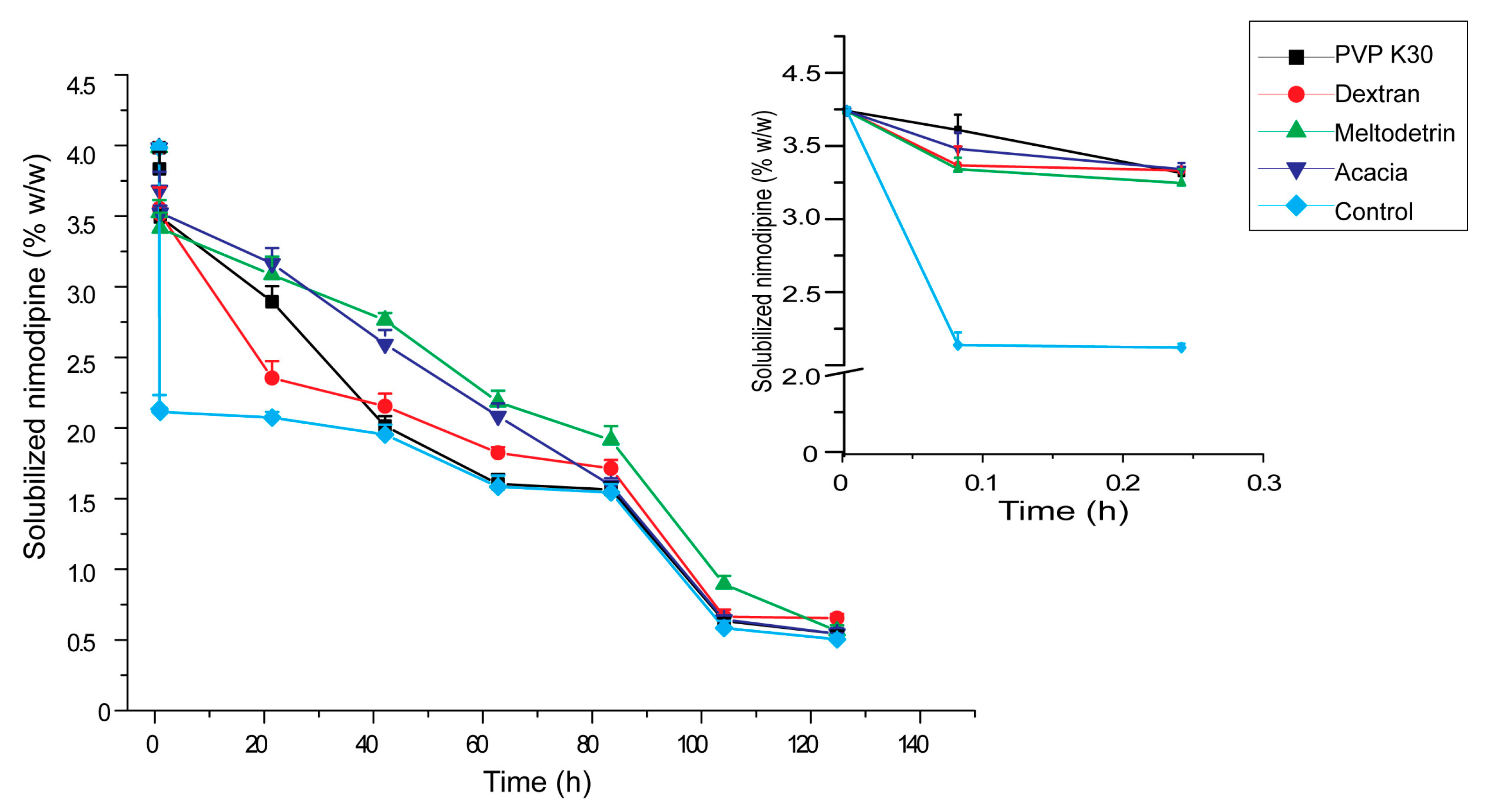
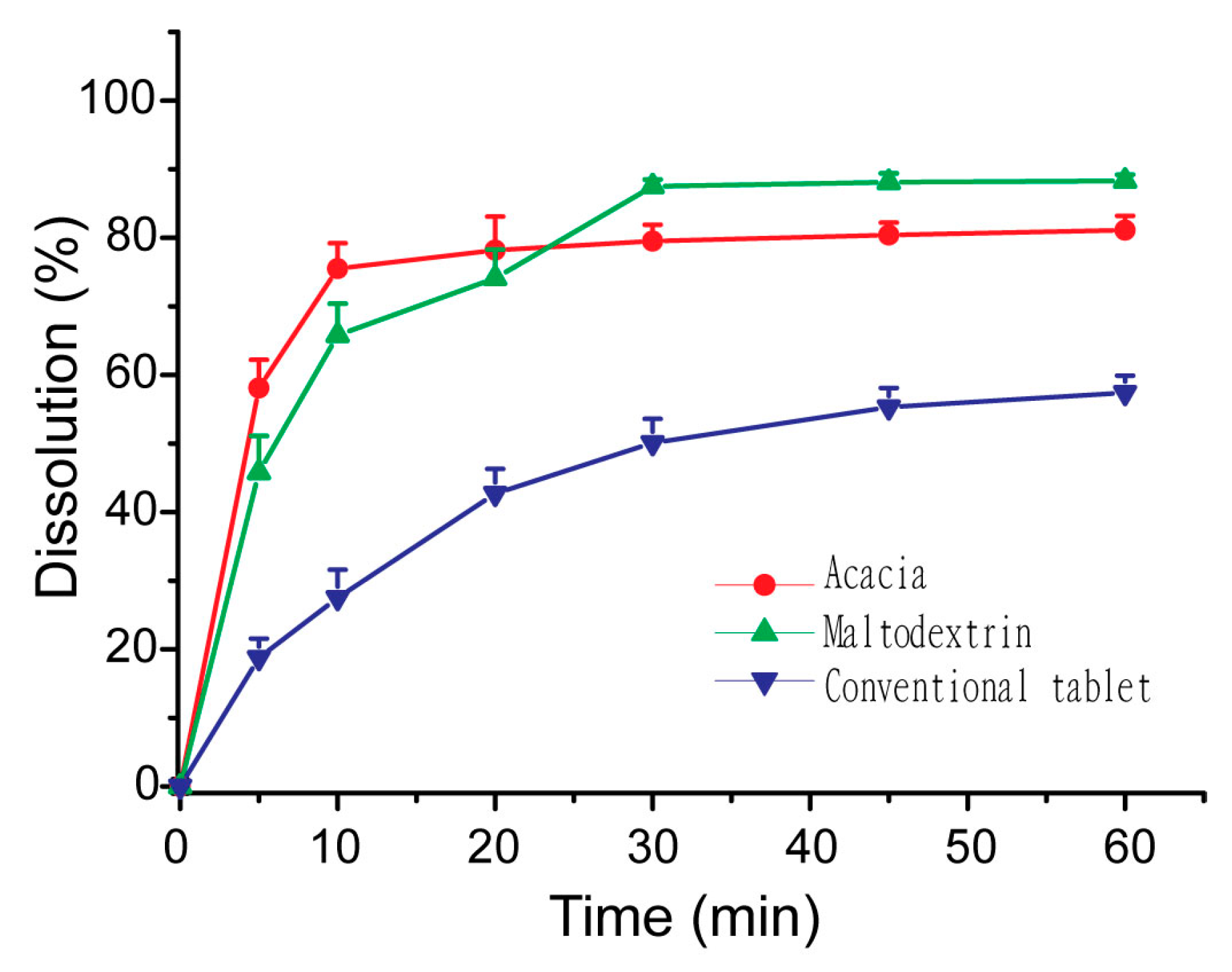
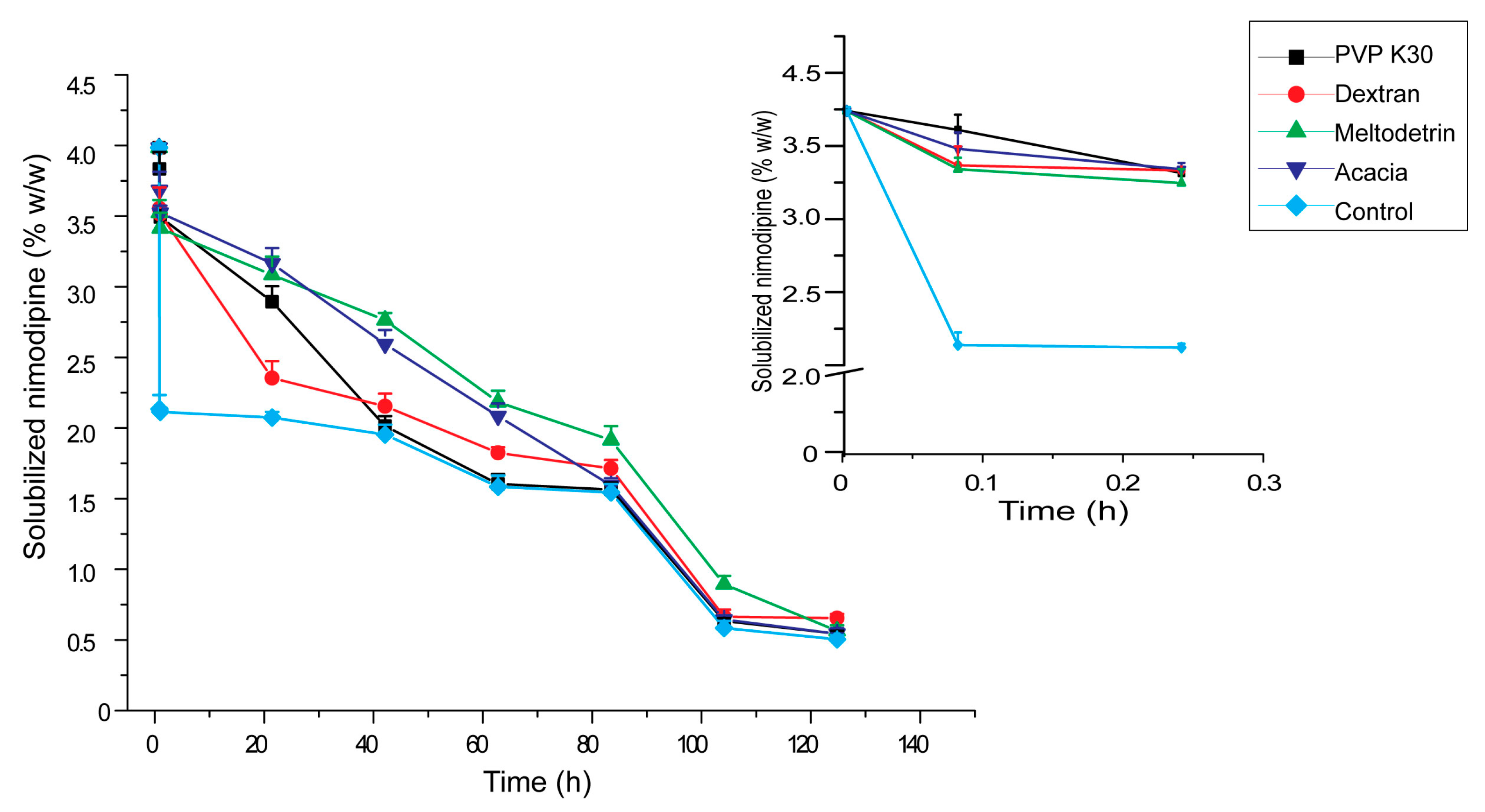
3.5. Influences of Carriers on in Vitro Dissolution of S-SMEDDS
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fu, Q.; Sun, J.; Zhang, D.; Li, M.; Wang, Y.J.; Ling, G.X.; Liu, X.H.; Sun, Y.H.; Sui, X.F.; Luo, C.; et al. Nimodipine nanocrystals for oral bioavailability improvement: Preparation, characterization and pharmacokinetic studies. Colloids Surf. B 2013, 109, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Chalikwar, S.S.; Belgamwar, V.S.; Talele, V.R.; Surana, S.J.; Patil, M.U. Formulation and evaluation of Nimodipine-loaded solid lipid nanoparticles delivered via lymphatic transport system. Colloids Surf. B 2012, 97, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Teng, Z.J.; Yu, M.; Ding, Y.; Zhang, H.Q.; Shen, Y.; Jiang, M.L.; Liu, P.X.; Opoku-Damoah, Y.; Webster, T.J.; Zhou, J.P. Preparation and characterization of nimodipine-loaded nanostructured lipid systems for enhanced solubility and bioavailability. Int. J. Nanomed. 2019, 14, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Ghareeb, M.M.; Neamah, A.J. Formulation and characterization of nimodipine nanoemulsion as ampoule for oral route. Int. J. Pharm. Sci. Res. 2017, 8, 591–602. [Google Scholar]
- Gursoy, R.N.; Benita, S. Self-emulsifying drug delivery systems (SMEDDS) for improved oral delivery of lipophilic drugs. Biomed. Pharmacother. 2004, 58, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Yu, B.; Zhao, Y.; Zhu, W.W.; Li, H.L.; Lou, H.X.; Zhai, G.X. Enhancement of oral absorption of curcumin by self-microemulsifying drug delivery systems. Int. J. Pharm. 2009, 371, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Duc Hanh, N.; Mitrevej, A.; Sathirakul, K.; Peungvicha, P.; Sinchaipanid, N. Development of phyllanthin-loaded self-microemulsifying drug delivery system for oral bioavailability enhancement. Drug Dev. Ind. Pharm. 2015, 41, 27–217. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Chen, J.X.; Chen, L.K.; Zheng, W.; Cao, Y.F.; Huang, T. Enhanced Oral Bioavailability of Chlormadinone Acetate through a Self-Microemulsifying Drug Delivery System for a Potential Dose Reduction. AAPS PharmSciTech 2018, 19, 3850–3858. [Google Scholar] [CrossRef]
- Dokania, S.; Joshi, A.K. Self-microemulsifying drug delivery system (SMEDDS)—Challenges and road ahead. Drug Deliv. 2015, 22, 675–690. [Google Scholar] [CrossRef] [PubMed]
- Kohli, K.; Chopra, S.; Dhar, D.; Arora, S.; Khar, R.K. Self-emulsifying drug delivery systems: An approach to enhance oral bioavailability. Drug Discov. Today 2010, 15, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Kanav, M.; Manju, N.; Garima, S.; Geeta, A. Prospectives of Solid Self-microemulsifying Systems in Novel Drug Delivery. Curr. Drug Deliv. 2017, 14, 1078–1096. [Google Scholar]
- Mandić, J.; Pobirk, A.Z.; Vrečer, F.; Gašperlin, M. Overview of solidification techniques for self-emulsifying drug delivery systems from industrial perspective. Int. J. Pharm. 2017, 533, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.H.; Kang, J.H.; Kim, D.W.; Lee, B.J.; Kim, J.O.; Yong, C.S.; Choi, H.G. Comparison of solid self-microemulsifying drug delivery system (solid SMEDDS) prepared with hydrophilic and hydrophobic solid carrier. Int. J. Pharm. 2011, 420, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Sermkaew, N.; Ketjinda, W.; Boonme, P.; Phadoongsombut, N.; Wiwattanapatapee, R. Liquid and solid self-microemulsifying drug delivery systems for improving the oral bioavailability of andrographolide from a crude extract of Andrographis paniculata. Eur. J. Pharm. Sci. 2013, 50, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Yeom, D.W.; Son, H.Y.; Kim, J.H.; Kim, S.R.; Lee, S.G.; Song, S.H.; RamChae, B.; WookChoi, Y. Development of a solidified self-microemulsifying drug delivery system (S-SMEDDS) for atorvastatin calcium with improved dissolution and bioavailability. Int. J. Pharm. 2016, 506, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Milović, M.; Djuriš, J.; Djekić, L.; Vasiljević, D.; Ibrić, S. Characterization and evaluation of solid self-microemulsifying drug delivery systems with porous carriers as systems for improved carbamazepine release. Int. J. Pharm. 2012, 436, 58–65. [Google Scholar] [CrossRef]
- Yi, T.; Wan, J.L.; Xu, H.B.; Yang, X.L. Controlled poorly soluble drug release from solid self-microemulsifying formulations with high viscosity hydroxypropylmethylcellulose. Eur. J. Pharm. Sci. 2008, 34, 274–280. [Google Scholar] [CrossRef]
- Čerpnjak, K.; Zvonar, A.; Vrečer, F.; Gašperlin, M. Development of a solid self-microemulsifying drug delivery system (SMEDDS) for solubility enhancement of naproxen. Drug Dev. Ind. Pharm. 2015, 41, 1548–1557. [Google Scholar] [CrossRef]
- Sun, C.J.; Gui, Y.; Hu, R.F.; Chen, J.Y.; Wang, B.; Guo, Y.X.; Lu, W.J.; Nie, X.J.; Shen, Q.; Gao, S.; et al. Preparation and pharmacokinetics evaluation of solid self-microemulsifying drug delivery system (S-SMEDDS) of osthole. AAPS PharmSciTech 2018, 19, 2301–2310. [Google Scholar] [CrossRef]
- Yi, T.; Wan, J.L.; Xu, H.B.; Yang, X.L. A new solid self-microemulsifying formulation prepared by spray-drying to improve the oral bioavailability of poorly water soluble drugs. Eur. J. Phar. Biopharm. 2008, 70, 439–444. [Google Scholar] [CrossRef]
- Joyce, P.; Dening, T.J.; Meola, T.R.; Schultz, H.B.; Holm, R.; Thomas, N.; Prestidge, C.A. Solidification to improve the biopharmaceutical performance of SEDDS: Opportunities and challenges. Adv. Drug Deliv. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Dening, T.J.; Rao, S.H.; Thomas, N.; Prestidge, C.A. Novel Nanostructured Solid Materials for Modulating Oral Drug Delivery from Solid-State Lipid-Based Drug Delivery Systems. AAPS J. 2016, 18, 23–40. [Google Scholar] [CrossRef]
- Tu, Q.R.; Zhu, J.B. Study on formulation of nimodipine self-microemulsifying drug delivery system. Chin. Pharm. J. 2005, 40, 43–46. [Google Scholar]
- He, Z.G.; Zhong, D.F.; Chen, X.Y.; Liu, X.H.; Tang, X.; Zhao, L.M. Development of a dissolution medium for nimodipine tablets based on bioavailability evaluation. Eur. J. Pharm. Sci. 2004, 21, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Kayali, I.; Qamhieh, K.; Fanun, M.; Wadaah, S.; Kunhatta, J.C.; Kanan, K. Transport properties of alternative fuel microemulsions based on sugar surfactant. J. Dispers. Sci. Technol. 2017, 28, 917–922. [Google Scholar] [CrossRef]
- Clarkson, M.T.; Smedley, S.I. Electrical conductivity and permittivity measurements near the percolation transition in a microemulsion I: Experiment. Phys. Rev. 1988, 37, 2070–2078. [Google Scholar] [CrossRef]
- Clarkson, M.T. Electrical conductivity and permittivity measurements near the percolation transition in a microemulsion II: Interpretation. Phys. Rev. 1988, 37, 2079–2090. [Google Scholar] [CrossRef]
- Lagües, M.; Ober, R.; Taupin, C. Study of structure and electrical conductivity in microemulsions: Evidence for percolation mechanism and phase inversion. J. Phys. Lett. 1978, 39, 487–491. [Google Scholar] [CrossRef]
- Podlogar, F.; Bester, R.M.; Gasperlin, M. The effect of internal structure of selected water-Tween 40(R)-Imwitor 308(R)-IPM microemulsions on ketoprofene release. Int. J. Pharm. 2005, 302, 68–77. [Google Scholar] [CrossRef]
- Schmidts, T.; Dobler, D.; Schlupp, P.; Nissing, C.; Garn, H.; Runkel, F. Development of multiple W/O/W emulsions as dermal carrier system for oligonucleotides: Effect of additives on emulsion stability. Int. J. Pharm. 2010, 398, 107–113. [Google Scholar] [CrossRef]
- Garti, N.; Avrahami, M.; Aserin, A. Improved solubilization of Celecoxib in U-type nonionic microemulsions and their structural transitions with progressive aqueous dilution. J. Colloid Interface Sci. 2006, 299, 352–365. [Google Scholar] [CrossRef] [PubMed]
- Pouton, C.W. Formulation of poorly water-soluble drugs for oral administration: Physicochemical and physiological issues and the lipid formulation classification system. Eur. J. Pharm. Sci. 2006, 29, 278–287. [Google Scholar] [CrossRef]
- Porter, C.J.H.; Pouton, C.W.; Cuine, J.F. Enhancing intestinal drug solubilisation using lipid-based delivery systems. Adv. Drug Deliv. Rev. 2008, 60, 673–691. [Google Scholar] [CrossRef] [PubMed]
- Feeney, O.M.; Crum, M.F.; McEvoy, C.L.; Trevaskis, N.L.; Williams, H.D.; Pouton, C.W.; Charman, W.N.; Bergström, C.A.S.; Porter, C.J.H. 50 years of oral lipid-based formulations: Provenance, progress and future perspectives. Adv. Drug Deliv. Rev. 2016, 101, 167–194. [Google Scholar] [CrossRef] [PubMed]
- Jo, K.; Kim, H.; Khadka, P.; Jang, T.; Kim, S.J.; Hwang, S.H.; Lee, J. Enhanced intestinal lymphatic absorption of saquinavir through supersaturated self-microemulsifying drug delivery systems. Asian J. Pharm. Sci. 2019. [Google Scholar] [CrossRef]
- McEvoy, C.L.; Trevaskis, N.L.; Feeney, O.M.; Edwards, G.A.; Perlman, M.E.; Ambler, C.M.; Porter, C.J.H. Correlating in Vitro Solubilization and Supersaturation Profiles with in Vivo Exposure for Lipid Based Formulations of the CETP Inhibitor CP-532,623. Pharmaceutics 2017, 14, 4525–4538. [Google Scholar] [CrossRef]
- Christensen, K.L.; Pedersen, G.P.; Kristensen, H.G. Physical stability of redispersible dry emulsions containing amorphous sucrose. Eur. J. Pharm. Biopharm. 2002, 53, 147–153. [Google Scholar] [CrossRef]
- Dollo, G.; Corre, P.L.; Guérin, A.; Chevanne, F.; Burgot, J.L.; Leverge, R. Spray-dried redispersible oil-in-water emulsion to improve oral bioavailability of poorly soluble drugs. Eur. J. Pharm. Sci. 2003, 19, 273–280. [Google Scholar] [CrossRef]
- Urbanetz, N.A.; Lippold, B.C. Solid dispersions of nimodipine and polyethylene glycol 2000: Dissolution properties and physico-chemical characterisation. Eur. J. Pharm. Biopharm. 2005, 59, 107–118. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carrier | Relative Molecular Mass of Carrier | Droplet Size of Reconstructed Emulsion (nm) | Polydispersity Index of Reconstructed Emulsion | Lipid Leak of S-SMEDDS | Particle Separation of S-SMEDDS | Crystallization of S-SMEDDS |
|---|---|---|---|---|---|---|
| None (SMEDDS) | - | 41.3 ± 5.7 | 0.13 ± 0.03 | - | - | - |
| Mannitol | 182 | 117.0 ± 7.2 | 0.16 ± 0.02 | Yes | - | - |
| Lactose | 342 | 124.0 ±10.6 | 0.14 ± 0.02 | Yes | - | - |
| Maltodextrin | 900–9000 | 139.5 ± 6.8 | 0.19 ± 0.05 | No | Bad | Yes |
| Dextran 40 [20] | 40,000 | 44.1 ± 4.7 | 0.25± 0.04 | No | Good | No |
| PVP K30 | 50,000 | 407.5 ± 3.9 | 0.42 ± 0.09 | No | - | - |
| Acacia | 240,000–580,000 | 177.6 ± 14.6 | 0.41 ± 0.07 | No | Good | No |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yi, T.; Zhang, J. Effects of Hydrophilic Carriers on Structural Transitions and In Vitro Properties of Solid Self-Microemulsifying Drug Delivery Systems. Pharmaceutics 2019, 11, 267. https://doi.org/10.3390/pharmaceutics11060267
Yi T, Zhang J. Effects of Hydrophilic Carriers on Structural Transitions and In Vitro Properties of Solid Self-Microemulsifying Drug Delivery Systems. Pharmaceutics. 2019; 11(6):267. https://doi.org/10.3390/pharmaceutics11060267
Chicago/Turabian StyleYi, Tao, and Jifen Zhang. 2019. "Effects of Hydrophilic Carriers on Structural Transitions and In Vitro Properties of Solid Self-Microemulsifying Drug Delivery Systems" Pharmaceutics 11, no. 6: 267. https://doi.org/10.3390/pharmaceutics11060267
APA StyleYi, T., & Zhang, J. (2019). Effects of Hydrophilic Carriers on Structural Transitions and In Vitro Properties of Solid Self-Microemulsifying Drug Delivery Systems. Pharmaceutics, 11(6), 267. https://doi.org/10.3390/pharmaceutics11060267