Microfluidics-Assisted Size Tuning and Biological Evaluation of PLGA Particles



Abstract
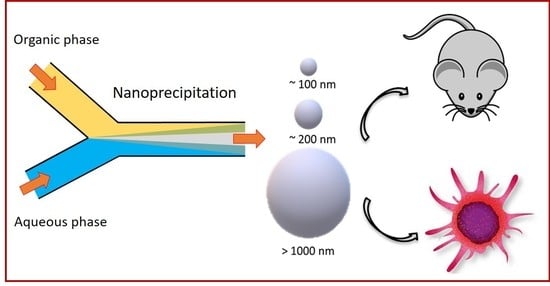
1. Introduction
2. Experimental
2.1. Materials
2.2. Equipment
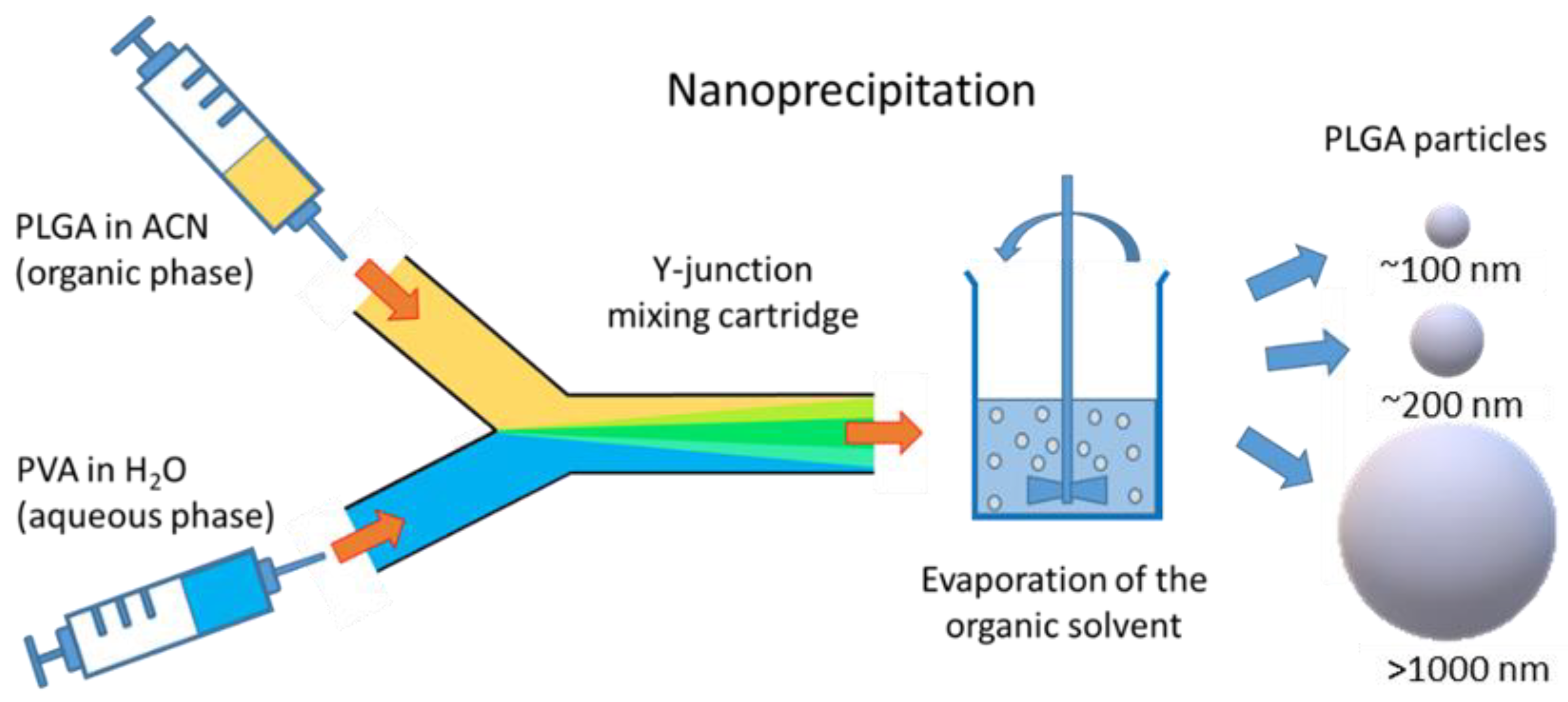
2.3. Preparation of PLGA Particles
2.4. Colloidal Characterization of PLGA Particles
2.5. Determination of Encapsulation Efficiency
2.6. In Situ Release Profile
3. In Vitro Cellular Uptake Experiments
3.1. Generation of Bone Marrow-Derived Dendritic Cells (BMDCs)
3.2. Isolation of Myeloid-Derived Suppressor Cells (mMDSCs) and Polymorphonuclear Myeloid-Derived Suppressor Cells (pmnMDSCs)
3.3. In Vitro Cellular Uptake
4. In Vivo Clearance Studies
Statistical Analysis
5. Results and Discussion
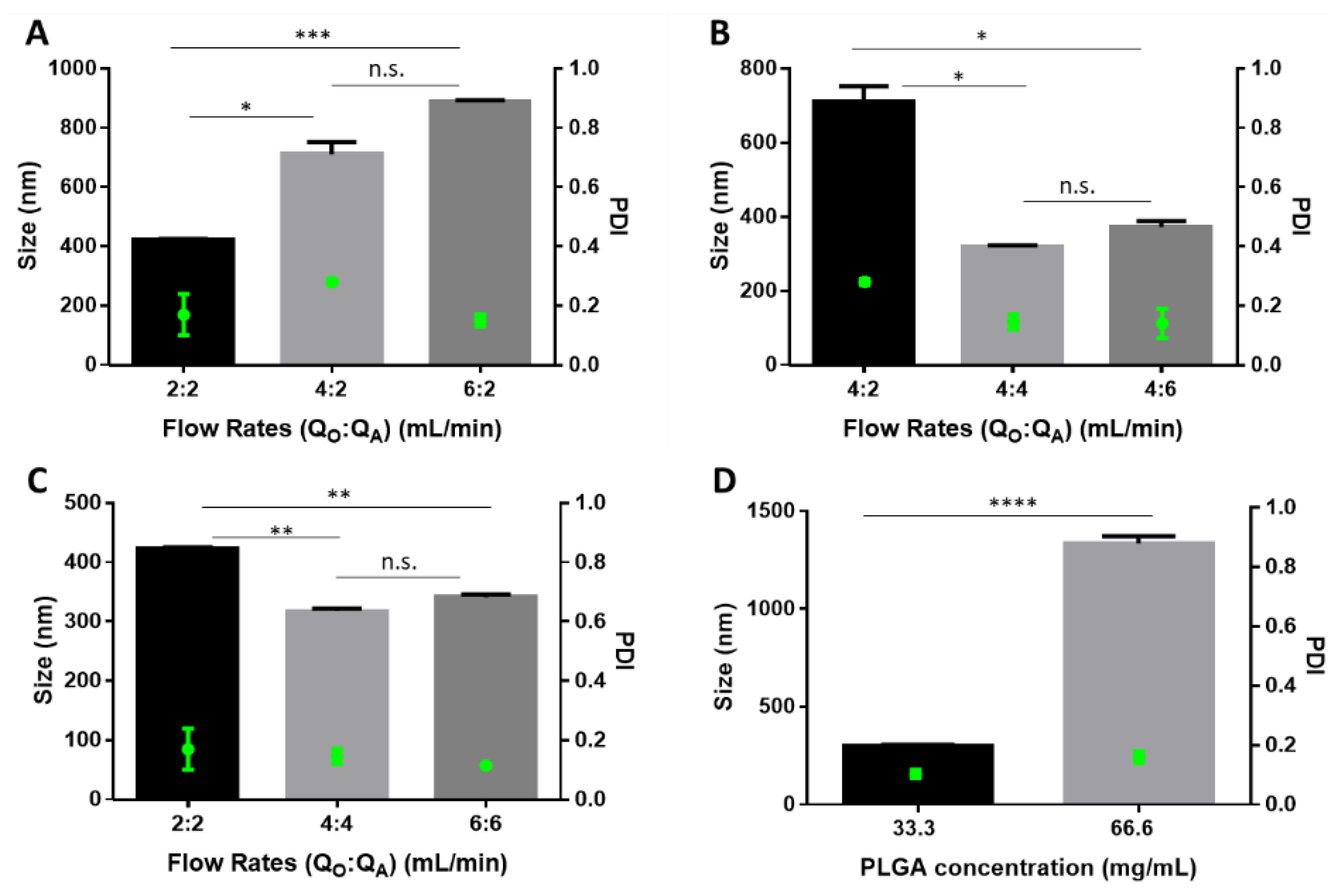
5.1. Preparation of Particles
5.2. Colloidal Characterization
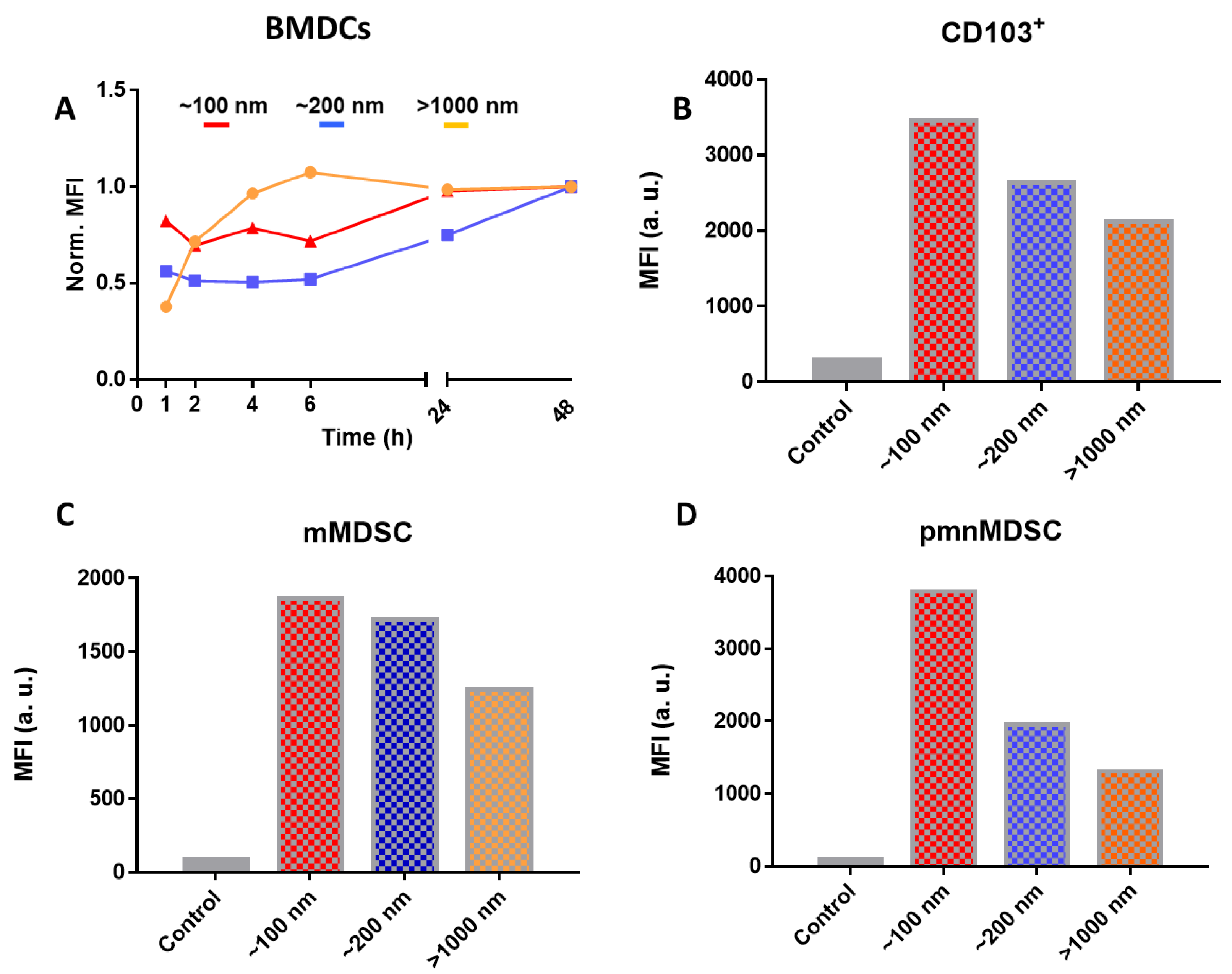
5.3. In Vitro Uptake Experiments
5.4. In Vivo Clearance of Particles
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Khalid, M.; El-Sawy, H.S. Polymeric nanoparticles: Promising platform for drug delivery. Int. J. Pharm. 2017, 528, 675–691. [Google Scholar]
- Kumari, A.; Yadav, S.K.; Yadav, S.C. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf. B Biointerfaces 2010, 75, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Ud Din, F.; Aman, W.; Ullah, I.; Qureshi, O.S.; Mustapha, O.; Shafique, S.; Zeb, A. Effective use of nanocarriers as drug delivery systems for the treatment of selected tumors. Int. J. Nanomed. 2017, 12, 7291. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.M.; Valencia, P.M.; Zhang, L.; Langer, R.; Farokhzad, O.C. Polymeric nanoparticles for drug delivery. In Cancer Nanotechnology; Springer: Berlin/Heidelberg, Germany, 2010; pp. 163–175. [Google Scholar]
- Hines, D.J.; Kaplan, D.L. Poly (lactic-co-glycolic) acid—controlled-release systems: Experimental and modeling insights. Crit. Rev. Ther. Drug Carr. Syst. 2013, 30, 257–276. [Google Scholar] [CrossRef]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Préat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef]
- Makadia, H.K.; Siegel, S.J. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef]
- Operti, M.C.; Fecher, D.; van Dinther, E.A.; Grimm, S.; Jaber, R.; Figdor, C.G.; Tagit, O. A comparative assessment of continuous production techniques to generate sub-micron size PLGA particles. Int. J. Pharm. 2018, 550, 140–148. [Google Scholar] [CrossRef]
- Derakhshandeh, K.; Erfan, M.; Dadashzadeh, S. Encapsulation of 9-nitrocamptothecin, a novel anticancer drug, in biodegradable nanoparticles: Factorial design, characterization and release kinetics. Eur. J. Pharm. Biopharm. 2007, 66, 34–41. [Google Scholar] [CrossRef]
- Fonseca, C.; Simoes, S.; Gaspar, R. Paclitaxel-loaded PLGA nanoparticles: Preparation, physicochemical characterization and in vitro anti-tumoral activity. J. Control. Release 2002, 83, 273–286. [Google Scholar] [CrossRef]
- Jose, S.; Juna, B.; Cinu, T.; Jyoti, H.; Aleykutty, N. Carboplatin loaded Surface modified PLGA nanoparticles: Optimization, characterization, and in vivo brain targeting studies. Colloids Surf. B Biointerfaces 2016, 142, 307–314. [Google Scholar] [CrossRef]
- Khan, I.; Gothwal, A.; Sharma, A.K.; Kesharwani, P.; Gupta, L.; Iyer, A.K.; Gupta, U. PLGA nanoparticles and their versatile role in anticancer drug delivery. Crit. Rev. Ther. Drug Carr. Syst. 2016, 33, 159–193. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-B.; Liu, P.; Shao, F.-M.; Miao, Q.-L. Formulation and evaluation of PLGA nanoparticles loaded capecitabine for prostate cancer. Int. J. Clin. Exp. Med. 2015, 8, 19670. [Google Scholar] [PubMed]
- Feczkó, T.; Tóth, J.; Dósa, G.; Gyenis, J. Optimization of protein encapsulation in PLGA nanoparticles. Chem. Eng. Process. Process. Intensif. 2011, 50, 757–765. [Google Scholar] [CrossRef]
- Mohammadi-Samani, S.; Taghipour, B. PLGA micro and nanoparticles in delivery of peptides and proteins; problems and approaches. Pharm. Dev. Technol. 2015, 20, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Pirooznia, N.; Hasannia, S.; Lotfi, A.S.; Ghanei, M. Encapsulation of alpha-1 antitrypsin in PLGA nanoparticles: In vitro characterization as an effective aerosol formulation in pulmonary diseases. J. Nanobiotechnol. 2012, 10, 20. [Google Scholar] [CrossRef] [PubMed]
- Rescignano, N.; Tarpani, L.; Tiribuzi, R.; Montesano, S.; Martino, S.; Latterini, L.; Kenny, J.M.; Armentano, I. Protein encapsulation in biodegradable polymeric nanoparticles: Morphology, fluorescence behaviour and stem cell uptake. Macromol. Biosci. 2013, 13, 1204–1212. [Google Scholar] [CrossRef]
- Santander-Ortega, M.J.; Csaba, N.; González, L.; Bastos-González, D.; Ortega-Vinuesa, J.L.; Alonso, M.J. Protein-loaded PLGA–PEO blend nanoparticles: Encapsulation, release and degradation characteristics. Colloid Polym. Sci. 2010, 288, 141–150. [Google Scholar] [CrossRef]
- Lü, J.-M.; Liang, Z.; Wang, X.; Gu, J.; Yao, Q.; Chen, C. New polymer of lactic-co-glycolic acid-modified polyethylenimine for nucleic acid delivery. Nanomedicine 2016, 11, 1971–1991. [Google Scholar] [CrossRef]
- Patil, Y.B.; Swaminathan, S.K.; Sadhukha, T.; Ma, L.; Panyam, J. The use of nanoparticle-mediated targeted gene silencing and drug delivery to overcome tumor drug resistance. Biomaterials 2010, 31, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Harguindey, A.; Domaille, D.W.; Fairbanks, B.D.; Wagner, J.; Bowman, C.N.; Cha, J.N. Synthesis and Assembly of Click-Nucleic-Acid-Containing PEG–PLGA Nanoparticles for DNA Delivery. Adv. Mater. 2017, 29, 1700743. [Google Scholar] [CrossRef]
- Cun, D.; Jensen, D.K.; Maltesen, M.J.; Bunker, M.; Whiteside, P.; Scurr, D.; Foged, C.; Nielsen, H.M. High loading efficiency and sustained release of siRNA encapsulated in PLGA nanoparticles: Quality by design optimization and characterization. Eur. J. Pharm. Biopharm. 2011, 77, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Colombo, S.; Cun, D.; Remaut, K.; Bunker, M.; Zhang, J.; Martin-Bertelsen, B.; Yaghmur, A.; Braeckmans, K.; Nielsen, H.M.; Foged, C. Mechanistic profiling of the siRNA delivery dynamics of lipid–polymer hybrid nanoparticles. J. Control. Release 2015, 201, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.-M.; Lee, S.J.; Kim, Y.S.; Lee, M.H.; Cha, G.S.; Jung, I.D.; Kang, T.H.; Han, H.D. Nanoparticle-based vaccine delivery for cancer immunotherapy. Immune Netw. 2013, 13, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Cody, V.; Saucier-Sawyer, J.K.; Saltzman, W.M.; Sasaki, C.T.; Edelson, R.L.; Birchall, M.A.; Hanlon, D.J. Polymer nanoparticles containing tumor lysates as antigen delivery vehicles for dendritic cell–based antitumor immunotherapy. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 1–10. [Google Scholar] [CrossRef]
- Ma, T.; Wang, L.; Yang, T.; Ma, G.; Wang, S. Homogeneous PLGA-lipid nanoparticle as a promising oral vaccine delivery system for ovalbumin. Asian J. Pharm. Sci. 2014, 9, 129–136. [Google Scholar] [CrossRef]
- Dölen, Y.; Kreutz, M.; Gileadi, U.; Tel, J.; Vasaturo, A.; van Dinther, E.A.; van Hout-Kuijer, M.A.; Cerundolo, V.; Figdor, C.G. Co-delivery of PLGA encapsulated invariant NKT cell agonist with antigenic protein induce strong T cell-mediated antitumor immune responses. Oncoimmunology 2016, 5, e1068493. [Google Scholar] [CrossRef]
- Clawson, C.; Huang, C.-T.; Futalan, D.; Seible, D.M.; Saenz, R.; Larsson, M.; Ma, W.; Minev, B.; Zhang, F.; Ozkan, M. Delivery of a peptide via poly (d, l-lactic-co-glycolic) acid nanoparticles enhances its dendritic cell–stimulatory capacity. Nanomed. Nanotechnol. Biol. Med. 2010, 6, 651–661. [Google Scholar] [CrossRef]
- Allahyari, M.; Mohit, E. Peptide/protein vaccine delivery system based on PLGA particles. Hum. Vaccines Immunother. 2016, 12, 806–828. [Google Scholar] [CrossRef]
- Hoshyar, N.; Gray, S.; Han, H.; Bao, G. The effect of nanoparticle size on in vivo pharmacokinetics and cellular interaction. Nanomedicine 2016, 11, 673–692. [Google Scholar] [CrossRef]
- Han, F.Y.; Thurecht, K.J.; Whittaker, A.K.; Smith, M.T. Bioerodable PLGA-based microparticles for producing sustained-release drug formulations and strategies for improving drug loading. Front. Pharmacol. 2016, 7, 185. [Google Scholar] [CrossRef]
- Hashizume, H.; Baluk, P.; Morikawa, S.; McLean, J.W.; Thurston, G.; Roberge, S.; Jain, R.K.; McDonald, D.M. Openings between defective endothelial cells explain tumor vessel leakiness. Am. J. Pathol. 2000, 156, 1363–1380. [Google Scholar] [CrossRef]
- Toy, R.; Hayden, E.; Shoup, C.; Baskaran, H.; Karathanasis, E. The effects of particle size, density and shape on margination of nanoparticles in microcirculation. Nanotechnology 2011, 22, 115101. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, S.M.; Parhamifar, L.; Ahmadvand, D.; Wibroe, P.P.; Andresen, T.; Farhangrazi, Z.; Hunter, A. Particulate systems for targeting of macrophages: Basic and therapeutic concepts. J. Innate Immun. 2012, 4, 509–528. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Bagby, T.R.; Cohen, M.S.; Forrest, M.L. Drug delivery to the lymphatic system: Importance in future cancer diagnosis and therapies. Expert Opin. Drug Deliv. 2009, 6, 785–792. [Google Scholar] [CrossRef]
- Bazile, D.; Ropert, C.; Huve, P.; Verrecchia, T.; Mariard, M.; Frydman, A.; Veillard, M.; Spenlehauer, G. Body distribution of fully biodegradable [14C]-poly (lactic acid) nanoparticles coated with albumin after parenteral administration to rats. Biomaterials 1992, 13, 1093–1102. [Google Scholar] [CrossRef]
- Verhoef, J.J.; Anchordoquy, T.J. Questioning the use of PEGylation for drug delivery. Drug Deliv. Transl. Res. 2013, 3, 499–503. [Google Scholar] [CrossRef]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef]
- Schöttler, S.; Becker, G.; Winzen, S.; Steinbach, T.; Mohr, K.; Landfester, K.; Mailänder, V.; Wurm, F.R. Protein adsorption is required for stealth effect of poly (ethylene glycol)-and poly (phosphoester)-coated nanocarriers. Nat. Nanotechnol. 2016, 11, 372. [Google Scholar] [CrossRef]
- Simon, J.; Müller, L.K.; Kokkinopoulou, M.; Lieberwirth, I.; Morsbach, S.; Landfester, K.; Mailänder, V. Exploiting the biomolecular corona: Pre-coating of nanoparticles enables controlled cellular interactions. Nanoscale 2018, 10, 10731–10739. [Google Scholar] [CrossRef]
- Morikawa, Y.; Tagami, T.; Hoshikawa, A.; Ozeki, T. The use of an efficient microfluidic mixing system for generating stabilized polymeric nanoparticles for controlled drug release. Biol. Pharm. Bull. 2018, 41, 899–907. [Google Scholar] [CrossRef]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Hasanzadeh Davarani, F.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M. Impact of particle size and polydispersity index on the clinical applications of lipidic nanocarrier systems. Pharmaceutics 2018, 10, 57. [Google Scholar] [CrossRef] [PubMed]
- Bahari, L.A.S.; Hamishehkar, H. The impact of variables on particle size of solid lipid nanoparticles and nanostructured lipid carriers; a comparative literature review. Adv. Pharm. Bull. 2016, 6, 143. [Google Scholar] [CrossRef] [PubMed]
- Chiesa, E.; Dorati, R.; Pisani, S.; Conti, B.; Bergamini, G.; Modena, T.; Genta, I. The microfluidic technique and the manufacturing of polysaccharide nanoparticles. Pharmaceutics 2018, 10, 267. [Google Scholar] [CrossRef] [PubMed]
- Whitesides, G.M. The origins and the future of microfluidics. Nature 2006, 442, 368. [Google Scholar] [CrossRef]
- Mark, D.; Haeberle, S.; Roth, G.; Von Stetten, F.; Zengerle, R. Microfluidic lab-on-a-chip platforms: Requirements, characteristics and applications. In Microfluidics Based Microsystems; Springer: Berlin/Heidelberg, Germany, 2010; pp. 305–376. [Google Scholar]
- Amoyav, B.; Benny, O. Controlled and tunable polymer particles’ production using a single microfluidic device. Appl. Nanosci. 2018, 8, 905–914. [Google Scholar] [CrossRef]
- Dashtimoghadam, E.; Fahimipour, F.; Davaji, B.; Hasani-Sadrabadi, M.; Tayebi, L. Microfluidic-directed synthesis of polymeric nanoparticles for bone cancer therapy. Dent. Mater. 2016, 1, e59–e60. [Google Scholar] [CrossRef]
- De Solorzano, I.O.; Uson, L.; Larrea, A.; Miana, M.; Sebastian, V.; Arruebo, M. Continuous synthesis of drug-loaded nanoparticles using microchannel emulsification and numerical modeling: Effect of passive mixing. Int. J. Nanomed. 2016, 11, 3397. [Google Scholar]
- Khan, I.U.; Serra, C.A.; Anton, N.; Vandamme, T.F. Production of nanoparticle drug delivery systems with microfluidics tools. Expert Opin. Drug Deliv. 2015, 12, 547–562. [Google Scholar] [CrossRef]
- Xie, H.; Smith, J.W. Fabrication of PLGA nanoparticles with a fluidic nanoprecipitation system. J. Nanobiotechnol. 2010, 8, 18. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, S.; Machado, A.; Lecommandoux, S.; Sandre, O.; Gu, F.; Colin, A. Controllable microfluidic production of drug-loaded PLGA nanoparticles using partially water-miscible mixed solvent microdroplets as a precursor. Sci. Rep. 2017, 7, 4794. [Google Scholar] [CrossRef]
- Gdowski, A.; Johnson, K.; Shah, S.; Gryczynski, I.; Vishwanatha, J.; Ranjan, A. Optimization and scale up of microfluidic nanolipomer production method for preclinical and potential clinical trials. J. Nanobiotechnol. 2018, 16, 12. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Zhang, C. Tuning the Size of Poly (lactic-co-glycolic Acid)(PLGA) Nanoparticles Fabricated by Nanoprecipitation. Biotechnol. J. 2018, 13, 1700203. [Google Scholar] [CrossRef] [PubMed]
- Halayqa, M.; Domańska, U. PLGA biodegradable nanoparticles containing perphenazine or chlorpromazine hydrochloride: Effect of formulation and release. Int. J. Mol. Sci. 2014, 15, 23909–23923. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Si, W.; Huang, C.; Ding, K.; Huang, W.; Chen, P.; Zhang, Q.; Dong, X. An aza-BODIPY photosensitizer for photoacoustic and photothermal imaging guided dual modal cancer phototherapy. J. Mater. Chem. B 2017, 5, 1566–1573. [Google Scholar] [CrossRef]
- Trofymchuk, K.; Valanciunaite, J.; Andreiuk, B.; Reisch, A.; Collot, M.; Klymchenko, A.S. BODIPY-loaded polymer nanoparticles: Chemical structure of cargo defines leakage from nanocarrier in living cells. J. Mater. Chem. B 2019, 7, 5199–5210. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, N.K.; Keliher, E.J.; Thurber, G.M.; Nahrendorf, M.; Weissleder, R. 18F labeled nanoparticles for in vivo PET-CT imaging. Bioconjug. Chem. 2009, 20, 397–401. [Google Scholar] [CrossRef]
- Elsabahy, M.; Heo, G.S.; Lim, S.-M.; Sun, G.; Wooley, K.L. Polymeric nanostructures for imaging and therapy. Chem. Rev. 2015, 115, 10967–11011. [Google Scholar] [CrossRef]
- Mura, S.; Hillaireau, H.; Nicolas, J.; Le Droumaguet, B.; Gueutin, C.; Zanna, S.; Tsapis, N.; Fattal, E. Influence of surface charge on the potential toxicity of PLGA nanoparticles towards Calu-3 cells. Int. J. Nanomed. 2011, 6, 2591. [Google Scholar]
- De Jesus Gomes, A.; Lunardi, C.N.; Caetano, F.H.; Lunardi, L.O.; da Hora Machado, A.E. Phagocytosis of PLGA microparticles in rat peritoneal exudate cells: A time-dependent study. Microsc. Microanal. 2006, 12, 399–405. [Google Scholar] [CrossRef]
- Pisani, E.; Fattal, E.; Paris, J.; Ringard, C.; Rosilio, V.; Tsapis, N. Surfactant dependent morphology of polymeric capsules of perfluorooctyl bromide: Influence of polymer adsorption at the dichloromethane–water interface. J. Colloid Interface Sci. 2008, 326, 66–71. [Google Scholar] [CrossRef]
- Honary, S.; Zahir, F. Effect of zeta potential on the properties of nano-drug delivery systems—A review (Part 1). Trop. J. Pharm. Res. 2013, 12, 255–264. [Google Scholar]
- Jyothi, N.V.N.; Prasanna, P.M.; Sakarkar, S.N.; Prabha, K.S.; Ramaiah, P.S.; Srawan, G. Microencapsulation techniques, factors influencing encapsulation efficiency. J. Microencapsul. 2010, 27, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Fredenberg, S.; Wahlgren, M.; Reslow, M.; Axelsson, A. The mechanisms of drug release in poly (lactic-co-glycolic acid)-based drug delivery systems—A review. Int. J. Pharm. 2011, 415, 34–52. [Google Scholar] [CrossRef] [PubMed]
- Siepmann, J.; Elkharraz, K.; Siepmann, F.; Klose, D. How autocatalysis accelerates drug release from PLGA-based microparticles: A quantitative treatment. Biomacromolecules 2005, 6, 2312–2319. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Palazzo, A.; Hennink, W.E.; Kok, R.J. Effect of particle size on drug loading and release kinetics of gefitinib-loaded PLGA microspheres. Mol. Pharm. 2016, 14, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Salifu, M.; Sirianni, R.W.; Stabenfeldt, S.E. Tailoring sub-micron PLGA particle release profiles via centrifugal fractioning. J. Biomed. Mater. Res. Part A 2016, 104, 688–696. [Google Scholar] [CrossRef]
- Moayedian, T.; Mosaffa, F.; Khameneh, B.; Tafaghodi, M. Combined effects of PEGylation and particle size on uptake of PLGA particles by macrophage cells. Nanomed. J. 2015, 2, 299–304. [Google Scholar]
- Swider, E.; Maharjan, S.; Houkes, K.; van Riessen, N.K.; Figdor, C.; Srinivas, M.; Tagit, O. Förster Resonance Energy Transfer-Based Stability Assessment of PLGA Nanoparticles in Vitro and in Vivo. ACS Appl. Bio Mater. 2019, 2, 1131–1140. [Google Scholar] [CrossRef]
- Foged, C.; Brodin, B.; Frokjaer, S.; Sundblad, A. Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model. Int. J. Pharm. 2005, 298, 315–322. [Google Scholar] [CrossRef]
- Tonigold, M.; Mailänder, V. Endocytosis and intracellular processing of nanoparticles in dendritic cells: Routes to effective immunonanomedicines. Future Med. 2016, 11, 2625–2630. [Google Scholar] [CrossRef]
- Cruz, L.J.; Tacken, P.J.; Fokkink, R.; Joosten, B.; Stuart, M.C.; Albericio, F.; Torensma, R.; Figdor, C.G. Targeted PLGA nano-but not microparticles specifically deliver antigen to human dendritic cells via DC-SIGN in vitro. J. Control. Release 2010, 144, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Helft, J.; Böttcher, J.; Chakravarty, P.; Zelenay, S.; Huotari, J.; Schraml, B.U.; Goubau, D.; e Sousa, C.R. GM-CSF mouse bone marrow cultures comprise a heterogeneous population of CD11c+ MHCII+ macrophages and dendritic cells. Immunity 2015, 42, 1197–1211. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Junca, A.; Driscoll, K.; Pellicciotta, I.; Du, S.; Lo, C.H.; Roy, R.; Parry, R.; Tenvooren, I.; Marquez, D.; Spitzer, M.H. Autocrine TGFβ is a Survival Factor for Monocytes and Drives Immunosuppressive Lineage Commitment. Cancer Immunol. Res. 2019, 7, 306–320. [Google Scholar] [CrossRef] [PubMed]
- Albanese, A.; Tang, P.S.; Chan, W.C. The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annu. Rev. Biomed. Eng. 2012, 14, 1–16. [Google Scholar] [CrossRef]
- Liu, X.; Huang, N.; Li, H.; Jin, Q.; Ji, J. Surface and size effects on cell interaction of gold nanoparticles with both phagocytic and nonphagocytic cells. Langmuir 2013, 29, 9138–9148. [Google Scholar] [CrossRef]
- Dreaden, E.C.; Austin, L.A.; Mackey, M.A.; El-Sayed, M.A. Size matters: Gold nanoparticles in targeted cancer drug delivery. Ther. Deliv. 2012, 3, 457–478. [Google Scholar] [CrossRef]
- Jeon, H.-J.; Jeong, Y.-I.; Jang, M.-K.; Park, Y.-H.; Nah, J.-W. Effect of solvent on the preparation of surfactant-free poly (DL-lactide-co-glycolide) nanoparticles and norfloxacin release characteristics. Int. J. Pharm. 2000, 207, 99–108. [Google Scholar] [CrossRef]
- Kreuter, J. Nanoparticle-based dmg delivery systems. J. Control. Release 1991, 16, 169–176. [Google Scholar] [CrossRef]
- Zuckerman, J.E.; Choi, C.H.J.; Han, H.; Davis, M.E. Polycation-siRNA nanoparticles can disassemble at the kidney glomerular basement membrane. Proc. Natl. Acad. Sci. USA 2012, 109, 3137–3142. [Google Scholar] [CrossRef]
- Kulkarni, S.A.; Feng, S.-S. Effects of particle size and surface modification on cellular uptake and biodistribution of polymeric nanoparticles for drug delivery. Pharm. Res. 2013, 30, 2512–2522. [Google Scholar] [CrossRef]
- Jindal, A.B. Nanocarriers for spleen targeting: Anatomo-physiological considerations, formulation strategies and therapeutic potential. Drug Deliv. Transl. Res. 2016, 6, 473–485. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | >1000 nm | ~200 nm | ~100 nm |
|---|---|---|---|
| PLGA (mg/mL) | 66.6 | 33.3 | 16.7 |
| PLGA:PEG-PLGA ratio | 70:30 | 70:30 | 70:30 |
| PVA (w/v%) | 3% | 1% | 1% |
| Flow rates (mL/min) (QO:QA) | 6:2 | 2:6 | 4:6 |
| Fluorescent dye (v/v%) | 1% | 1% | 1% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Operti, M.C.; Dölen, Y.; Keulen, J.; van Dinther, E.A.W.; Figdor, C.G.; Tagit, O. Microfluidics-Assisted Size Tuning and Biological Evaluation of PLGA Particles. Pharmaceutics 2019, 11, 590. https://doi.org/10.3390/pharmaceutics11110590
Operti MC, Dölen Y, Keulen J, van Dinther EAW, Figdor CG, Tagit O. Microfluidics-Assisted Size Tuning and Biological Evaluation of PLGA Particles. Pharmaceutics. 2019; 11(11):590. https://doi.org/10.3390/pharmaceutics11110590
Chicago/Turabian StyleOperti, Maria Camilla, Yusuf Dölen, Jibbe Keulen, Eric A. W. van Dinther, Carl G. Figdor, and Oya Tagit. 2019. "Microfluidics-Assisted Size Tuning and Biological Evaluation of PLGA Particles" Pharmaceutics 11, no. 11: 590. https://doi.org/10.3390/pharmaceutics11110590
APA StyleOperti, M. C., Dölen, Y., Keulen, J., van Dinther, E. A. W., Figdor, C. G., & Tagit, O. (2019). Microfluidics-Assisted Size Tuning and Biological Evaluation of PLGA Particles. Pharmaceutics, 11(11), 590. https://doi.org/10.3390/pharmaceutics11110590