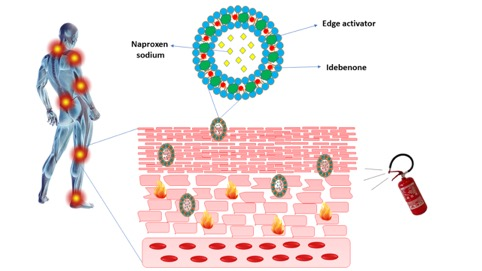
Development and In Vivo Evaluation of Multidrug Ultradeformable Vesicles for the Treatment of Skin Inflammation

 ,
, 
 ,
,  ,
,  and
and 
Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Ultradeformable Vesicles Preparation
2.3. Physicochemical Characterization
2.4. Deformability Index (DI) Evaluation
2.5. Evaluation of Encapsulation Efficiency (EE)
2.6. HPLC Determination of NS and IDE
2.7. Ex Vivo Skin Penetration and Permeation Studies
2.7.1. Preparation of Stratum Corneum Epidermis (SCE) Membranes
2.7.2. In Vitro Evaluation of the Permeation of Compounds from Ultradeformable Carriers
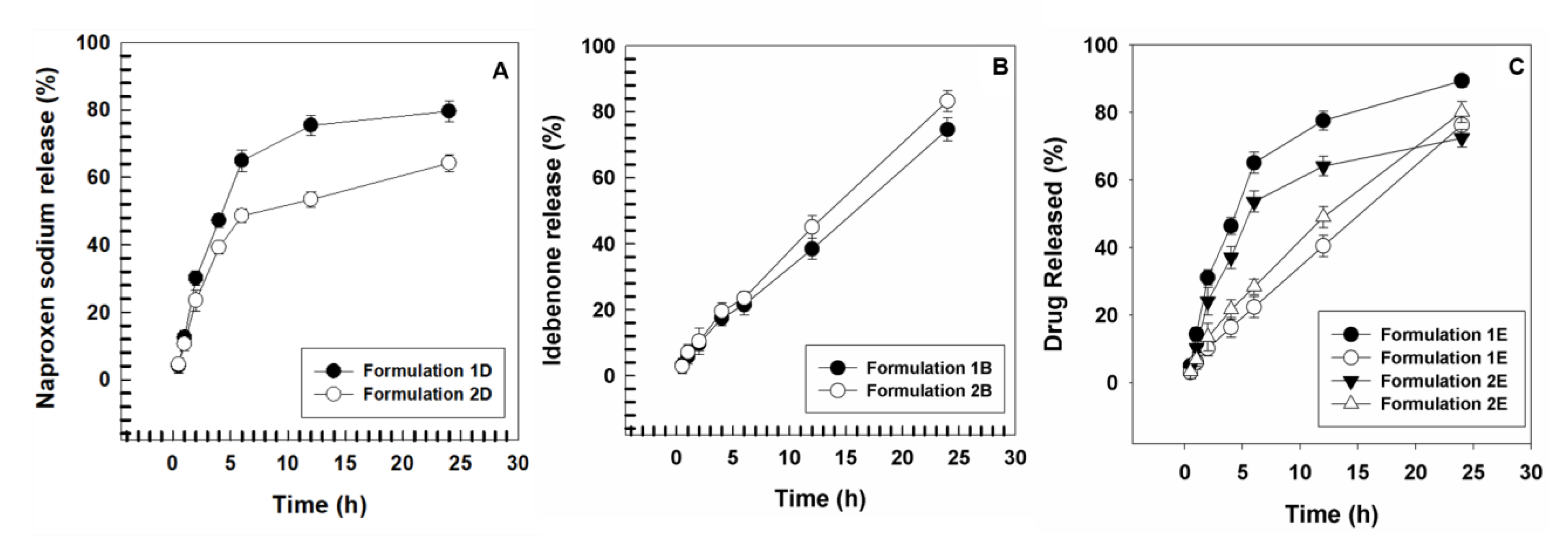
2.8. Evaluation of Release Profiles of Drugs from Vesicular Carriers
2.9. In Vivo Tolerability on Human Volunteers
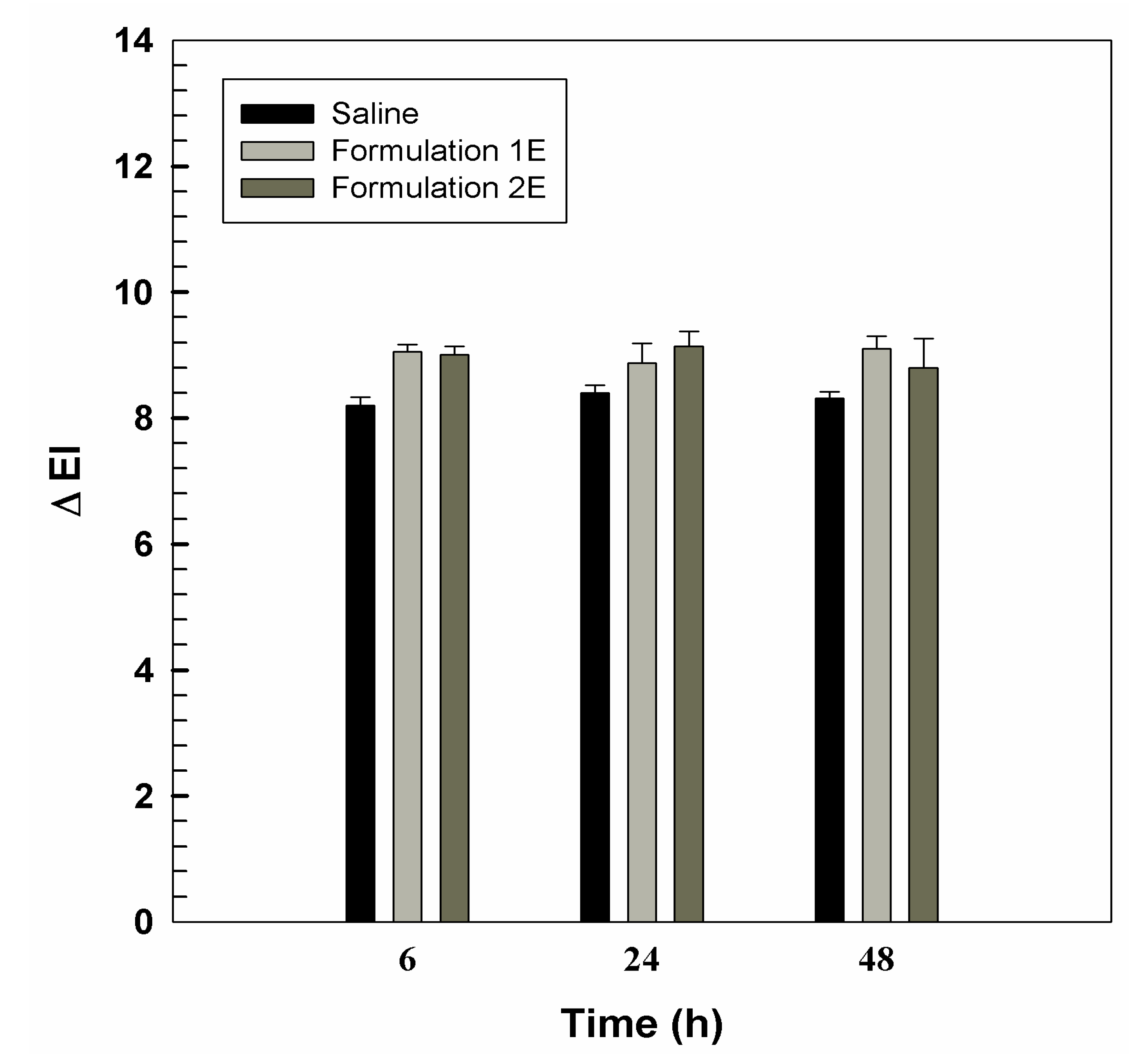
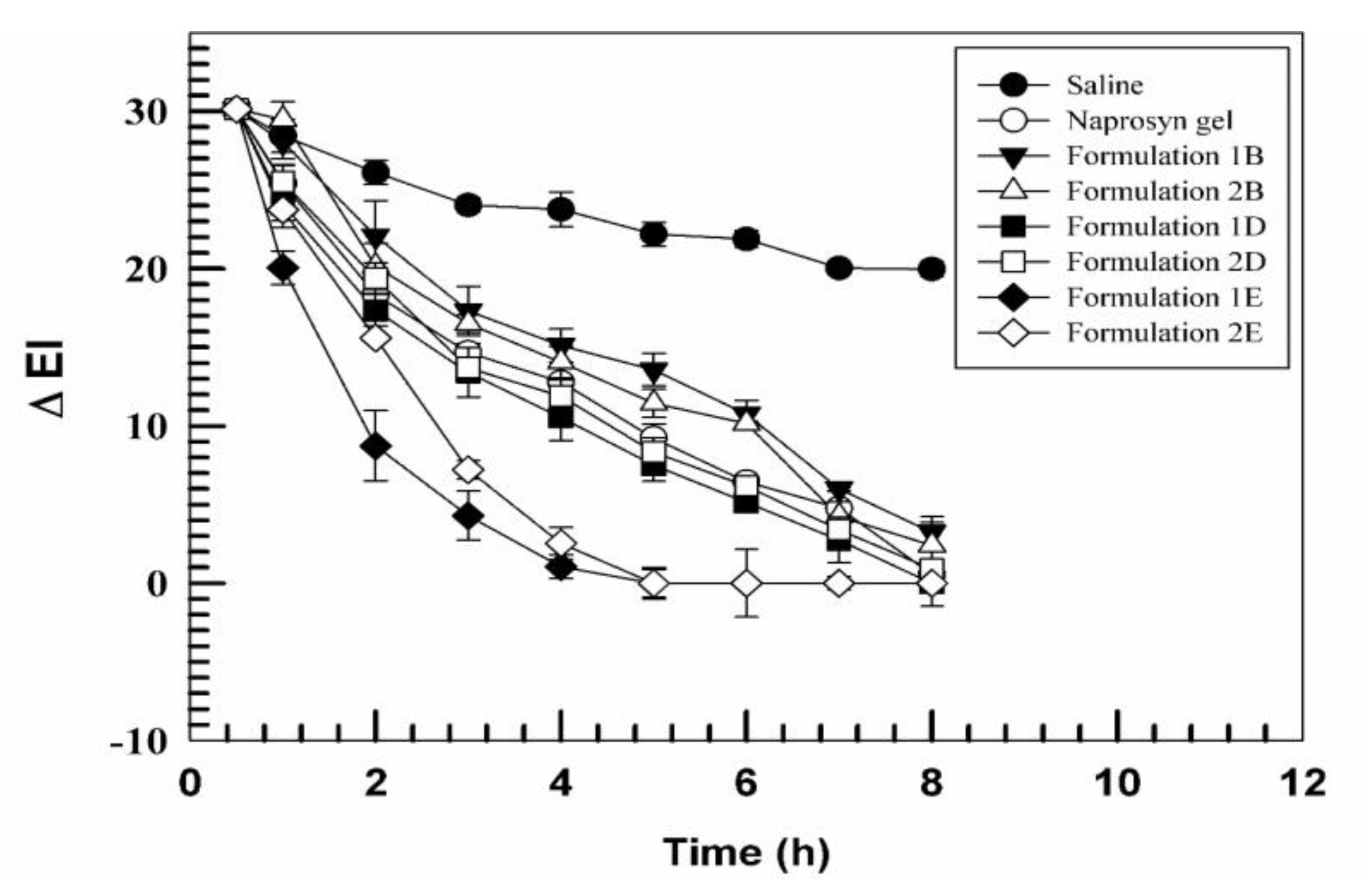
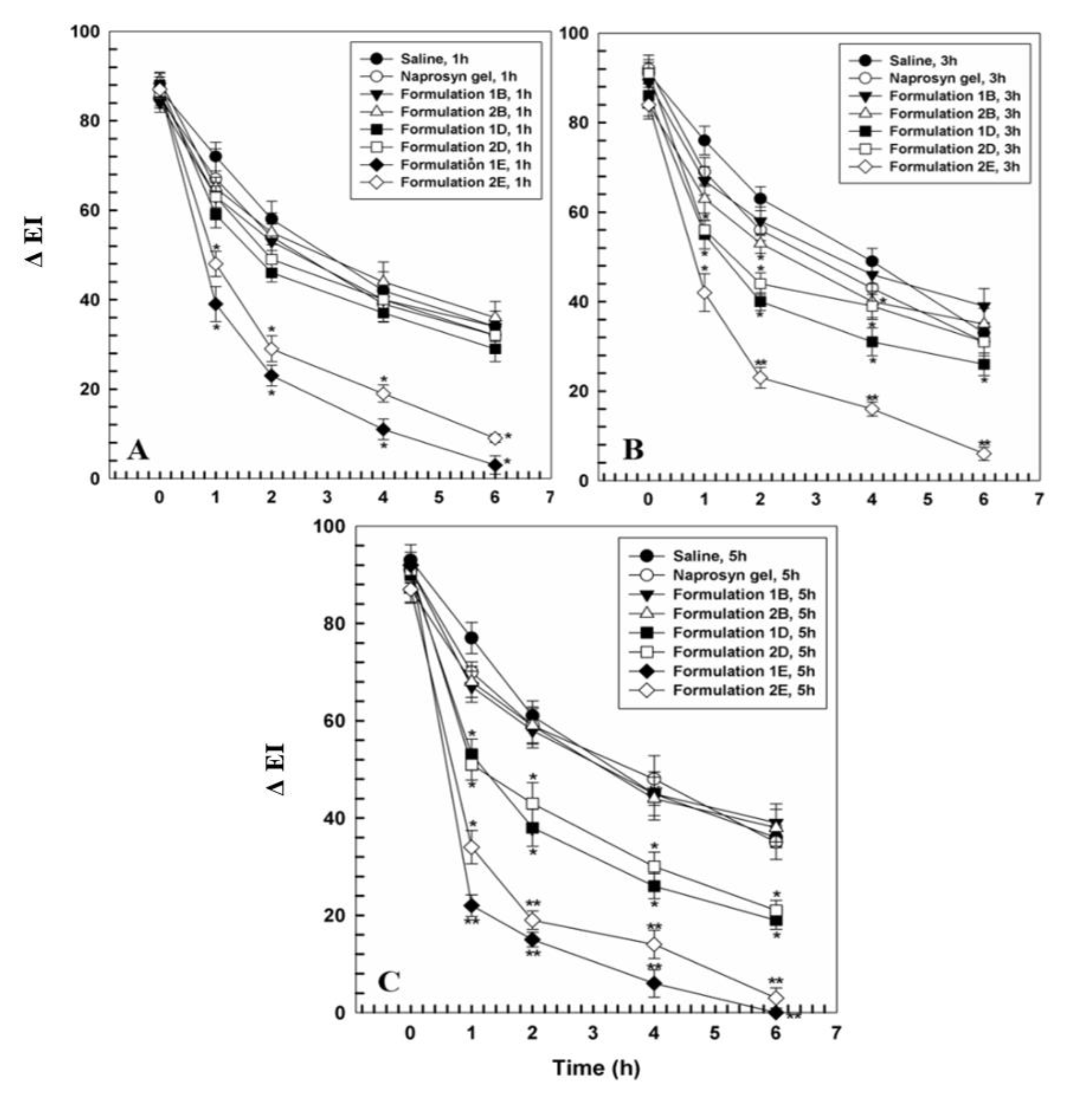
2.10. In Vivo Anti-Inflammatory Activity
2.11. Statistical Analysis
3. Results and Discussion
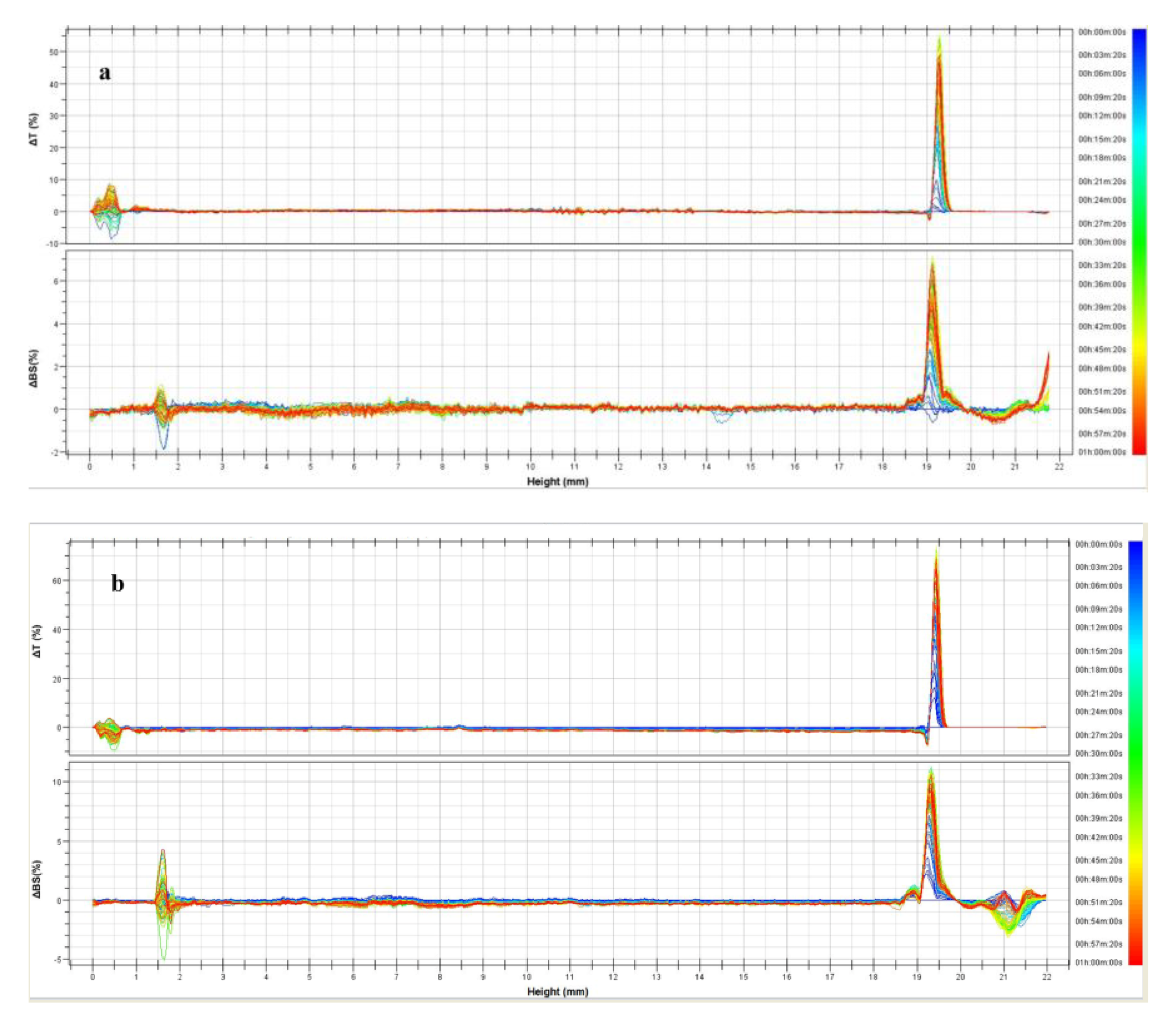
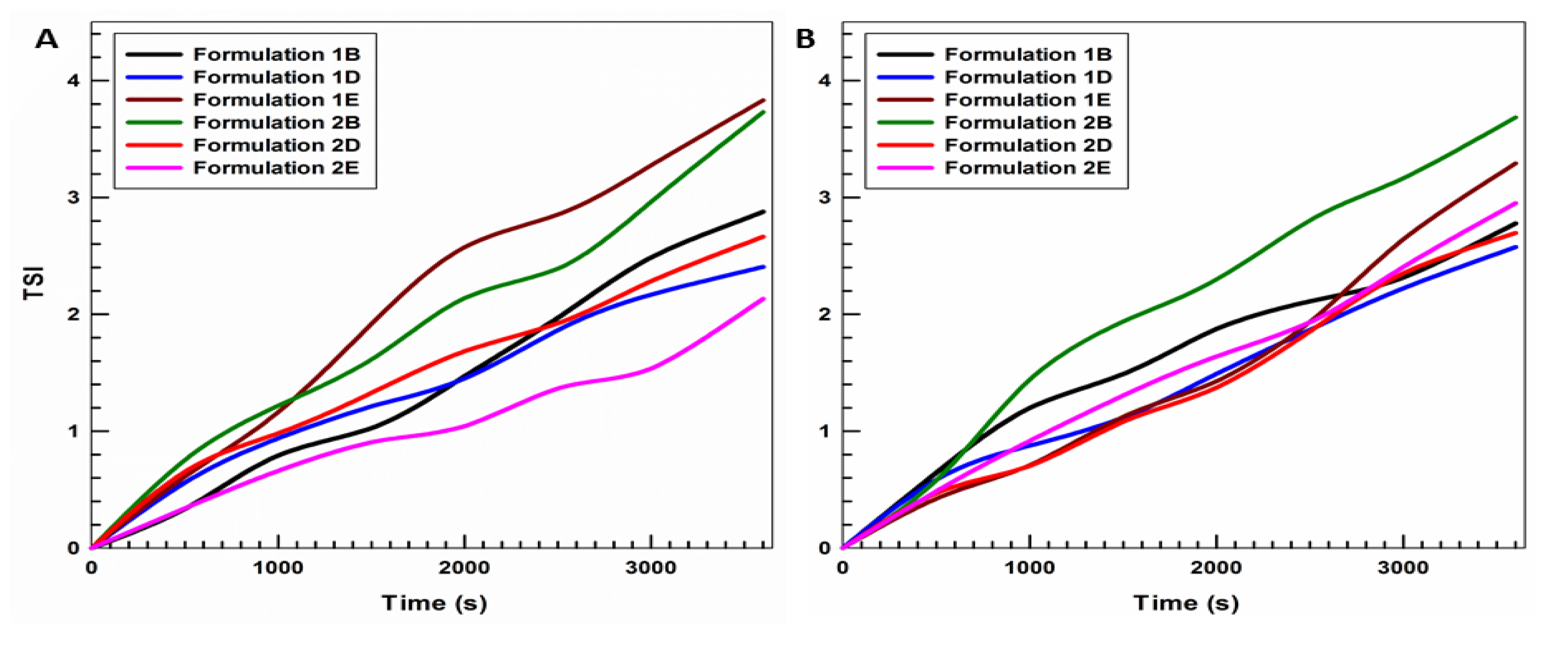
3.1. Physicochemical Characterization of UVs
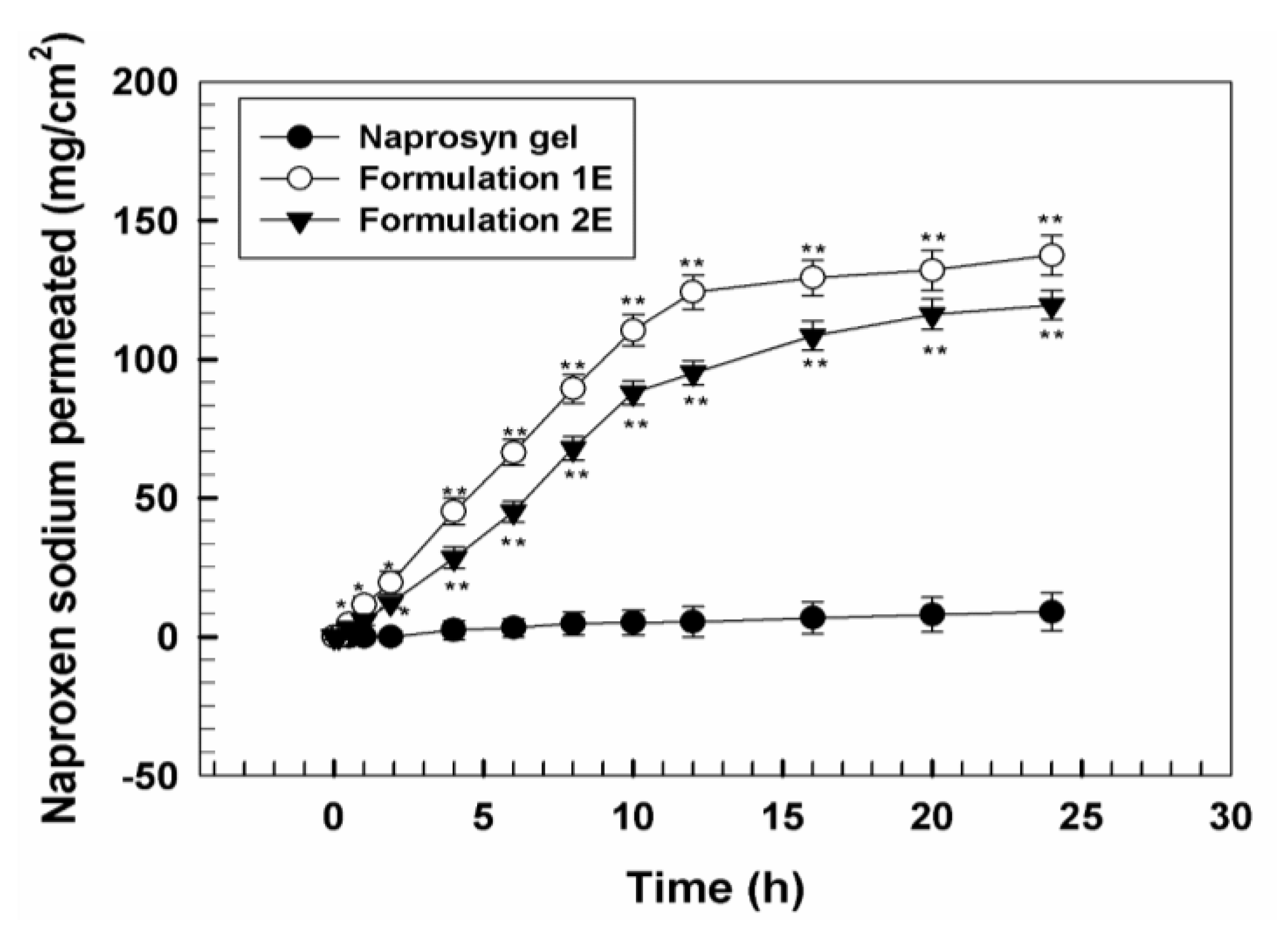
3.2. In Vivo Tolerability on Human Volunteers and Evaluation of Anti-Inflammatory Activity
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kumar, L.; Verma, S.; Singh, M.; Chalotra, T.; Utreja, P. Advanced Drug Delivery Systems for Transdermal Delivery of Non-Steroidal Anti-Inflammatory Drugs: A Review. Curr. Drug Deliv. 2018, 15, 1087–1099. [Google Scholar] [CrossRef] [PubMed]
- Barba, C.; Alonso, C.; Martí, M.; Carrer, V.; Yousef, I.; Coderch, L. Selective modification of skin barrier lipids. J. Pharm. Biomed. Anal. 2019, 172, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Singh Malik, D.; Mital, N.; Kaur, G. Topical drug delivery systems: a patent review. Expert Opin. Ther. Pat. 2016, 26, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Munch, S.; Wohlrab, J.; Neubert, R.H.H. Dermal and transdermal delivery of pharmaceutically relevant macromolecules. Eur. J. Pharm. Biopharm. 2017, 119, 235–242. [Google Scholar] [CrossRef]
- Cosco, D.; Paolino, D.; Maiuolo, J.; Di Marzio, L.; Carafa, M.; Ventura, C.A.; Fresta, M. Ultradeformable liposomes as multidrug carrier of resveratrol and 5-fluorouracil for their topical delivery. Int. J. Pharm. 2015, 489, 1–10. [Google Scholar] [CrossRef]
- Sala, M.; Diab, R.; Elaissari, A.; Fessi, H. Lipid nanocarriers as skin drug delivery systems: Properties, mechanisms of skin interactions and medical applications. Int. J. Pharm. 2018, 535, 1–17. [Google Scholar] [CrossRef]
- Bartelds, R.; Nematollahi, M.H.; Pols, T.; Stuart, M.C.A.; Pardakhty, A.; Asadikaram, G.; Poolman, B. Niosomes, an alternative for liposomal delivery. PLoS ONE 2018, 13, e0194179. [Google Scholar] [CrossRef]
- Paolino, D.; Cosco, D.; Muzzalupo, R.; Trapasso, E.; Picci, N.; Fresta, M. Innovative bola-surfactant niosomes as topical delivery systems of 5-fluorouracil for the treatment of skin cancer. Int. J. Pharm. 2008, 353, 233–242. [Google Scholar] [CrossRef]
- Nakazawa, H.; Imai, T.; Suzuki, M.; Akakabe, N.; Hatta, I.; Kato, S. Simultaneous Measurements of Structure and Water Permeability in an Isolated Human Skin Stratum Corneum Sheet. Polymers 2019, 11, 829. [Google Scholar] [CrossRef]
- El Zaafarany, G.M.; Awad, G.A.S.; Holayel, S.M.; Mortada, N.D. Role of edge activators and surface charge in developing ultradeformable vesicles with enhanced skin delivery. Int. J. Pharm. 2010, 397, 164–172. [Google Scholar] [CrossRef]
- Cevc, G.; Vierl, U. Nanotechnology and the transdermal route: A state of the art review and critical appraisal. J. Control. Release 2010, 141, 277–299. [Google Scholar] [CrossRef] [PubMed]
- Calienni, M.N.; Febres-Molina, C.; Llovera, R.E.; Zevallos-Delgado, C.; Tuttolomondo, M.E.; Paolino, D.; Fresta, M.; Barazorda-Ccahuana, H.L.; Gomez, B.; Alonso, S.D.V.; et al. Nanoformulation for potential topical delivery of Vismodegib in skin cancer treatment. Int. J. Pharm. 2019, 565, 108–122. [Google Scholar] [CrossRef] [PubMed]
- Celia, C.; Locatelli, M.; Cilurzo, F.; Cosco, D.; Gentile, E.; Scalise, D.; Carafa, M.; Ventura, C.A.; Fleury, M.; Tisserand, C.; et al. Long Term Stability Evaluation of Prostacyclin Released from Biomedical Device through Turbiscan Lab Expert. Med. Chem. 2015, 11, 391–399. [Google Scholar] [CrossRef]
- Das Kurmi, B.; Tekchandani, P.; Paliwal, R.; Paliwal, S.R. Transdermal Drug Delivery: Opportunities and Challenges for Controlled Delivery of Therapeutic Agents Using Nanocarriers. Curr. Drug Metab. 2017, 18, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Mir-Palomo, S.; Nácher, A.; Díez-Sales, O.; Ofelia Vila Busó, M.A.; Caddeo, C.; Manca, M.L.; Manconi, M.; Fadda, A.M.; Saurí, A.R. Inhibition of skin inflammation by baicalin ultradeformable vesicles. Int. J. Pharm. 2016, 511, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Zeb, A.; Qureshi, O.S.; Yu, C.-H.; Akram, M.; Kim, H.-S.; Kim, M.-S.; Kang, J.-H.; Majid, A.; Chang, S.-Y.; Bae, O.-N.; et al. Enhanced anti-rheumatic activity of methotrexate-entrapped ultradeformable liposomal gel in adjuvant-induced arthritis rat model. Int. J. Pharm. 2017, 525, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Pleguezuelos-Villa, M.; Mir-Palomo, S.; Díez-Sales, O.; Buso, M.A.O.V.; Sauri, A.R.; Nácher, A. A novel ultradeformable liposomes of Naringin for anti-inflammatory therapy. Coll. Surf. B Biointerfaces 2018, 162, 265–270. [Google Scholar] [CrossRef]
- Wu, P.-S.; Li, Y.-S.; Kuo, Y.-C.; Tsai, S.-J.J.; Lin, C.-C. Preparation and Evaluation of Novel Transfersomes Combined with the Natural Antioxidant Resveratrol. Molecules 2019, 24, 600. [Google Scholar] [CrossRef]
- Tosato, M.G.; Maya Girón, J.V.; Martin, A.A.; Krishna Tippavajhala, V.; Fernández Lorenzo de Mele, M.; Dicelio, L. Comparative study of transdermal drug delivery systems of resveratrol: High efficiency of deformable liposomes. Mater. Sci. Eng. C 2018, 90, 356–364. [Google Scholar] [CrossRef]
- Sultana, S.S.; Sailaja, A.K. Preparation and Evaluation of Naproxen Sodium Loaded Liposomes, Ethosomes and Transferosomes. J. Bionanosci. 2017, 11, 284–291. [Google Scholar] [CrossRef]
- Zhang, Z.-W.; Xu, X.-C.; Liu, T.; Yuan, S. Mitochondrion-Permeable Antioxidants to Treat ROS-Burst-Mediated Acute Diseases. Oxid. Med. Cell. Longev. 2016, 2016, 6859523. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, V.; Petronilli, V.; Ghelli, A.; Carelli, V.; Rugolo, M.; Lenaz, G.; Bernardi, P. The effects of idebenone on mitochondrial bioenergetics. Biochim. Biophys. Acta 2012, 1817, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, A.; Bonacci, S.; Paolino, D.; Celia, C.; Procopio, A.; Fresta, M.; Cosco, D. Paclitaxel-loaded sodium deoxycholate-stabilized zein nanoparticles: characterization and in vitro cytotoxicity. Heliyon 2019, 5, e02422. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, A.; Paolino, D.; Iannone, M.; Palma, E.; Fresta, M.; Cosco, D. Sodium deoxycholate-decorated zein nanoparticles for a stable colloidal drug delivery system. Int. J. Nanomed. 2018, 13, 601–614. [Google Scholar] [CrossRef]
- Caddeo, C.; Manconi, M.; Sinico, C.; Valenti, D.; Celia, C.; Monduzzi, M.; Fadda, A.M. Penetration Enhancer-Containing Vesicles: Does the Penetration Enhancer Structure Affect Topical Drug Delivery? Curr. Drug Targets 2015, 16, 1438–1447. [Google Scholar] [CrossRef]
- Paolino, D.; Cosco, D.; Cilurzo, F.; Trapasso, E.; Morittu, V.M.; Celia, C.; Fresta, M. Improved in vitro and in vivo collagen biosynthesis by asiaticoside-loaded ultradeformable vesicles. J. Control. Release 2012, 162, 143–151. [Google Scholar] [CrossRef]
- Kligman, A.M.; Christophers, E. Preparation of Isolated Sheets of Human Stratum Corneum. Arch. Dermatol. 1963, 88, 702–705. [Google Scholar] [CrossRef]
- Bronaugh, R.L.; Stewart, R.F.; Simon, M. Methods for in vitro percutaneous absorption studies. VII: Use of excised human skin. J. Pharm. Sci. 1986, 75, 1094–1097. [Google Scholar] [CrossRef]
- Ameen, D.; Michniak-Kohn, B. Development and in vitro evaluation of pressure sensitive adhesive patch for the transdermal delivery of galantamine: Effect of penetration enhancers and crystallization inhibition. Eur. J. Pharm. Biopharm. 2019, 139, 262–271. [Google Scholar] [CrossRef]
- Nguyen, H.X.; Puri, A.; Bhattaccharjee, S.A.; Banga, A.K. Qualitative and quantitative analysis of lateral diffusion of drugs in human skin. Int. J. Pharm. 2018, 544, 62–74. [Google Scholar] [CrossRef]
- Paolino, D.; Ventura, C.A.; Nistico, S.; Puglisi, G.; Fresta, M. Lecithin microemulsions for the topical administration of ketoprofen: Percutaneous adsorption through human skin and in vivo human skin tolerability. Int. J. Pharm. 2002, 244, 21–31. [Google Scholar] [CrossRef]
- Paolino, D.; Lucania, G.; Mardente, D.; Alhaique, F.; Fresta, M. Ethosomes for skin delivery of ammonium glycyrrhizinate: In vitro percutaneous permeation through human skin and in vivo anti-inflammatory activity on human volunteers. J. Control. Release 2005, 106, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Cosco, D.; Federico, C.; Maiuolo, J.; Bulotta, S.; Molinaro, R.; Paolino, D.; Tassone, P.; Fresta, M. Physicochemical features and transfection properties of chitosan/poloxamer 188/poly(d,l-lactide-co-glycolide) nanoplexes. Int. J. Nanomed. 2014, 9, 2359. [Google Scholar] [CrossRef] [PubMed]
- Cosco, D.; Paolino, D.; De Angelis, F.; Cilurzo, F.; Celia, C.; Di Marzio, L.; Russo, D.; Tsapis, N.; Fattal, E.; Fresta, M. Aqueous-core PEG-coated PLA nanocapsules for an efficient entrapment of water soluble anticancer drugs and a smart therapeutic response. Eur. J. Pharm. Biopharm. 2015, 89, 30–39. [Google Scholar] [CrossRef]
- Molinaro, R.; Evangelopoulos, M.; Hoffman, J.R.; Corbo, C.; Taraballi, F.; Martinez, J.O.; Hartman, K.A.; Cosco, D.; Costa, G.; Romeo, I.; et al. Design and Development of Biomimetic Nanovesicles Using a Microfluidic Approach. Adv. Mater. 2018, 30, e1702749. [Google Scholar] [CrossRef]
- Jain, S.; Jain, P.; Umamaheshwari, R.B.; Jain, N.K. Transfersomes—A novel vesicular carrier for enhanced transdermal delivery: Development, characterization, and performance evaluation. Drug Dev. Ind. Pharm. 2003, 29, 1013–1026. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | P90G 1 (% w/w) | SDC 2 (% w/w) | T80 3 (% w/w) | IDE 4 (% w/w) | NS 5 (% w/v) |
|---|---|---|---|---|---|
| 1 | 85 | - | 15 | - | |
| 1A | 80 | - | 15 | 5 | - |
| 1 B | 75 | - | 15 | 10 | - |
| 1C | 85 | - | 15 | - | 0.4 |
| 1D | 85 | - | 15 | - | 1 |
| 1E | 75 | - | 15 | 10 | 1 |
| 2 | 90 | 10 | - | - | - |
| 2A | 85 | 10 | - | 5 | - |
| 2B | 80 | 10 | - | 10 | - |
| 2C | 90 | 10 | - | - | 0.4 |
| 2D | 90 | 10 | - | - | 1 |
| 2E | 80 | 10 | - | 10 | 1 |
| Sample | Mean Sizes (nm) | Polydispersity Index | Zeta Potential (mV) |
|---|---|---|---|
| 1 | 136 ± 3 | 0.13 ± 0.01 | −26 ± 2 |
| 1A | 137 ± 2 | 0.10 ± 0.01 | −33 ± 1 |
| 1B | 134 ± 3 | 0.08 ± 0.03 | −36 ± 2 |
| 1C | 141 ± 3 | 0.09 ± 0.01 | −24 ± 1 |
| 1D | 131 ± 2 | 0.07 ± 0.01 | −26 ± 1 |
| 1E | 128 ± 3 | 0.09 ± 0.01 | −20 ± 1 |
| 2 | 137 ± 2 | 0.11 ± 0.01 | −37 ± 1 |
| 2A | 144 ± 4 | 0.11 ± 0.01 | −33 ± 1 |
| 2B | 140 ± 3 | 0.08 ± 0.01 | −40 ± 1 |
| 2C | 137 ± 3 | 0.07 ± 0.02 | −37 ± 1 |
| 2D | 126 ± 2 | 0.12 ± 0.00 | −39 ± 1 |
| 2E | 132 ± 2 | 0.08 ± 0.01 | −27 ± 1 |
| Formulation | IDE EE% ± SD | NS EE% ± SD |
|---|---|---|
| 1 | - | - |
| 1A | 73 ± 1 | - |
| 1B | 82 ± 1 | - |
| 1C | - | 59 ± 1 |
| 1D | - | 30 ± 1 |
| 1E | 81 ± 1 | 40 ± 1 |
| 2 | - | - |
| 2A | 51 ± 1 | - |
| 2B | 72 ± 1 | - |
| 2C | - | 44 ± 1 |
| 2D | - | 20 ± 1 |
| 2E | 72 ± 1 | 29 ± 1 |
| Formulation | d0 (nm) | d1 (nm) | DI |
|---|---|---|---|
| CTRL | 165 ± 1 | 90 ± 1 | 17.75 |
| 1 | 136 ± 1 | 126 ± 1 | 109.73 |
| 1A | 141 ± 1 | 133 ± 1 | 142.21 |
| 1B | 131 ± 3 | 125 ± 1 | 176.16 |
| 1C | 137 ± 1 | 125 ± 2 | 92.12 |
| 1D | 134 ± 1 | 121 ± 1 | 83.17 |
| 1E | 128 ± 2 | 122 ± 1 | 172.56 |
| 2 | 140 ± 1 | 110 ± 2 | 37.65 |
| 2A | 137 ± 1 | 115 ± 1 | 50.25 |
| 2B | 126 ± 1 | 113 ± 1.2 | 78.20 |
| 2C | 144 ± 1 | 112 ± 0.4 | 36.31 |
| 2D | 140 ± 2 | 108 ± 1.1 | 35.30 |
| 2E | 132 ± 1 | 118 ± 0.4 | 76.08 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molinaro, R.; Gagliardi, A.; Mancuso, A.; Cosco, D.; Soliman, M.E.; Casettari, L.; Paolino, D. Development and In Vivo Evaluation of Multidrug Ultradeformable Vesicles for the Treatment of Skin Inflammation. Pharmaceutics 2019, 11, 644. https://doi.org/10.3390/pharmaceutics11120644
Molinaro R, Gagliardi A, Mancuso A, Cosco D, Soliman ME, Casettari L, Paolino D. Development and In Vivo Evaluation of Multidrug Ultradeformable Vesicles for the Treatment of Skin Inflammation. Pharmaceutics. 2019; 11(12):644. https://doi.org/10.3390/pharmaceutics11120644
Chicago/Turabian StyleMolinaro, Roberto, Agnese Gagliardi, Antonia Mancuso, Donato Cosco, Mahmoud E. Soliman, Luca Casettari, and Donatella Paolino. 2019. "Development and In Vivo Evaluation of Multidrug Ultradeformable Vesicles for the Treatment of Skin Inflammation" Pharmaceutics 11, no. 12: 644. https://doi.org/10.3390/pharmaceutics11120644
APA StyleMolinaro, R., Gagliardi, A., Mancuso, A., Cosco, D., Soliman, M. E., Casettari, L., & Paolino, D. (2019). Development and In Vivo Evaluation of Multidrug Ultradeformable Vesicles for the Treatment of Skin Inflammation. Pharmaceutics, 11(12), 644. https://doi.org/10.3390/pharmaceutics11120644